

Abstract
Background
Pancreatic acinar cell carcinoma (ACC) is a rare tumor entity with an unfavorable prognosis. Recent whole-exome sequencing identified p53 mutations in a subset of human ACC. Activation of the mammalian target of rapamycin (mTOR) pathway is associated with various pancreatic neoplasms. We thus aimed at analyzing whether activation of mTOR with a concomitant loss of p53 may initiate ACC.
Methods
We generated transgenic mouse models in which mTOR was hyperactivated through pancreas-specific, homozygous tuberous sclerosis 1 (Tsc1) deficiency, with or without deletion of p53 (Tsc1-/- and Tsc1-/-; p53-/-). Activity of mTOR signaling was investigated using mouse tissues and isolated murine cell lines. Human ACC specimens were used to corroborate the findings from the transgenic mouse models.
Results
Hyperactive mTOR signaling in Tsc1-/- mice was not oncogenic but rather induced a near-complete loss of the pancreatic acinar compartment. Acinar cells were lost as a result of apoptosis which was associated with p53 activation. Concomitantly, ductal cells were enriched. Ablation of p53 in Tsc1-deficient mice prevented acinar cell death but promoted formation of acinar cells with severe nuclear abnormalities. One out of seven Tsc1-/-; p53-/- animals developed pancreatic tumors showing a distinctive tumor morphology, reminiscent of human ACC. Hyperactive mTOR signaling was also detected in a subset of human ACC.
Conclusion
Hyperactive mTOR signaling combined with loss of p53 in mice induces tumors similar to human ACC.
Electronic supplementary material
The online version of this article (doi:10.1186/s12943-015-0483-1) contains supplementary material, which is available to authorized users.
Keywords: Pancreatic cancer, p53, mTOR, ACC, Organ involution, Tissue homeostasis
Background
Pancreatic acinar cell carcinoma (ACC) is a rare exocrine cancer which accounts for 1–2 % of pancreatic malignancies [1, 2]. On histological and molecular levels, ACCs display certain features of normal pancreatic acini by expressing exocrine enzymes (e.g., trypsin) and by forming small glandular structures [3]. In comparison to pancreatic ductal adenocarcinoma (PDAC), ACCs tend to have a relatively favorable tumor biology which is more likely to respond to the available therapies [4–7]. Historically, it is conceived that ACCs do not share typical mutations of PDAC such as p53 [3, 8]. However, this view has recently been updated by whole-exome sequencing of ACCs [9]. Here, 13 % of ACC samples contained somatic p53 mutations and 39 % of them had a loss of heterozygosity at the p53 locus [9]. Therefore, it is likely that the p53 pathway actively participates in the development of a subset of ACCs.
The mammalian target of rapamycin (mTOR) is an atypical serine/threonine kinase that interacts with other proteins to form two functional complexes: mTORC1 and mTORC2 [10]. Particularly, mTORC1 (referred to hereinafter as mTOR) integrates a variety of intracellular and extracellular cues and is a key regulator of protein synthesis. It potentially influences carcinogenesis at various stages including cancer initiation and metastasis [11, 12]. The tuberous sclerosis 1 and 2 complex (Tsc1-Tsc2 complex) negatively regulates mTOR activity [13–16].
There is increasing evidence that mTOR activation is a common event in the development of various pancreatic tumors such as PDAC, a subset of cystic tumors and pancreatic neuroendocrine tumors (PNETs, [17–19]). Furthermore, we have previously demonstrated that the oncogenic effect of mTOR largely relies on its activation pattern: PI3K/Akt (phosphatidylinositol-4, 5-bisphosphate 3-kinase/protein kinase B)-mediated mTOR signaling induced cystic pancreatic lesions whereas oncogenic Mek/Erk-induced mTOR signaling promoted metastatic PDACs [18]. Since p53 interacts with mTOR signaling at multiple levels via the Tsc1-Tsc2 complex [20, 21], we aimed to analyze whether hyperactivated mTOR signaling with a loss of p53 may specify ACC.
Results
Tsc1 deficiency triggers loss of acinar cells in the exocrine pancreas
Previously, we generated pancreas-specific Tsc1-haploinsufficient (p48Cre/+; Tsc1fl/+) mice by crossing the Ptf1aCre/+ line (also known as p48Cre/+) with the Tsc1fl/fl line in which one allele of Tsc1 is specifically ablated in pancreatic progenitors during embryonic development and continuously deleted in pancreatic acinar cells in adulthood. However, neither overt pathologies nor hyperactive mTOR signaling was observed in the pancreata of these animals [18]. To further investigate the role of mTOR signaling in pancreatic exocrine physiology, we thus generated transgenic p48Cre/+; Tsc1fl/fl mice (herein referred to as Tsc1-/-). A complete loss of Tsc1 expression in the pancreata of these transgenic animals was previously confirmed [18]. The p48Cre/+; Tsc1fl/+ mice (herein referred to as Tsc1+/-) were used as controls.
All Tsc1-/- mice died between the age of 100 days and 224 days (n = 5; median survival: 133 days; Fig. 1a). Histological examination of Tsc1-/- pancreata revealed a destruction of the normal pancreatic acinar cell compartment (50–80 %) with a relative enrichment of ductal cells. However, no such changes were observed in Tsc1-/+ (n = 14) pancreata (Fig. 1b). The number of acinar cells was reduced and the residual acinar cells were surrounded by ductal cells in Tsc1-/- pancreata (α-amylase and Krt19 double-IF; Fig. 1c). Similar to previous observations in Tsc2-deficient mice [22], hyperglycemic episodes were observed in two Tsc1-/- mice by the time of death/sacrifice and the number of β cells was also significantly reduced (blood glucose levels of 370 and 590 mg/dl, respectively; normal: 160 to 200 mg/dl, insulin and glucagon double-IF; Fig. 1d). These data suggest that the Tsc1-/- mice suffered from both exocrine and endocrine insuffiency by the time of sacrifice. However, the Tsc1-/+ pancreata were histologically normal. Taken together, Tsc1 deficiency caused loss of functional parenchymal cells in both the exocrine and endocrine compartment of the pancreas. However, Tsc1 haploinsufficiency did not result in a gross pathological phenotype.
Fig. 1.
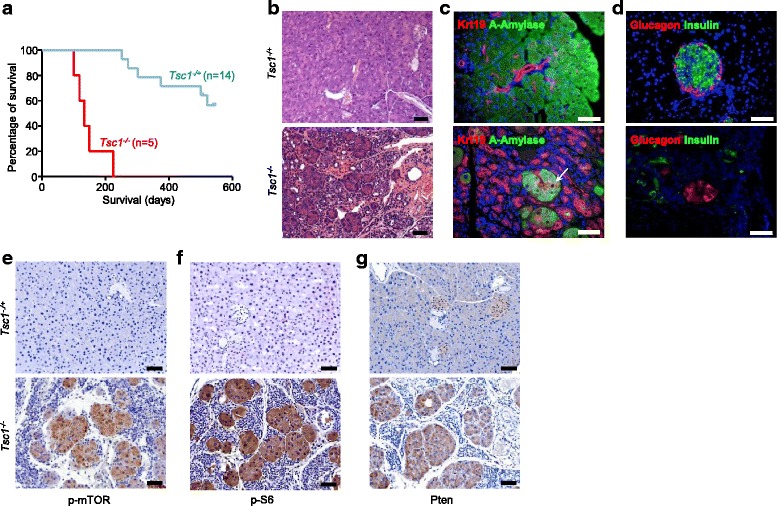
Tsc1 deficiency triggers loss of acinar cells in the exocrine pancreas. (a) Kaplan–Meier survival analysis shows Tsc1 -/- mice survival time (median survival: 131 days; n = 5); (b), Representative H&E-stained sections of Tsc1 -/+ and Tsc1 -/- pancreata show significant loss parenchymal cells in Tsc1 -/ -mice, but not in Tsc1 -/+ mice; scale bar: 50 μm (c-d), Co-IF for Krt19, α-amylase, glucagon and insulin show the loss of tissue homeostasis in the endocrine and exocrine compartment of Tsc1 -/- pancreata; scale bars: 50 μm; (e-g), Representative IHC pictures show distinct in vivo activation of mTOR signaling (p-mTORSer2448 and p-S6Ser235/236) and induction of Pten in pancreatic tissues from Tsc1 -/- mice, scale bar: 50 μm
Next, we aimed to determine whether Tsc1 deficiency resulted in hyperactive mTOR signaling in the pancreas. IHC studies revealed that residual acinar cells in Tsc1-/- animals were strongly positive for p-mTORSer2448 and p-S6Ser235/236 while the acinar cells in Tsc1-/+ mice were only weakly positive (Fig. 1e, f). As a feedback response to hyperactive mTOR signaling, Pten was induced in residual acinar cells (Fig. 1g). No such differences were observed in Tsc1-/+ pancreata.
Hyper-activated mTOR signaling induces p53 and apoptosis of acinar cells
We then set out to investigate potential reasons for the loss of acinar cells induced by hyper-activated mTOR signaling. Previous studies in mouse embryonic fibroblasts (MEFs) showed that hyper-activated mTOR rendered these cells susceptible to p53-dependent apoptosis [23]. Thus, expression of cleaved-caspase 3 (a marker of apoptosis) and p53 was analyzed, revealing that the residual acinar cells in Tsc1-/ - pancreata were apoptotic and were strongly positive for nuclear p53 (Fig. 2a, b). In addition, an increased proliferation rate was observed in these residual acinar cells (Fig. 2c, d). Such changes were not seen in Tsc1-/+ pancreata; Fig. 2a, b and c).
Fig. 2.
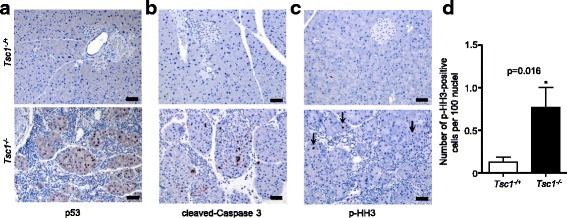
Hyperactivated mTOR signaling induces p53 and apoptosis in acinar cells. (a-c) Representative IHC pictures show in vivo activation of p53, apoptosis (cleaved-caspase 3), increased proliferation (p-Histone H3 (p-HH3)) in pancreatic tissues from Tsc1 -/- mice, scale bar: 50 μm; (d) Quantification of p-HH3-positive cells in Tsc1 -/- (n = 4) and Tsc1 -/+ (n = 4) pancreata; *: p < 0.05
Loss of p53 and Tsc1 promote acinar cell transformation
To confirm that acinar cell apoptosis was p53-dependent, we inactivated p53 in the context of Tsc1 deficiency. Thus, p48Cre/+; p53fl/fl; Tsc1fl/fl (referred to hereinafter as: p53-/-; Tsc1-/-) mice were generated. Surprisingly, additional ablation of p53 did not prolong the survival of Tsc1-/- mice: all p53-/-; Tsc1-/- mice died between the age of 84 days and 237 days (n = 7; median survival: 101 days vs. 133 days in Tsc1-/- mice, n = 5; Fig. 3a); however, neither significant loss of acinar cells nor death of acinar cells was observed. Instead, histological examination of p53-/-; Tsc1-/- pancreata revealed occurrence of dysplastic acinar cells in all animals (Fig. 3b, upper panel). These cells were characterized by an increased nuclear-cytoplasmic ratio with moderate to severe nuclear atypia with coarse granular chromatin, multiple nucleoli and single atypical mitoses. Additionally, nodular hyperplasia in acinar cells (circumscribed aggregates of acinar cells with distinct tinctorial differences to the surrounding acinar cells) were observed in 4 mice (Fig. 3b, lower panel); further, a large pancreatic tumor (Fig. 3c, Additional file 1: Figure S1A) was seen in one out of seven mice. Histological examination of the tumor revealed that the tumor cells were highly proliferative and showed distinctive tumor morphology reminiscent of human ACC (Fig. 3c) with an acinar to solid growth pattern and neoplastic cells with an amphophilic to eosinophilic granular cytoplasm. Positivity of the tumor cells for secretory enzymes such as Trypsin 3 (protease, serine 3) and α-Amylase (Additional file 1: Figure S1B) underscored the acinar origin of the neoplasia.
Fig. 3.
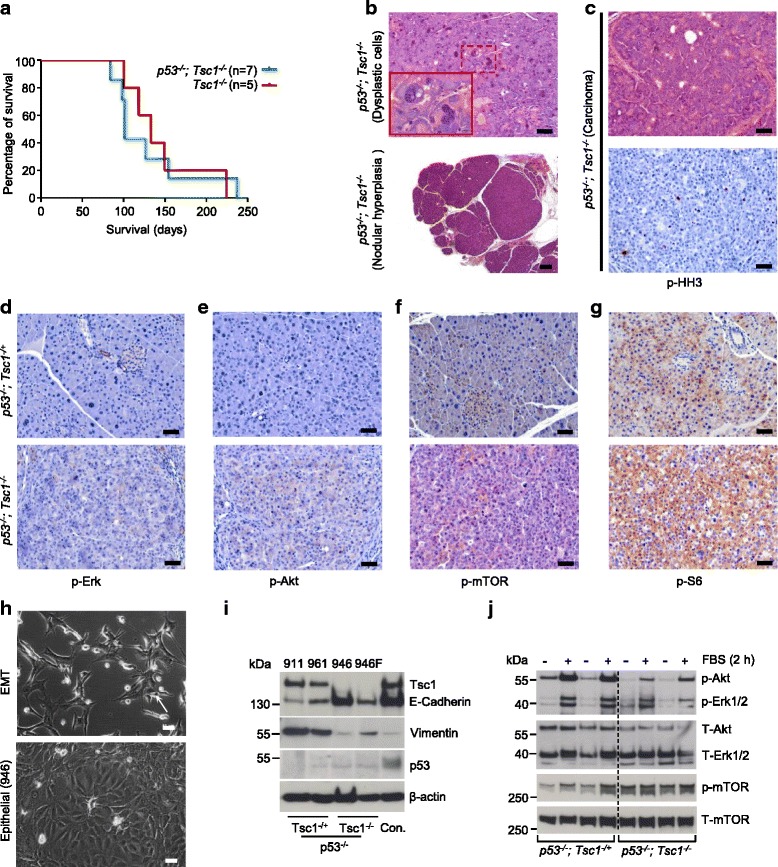
Loss of p53 and Tsc1 promote acinar cell transformation. (a) Kaplan–Meier survival analysis shows p53 -/-; Tsc1 -/- mice survival time (median survival: 101 days; n = 7) which is not different from that of Tsc1 -/- mice (median survival: 133 days; n = 5); (b) Representative H&E-stained sections of p53 -/-; Tsc1 -/+ and p53 -/-; Tsc1 -/- pancreata show pancreatic acinar cells with nuclear abnormalities (upper panel) and nodular hyperplasia (lower panel); scale bar: 50 μm (upper panel) and 200 μm (lower panel); (c) Representative H&E-stained sections and IHC pictures show the histology of one ACC-like tumor with a high proliferative index (p-HH3); (d-g) Representative IHC pictures show distinct in vivo activation of mTOR signaling (p-mTORSer2448 and p-S6Ser235/236), but inactivation of Erk (p-Erk1/2Thr202/Tyr204) and Akt (p-AktSer473) in tumor cells (and atypical acinar cells) of pancreatic tissues from p53 -/-; Tsc1 -/- mice, scale bar: 50 μm; (h) Phase contrast images of isolated cell lines from p53 -/- ; Tsc1 -/+ (911 and 961) and p53 -/- ; Tsc1 -/- (946 and 946 F) mice show fibroblast-like (961, 911 and 946 F) and epithelial (946) morphologies, scale bar: 50 μm; (i) Western-blot analysis demonstrates expression of Tsc1, E-Cadherin, Vimentin and p53 in p53 -/- ; Tsc1 -/+ and p53 -/- ; Tsc1 -/- cells; previously described cystic cells lines isolated from P48 Cre/+ ; Pten -/- ; Tsc1 -/+ mice were used as used as external controls [18]; (j) Western-blot analysis demonstrates phosphorylation levels of p-AktSer473, p-Erk1/2Thr202/Tyr204 and p-mTORSer2448 in p53-/- ; Tsc1 -/+ and p53 -/- ; Tsc1 -/- cells after fetal bovine serum (FBS) treatment for 2 h; one of the three independent experiments is shown
In regard to the endocrine compartment, however, we observed a phenotype as described for Tsc1-/- pancreata: no islets were seen in any of the p53-/-; Tsc1-/- pancreata, except for one with a large ACC-like tumor. Since no difference in survival of p53-/-; Tsc1-/- and Tsc1-/- mice was found, uncontrolled diabetes mellitus seems to be the cause of death in these animals. Such changes were not seen in p53-/-; Tsc1-/+ pancreata (Additional file 1: Figure S1C).
To investigate signal changes occurring in p53-/-; Tsc1-/+ and p53-/-; Tsc1-/- pancreata, we performed a set of phosphorylation stainings for Erk, Akt, mTOR and S6. This analysis revealed that pancreatic epithelial cells in p53-/-; Tsc1-/+ mice and tumor cells (and atypical acinar cells) in p53-/-; Tsc1-/- mice were generally devoid of p-AktSer473 and p-Erk1/2Thr202/Tyr204 (Fig. 3d, e); pancreatic acinar cells of p53-/-; Tsc1-/+ mice had detectable levels of p-mTORSer2448 and p-S6Ser235/236; however, the staining intensities in the tumor cells (and atypical acinar cells) of p53-/-; Tsc1-/- mice were more pronounced (Fig. 3f, g). These data suggest that hyper-activated mTOR signaling in p53-/-; Tsc1-/- pancreata was not dependent on either the Mek/Erk nor the PI3K/Akt cascade.
In a next step, we established a set of mouse cell lines from p53-/-; Tsc1-/+ (911 and 961 cells) and p53-/-; Tsc1-/- mice (946 F and 946 cells from ACC-like tumors). As previously reported, a set of p53-deficient cell lines underwent epithelial-to-mesenchymal transition (EMT) and exhibited fibroblast-like morphologies in vitro (Fig. 3h, upper panel) [24], except for one clone (946), which showed a stable epithelial morphology (Fig. 3h, lower panel). Cell lines that had undergone EMT (911, 961 and 946 F) expressed low levels of E-Cadherin but high levels of vimentin, compared to those without EMT features (946 cells, Fig. 3i). Neither was Tsc1 detected in the Tsc1-deficient cell lines (946 and 946 F) nor was p53 found in p53-deficient cells (911, 961, 946 and 946 F cells, Fig. 3i, and IHC of p53: Additional file 1: Figure S2A). To test clonogenicity and tumorigenicity of these cell lines, we performed orthotopic implantation into wild-type (WT) mice and subcutaneous injection into BALB/c nude mice. None of the tested cells lines were able to form tumors in WT mice (n = 24). However, tumors formed at 67 % of the sites (16/24) injected with cells with EMT features (cell lines 911, 961 and 946 F), while no tumors were found at the sites (0/3) injected with the cells without EMT features (946; BALB/c nude mice). An overview of the transplanted cells, mouse numbers and tumor formation is provided in Additional file 2: Table S1. No difference in proliferation of these cell lines in vivo was detected by quantifying the number of phospho-Histone H3 (pHH3) positive cells (Additional file 1: Figure S2B).
To further confirm the signal changes in vitro, we cultured p53-/-; Tsc1-/+ and p53-/-; Tsc-/- cell lines under serum deprivation conditions. Only weak constitutively active Akt or Erk signaling (as demonstrated by p-AktSer473 and p-Erk1/2Thr202/Tyr204 staining) was observed in these cell lines, but serum treatment (20 % FBS) induced pronounced Akt and Erk activation in these cells; though to a lesser extent in the p53-/-; Tsc1-/- cell lines. The p53-/-; Tsc1-/+ cell lines had relatively low levels of basal mTOR signaling (as demonstrated by p-mTORSer2448) which was slightly increased by serum treatment. In contrast, mTOR signaling in the p53-/-; Tsc1-/- cells was constantly active and serum treatment had marginal additional effects (Fig. 3j).
mTOR signaling is activated while p53 signaling is inactivated in human ACCs
To investigate mTOR and p53 signaling in human ACCs, 14 ACC samples were stained (Fig. 4a) with p-mTORSer2448, p-S6Ser235/236 and p53 antibodies. This analysis revealed that 43 % (6/14) and 64 % (9/14) of the samples were positive for p-mTORSer2448 and p-S6Ser235/236, respectively (Fig. 4b, c). Notably, no nuclear p53 staining was detected in any of tested samples, indicating a universal inactivation of p53 signalling in human ACC. As a control, PDAC sections were stained with p53 antibodies (Fig. 4d).
Fig. 4.
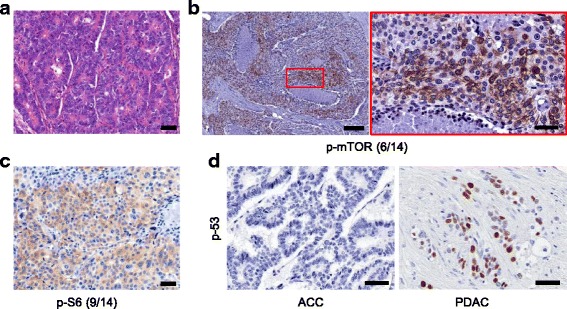
mTOR signaling is activated while p53 signaling is inactivated in human ACCs. (a) Representative H&E-stained sections of the histology of human ACCs; (b-c) Representative IHC pictures show distinct in vivo activation of mTOR signaling (p-mTORSer2448 and p-S6Ser235/236) in a subset of human ACC, but universal inactivation of p53 signalling (nuclear staining for p53), scale bar in (b) (right panel) and (c): 50 μm; scale bar in (b) (left panel): 200 μm; (d) Representative IHC pictures show inactivation of p53 signalling (nuclear staining for p53) in ACCs, but not in PDACs; scale bar: 50 μm
Discussion
The Tsc/mTOR/p53 signaling loop is a delicate feedback system by which cells are able to cope with genotoxic and nutritional stress [25]. mTOR activates p53 by either increasing its protein synthesis or its stability, which increases the GAP activity of the Tsc1-Tsc2 complex via AMPK and sestrin 1/2 [20, 21]. Hence, the Tsc1-Tsc2 complex constitutes an essential component of the signaling loop. It has been reported that hyperactive mTOR signaling - caused by loss of Tsc1 or Tsc2 - induced accumulation of p53 and apoptosis in mouse embryonic fibroblasts upon glucose starvation [23]. Consistently, we demonstrate that pancreatic acinar cells do not tolerate hyperactive mTOR signaling induced by Tsc1 deficiency and undergo apoptosis, likely in a p53-dependent way. Additional ablation of p53 eliminated cell death, but simultaneously promoted formation of dysplastic acinar cells and induced a malignancy resembling ACC. However, degeneration of the endocrine compartment was not affected by the p53 status. These data suggest that mTOR hyperactivation-induced apoptosis of endocrine cells is not a p53-dependent process. This argument is in line with published data showing that impairment of autophagy and induction of endoplasmic reticulum (ER) stress are potentially responsible for cell death [26]. The exact mechanisms underlying such effects remain yet to be defined.
By analysing pancreas tissues of p53-/-;Tsc1-/- mice, we observed that all samples displayed dysplastic acinar cells and four out of them showed nodular hyperplasia in the exocrine pancreas. These data suggest that the presumed precancerous lesions indeed developed in those mice without established ACC-like tumors. However, due to the general loss of islets in these mice, uncontrolled diabetes mellitus seems to have caused the early death of these animals. Thus, this “compound” phenotype in the endocrine compartment does not allow for a sufficient follow-up of ACC-like tumor development in our mouse models. Following this reasoning, the only mouse developing an ACC-like tumor had the longest survival (237 days). To resolve this issue, it is worthwhile to specifically ablate p53 and Tsc1 in pancreatic acinar cells in the adult organ using an inducible system.
Recently, Ding and co-workers reported that Tsc1 ablation mediated by Neurogenin 3-Cre (Neurog3Cre; Tsc1-/- mice) also induced “adenocarcinoma-like” lesions showing features of ACCs [27]. Interestingly, no massive acinar cell apoptosis/involution was described in the pancreas of Neurog3Cre; Tsc1-/-mice. Furthermore, ACC-like tumors developed at a relatively long latency. Of note, the Neurog3Cre line is not a “usual” Cre line for studying pancreatic exocrine malignancies because it mainly mediates genetic recombination in the endocrine compartment. Lineage tracing experiments revealed that only a small percentage of acinar cells (around 0.8 %) in the adult organ were Cre-positive in the Neurog3Cre line [28]. Thus, the majority of acinar cells in Neurog3Cre; Tsc1-/- mice are spared from genetic recombination at the Tsc1 locus. This explains why no massive acinar cell apoptosis was observed in Neurog3Cre; Tsc1-/- mice. This lack of organ involution in the acinar cell compartment allows for long-term monitoring of tumor development in these mice. Indeed, all Neurog3Cre; Tsc1-/- mice developed ACC-like tumors by the age of 10 months. Given the continuous DNA damage in Tsc1-deficient cells, it remains unknown how many of the ACC-like tumors have acquired a genetic alteration of p53 during tumor evolution. Due to the “premature” demise of the p53-/-; Tsc1-/- mice, the real penetrance of the mTOR-p53 axis in initiating ACC-like tumors remains unclear. Nevertheless, two studies now provide consistent evidence that hyperactive mTOR is the driving force for the development of - at least a subset - of ACCs.
Conclusions
The mTOR-p53 axis constitutes an important regulatory machinery which controls tissue homeostasis of the pancreas. Dysregulation of this axis contributes to the development of ACCs. Preclinical studies testing the effectiveness of anti-mTOR therapies in ACC mouse models are warranted.
Methods
Patient material and tissue collection
ACC tissues were from patients who had undergone pancreatic resections. All sample diagnoses were confirmed histologically. Samples were fixed in paraformaldehyde solution for 24 h and subsequently paraffin-embedded for histological analysis. The use of tissue for analysis was approved by the local Ethics Committee (Technical University Munich, #1926/07) and written informed consent was obtained from the patients prior to surgery (Department of Surgery, Klinikum rechts der Isar).
Mouse lines
Mice containing a floxed allele of Tsc1 (005680) and of p53 (008462) were obtained from The Jackson Laboratory (Bar Harbor, USA). The pancreas-specific Cre recombinase line Ptf1aCre/+ (also known as p48Cre/+) was a kind gift from Roland M. Schmid and Jens T. Siveke (Dept. of Gastroenterology, TU Munich). The wild type (WT; C57BL/6 J) and thhe BALB/c nude mice were obtained from Charles River Laboratory (Sulzfeld, Germany).
Mouse breeding
Mouse breeding was performed and husbandry was maintained at the specific pathogen free (SPF) mouse facility at the Technical University of Munich. The compound transgenic mice were maintained on a mixed background. All mouse experiments and procedures were approved by the Institutional Animal Care and Use Committees of the Technical University of Munich. All procedures were in accordance with the Office of Laboratory Animal Welfare and the German Federal Animal Protection Laws. All mouse experiments and procedures were approved by Bavarian Government (No. 55.2-1-54-2532-42-13; 55.2-1-54-2531-33-08). Unless otherwise stated, all animals were followed up for up to 1.5 years or sacrificed for histological analysis if they showed any sign of disease.
Primary cell isolation
Freshly dissected sterile tumor tissues were washed twice with ice-cold PBS, cut into small cubes (approximately 1 mm) and dispensed into 5 ml of complete medium containing collagenase (1.2 mg/ml). The resulting solution (mixed with tissue blocks) was incubated at 37 °C for 0.5 h. After centrifugation at 300 rpm for 5 min, the small tissue blocks were washed twice with collagenase-free medium, followed by incubation at 37 °C with medium containing collagenase for an additional 0.5 h. After passing the undigested tissue blocks through a 100 μm nylon mesh, cell suspensions were obtained. These cell suspensions were washed two times with complete medium and seeded into a 10 cm2 dish.
Cell transplantation experiments
For mouse cell transplantation experiments into BALB/c nude mice, 106 tumor cells were resuspended in 50 μl of PBS and were injected into the left lower and right upper flank. Tumor growth was monitored macroscopically every week. Orthotopic transplantations of mouse cells were carried out as described previously [29, 30]. Briefly, mice were anesthetized, and a left-lateral incision of the abdomen was made to visualize the tail region of the pancreas. 106 tumor cells were suspended in 50 μl of PBS and were carefully injected into the pancreatic tail. The abdominal wall and skin were closed using running sutures. All mice were sacrificed for histological evaluation after 4 weeks.
Further materials and methods
A detailed materials and methods section is provided as a Additional file 3.
Statistical analysis
GraphPad Prism 5 Software (GraphPad, San Diego, CA, USA) was used for the statistical (survival) analysis. Statistical significance was set at p < 0.05.
Additional files
Figure S1. A, Gross pathology of one ACC-like tumor in p53 -/- ; Tsc1 -/- mice; B, IHC demonstrates expression of α-Amylase and Trypsin 3 in tumors cells and acinar cells of p53 -/- ; Tsc1 -/- and p53 -/- ; Tsc1 -/+ mice, respectively; scale bar: 50 μm; C, Representative H&E stained section shows normal histology of p53 -/- ; Tsc1 -/+ pancreata; scale bar: 50 μm. Figure S2. A, Representative IHC pictures demonstrate the knockout status of p53 in p53 -/- ; Tsc1 -/- and p53 -/- ; Tsc1 -/+ pancreata, respectively; scale bar: 100 μm; B, Quantification of proliferating cells (marked by p-HH3 staining) in tumors that developed after implantation of p53 -/- ; Tsc1 -/- (946 F) and p53 -/- ; Tsc1 -/+ (911 and 961) cell lines into BALB/c nude mice shows no differences; n.s.: no significant change tested by one-way ANOVA. (PPT 38925 kb)
Tumor formation by p53 -/- ; Tsc1 -/- or p53 -/- ; Tsc1 -/+ cells in wild-type (WT) and BALB/c nude mice. (DOC 28 kb)
Supplementary Materials and Methods. (DOC 59 kb)
Acknowledgements
This project was supported in part by a grant from the TU Munich commission for clinical research (KKF C21-11, to BK), the Deutsche Forschungsgemeinschaft (MI 1173/5-1, to CWM, BK and JK), the European Union (FP7, PacaNet, to CWM and JK). We would like to thank Manja Thorwirth and Nadja Maeritz for their excellent technical support.
Abbreviations
- ACC
acinar cell carcinoma
- mTOR
mammalian target of rapamycin
- PI3K
phosphatidylinositol-4, 5-bisphosphate 3-kinase
- Tsc
Tuberous sclerosis complex
- Tsc1
hamartin
Footnotes
Bo Kong, Tao Cheng, Chengjia Qian contributed equally to this work.
Jörg Kleeff and Christoph Michalski jointly supervised the work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BK, CWM and JK designed the study. BK, CQ, TC, WW, JC, IR and SR performed experiments and acquired data. KS, AMS and IE provided expertise in pathological analysis. BK, JK and CWM drafted the manuscript. ME and HF revised the manuscript critically for important intellectual content. All authors approved the final version of the manuscript.
References
- 1.Klimstra DS, Heffess CS, Oertel JE, Rosai J. Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol. 1992;16(9):815–37. doi: 10.1097/00000478-199209000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Chen J, Baithun SI, Ramsay MA. Histogenesis of pancreatic carcinomas: a study based on 248 cases. J Pathol. 1985;146(1):65–76. doi: 10.1002/path.1711460108. [DOI] [PubMed] [Google Scholar]
- 3.Abraham SC, Wu TT, Hruban RH, Lee JH, Yeo CJ, Conlon K, et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol. 2002;160(3):953–62. doi: 10.1016/S0002-9440(10)64917-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lowery MA, Klimstra DS, Shia J, Yu KH, Allen PJ, Brennan MF, et al. Acinar cell carcinoma of the pancreas: new genetic and treatment insights into a rare malignancy. Oncologist. 2011;16(12):1714–20. doi: 10.1634/theoncologist.2011-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schempf U, Sipos B, Konig C, Malek NP, Bitzer M, Plentz RR. FOLFIRINOX as first-line treatment for unresectable acinar cell carcinoma of the pancreas: a case report. Z Gastroenterol. 2014;52(2):200–3. doi: 10.1055/s-0033-1356439. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto T, Ohzato H, Fukunaga M, Imamura H, Furukawa H. Acinar cell carcinoma of the pancreas: a possible role of S-1 as chemotherapy for acinar cell carcinoma. A case report. Jop. 2012;13(1):87–90. [PubMed] [Google Scholar]
- 7.Cananzi FC, Jayanth A, Lorenzi B, Belgaumkar A, Mochlinski K, Sharma A, et al. “Chronic” metastatic pancreatic acinar cell carcinoma. Pancreatology. 2013;13(5):549–52. doi: 10.1016/j.pan.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiao Y, Yonescu R, Offerhaus GJ, Klimstra DS, Maitra A, Eshleman JR, et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol. 2014;232(4):428–35. doi: 10.1002/path.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–30. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 11.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle J, Snell R, van den Ouweland A, et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet. 1998;7(6):1053–7. doi: 10.1093/hmg/7.6.1053. [DOI] [PubMed] [Google Scholar]
- 14.Plank TL, Yeung RS, Henske EP. Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer Res. 1998;58(21):4766–70. [PubMed] [Google Scholar]
- 15.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 16.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331(6021):1199–203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong B, Wu W, Cheng T, Schlitter AM, Qian C, Bruns P, et al. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut. 2015. [DOI] [PubMed]
- 19.Garcia-Carracedo D, Turk AT, Fine SA, Akhavan N, Tweel BC, Parsons R, et al. Loss of PTEN expression is associated with poor prognosis in patients with intraductal papillary mucinous neoplasms of the pancreas. Clin Cancer Res. 2013;19(24):6830–41. doi: 10.1158/1078-0432.CCR-13-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67(7):3043–53. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 22.Shigeyama Y, Kobayashi T, Kido Y, Hashimoto N, Asahara S, Matsuda T, et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol. 2008;28(9):2971–9. doi: 10.1128/MCB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CH, Inoki K, Karbowniczek M, Petroulakis E, Sonenberg N, Henske EP, et al. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. Embo J. 2007;26(23):4812–23. doi: 10.1038/sj.emboj.7601900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinho AV, Rooman I, Real FX. p53-dependent regulation of growth, epithelial-mesenchymal transition and stemness in normal pancreatic epithelial cells. Cell Cycle. 2011;10(8):1312–21. doi: 10.4161/cc.10.8.15363. [DOI] [PubMed] [Google Scholar]
- 25.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102(23):8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartolome A, Kimura-Koyanagi M, Asahara S, Guillen C, Inoue H, Teruyama K, et al. Pancreatic beta-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes. 2014;63(9):2996–3008. doi: 10.2337/db13-0970. [DOI] [PubMed] [Google Scholar]
- 27.Ding L, Han L, Li Y, Zhao J, He P, Zhang W. Neurogenin 3-directed cre deletion of tsc1 gene causes pancreatic acinar carcinoma. Neoplasia. 2014;16(11):909–17. doi: 10.1016/j.neo.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schonhoff SE, Giel-Moloney M, Leiter AB. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol. 2004;270(2):443–54. doi: 10.1016/j.ydbio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 29.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–47. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong B, Cheng T, Wu W, Regel I, Raulefs S, Friess H, et al. Hypoxia-induced endoplasmic reticulum stress characterizes a necrotic phenotype of pancreatic cancer. Oncotarget. 2015;6(31):32154–60. doi: 10.18632/oncotarget.5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A, Gross pathology of one ACC-like tumor in p53 -/- ; Tsc1 -/- mice; B, IHC demonstrates expression of α-Amylase and Trypsin 3 in tumors cells and acinar cells of p53 -/- ; Tsc1 -/- and p53 -/- ; Tsc1 -/+ mice, respectively; scale bar: 50 μm; C, Representative H&E stained section shows normal histology of p53 -/- ; Tsc1 -/+ pancreata; scale bar: 50 μm. Figure S2. A, Representative IHC pictures demonstrate the knockout status of p53 in p53 -/- ; Tsc1 -/- and p53 -/- ; Tsc1 -/+ pancreata, respectively; scale bar: 100 μm; B, Quantification of proliferating cells (marked by p-HH3 staining) in tumors that developed after implantation of p53 -/- ; Tsc1 -/- (946 F) and p53 -/- ; Tsc1 -/+ (911 and 961) cell lines into BALB/c nude mice shows no differences; n.s.: no significant change tested by one-way ANOVA. (PPT 38925 kb)
Tumor formation by p53 -/- ; Tsc1 -/- or p53 -/- ; Tsc1 -/+ cells in wild-type (WT) and BALB/c nude mice. (DOC 28 kb)
Supplementary Materials and Methods. (DOC 59 kb)