

Abstract
Polyhydroxybutyrate (PHB) synthases (PhaCs) catalyze the conversion of 3-(R)-hydroxybutyryl CoA (HBCoA) to PHB, which is deposited as granules in the cytoplasm of microorganisms. The class I PhaC from Caulobacter crescentus (PhaCCc) is a highly soluble protein with a turnover number of 75 s–1 and no lag phase in coenzyme A (CoA) release. Studies with [1-14C]HBCoA and PhaCCc monitored by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and autoradiography reveal that the rate of elongation is much faster than the rate of initiation. Priming with the artificial primer [3H]sTCoA and monitoring for CoA release reveal a single CoA/PhaC, suggesting that the protein is uniformly loaded and that the elongation process could be studied. Reaction of sT-PhaCCc with [1-14C]HBCoA revealed that priming with sTCoA increased the uniformity of elongation, allowing distinct polymerization species to be observed by SDS–PAGE and autoradiography. However, in the absence of HBCoA, [3H]sT-PhaC unexpectedly generates [3H]sDCoA with a rate constant of 0.017 s–1. We propose that the [3H]sDCoA forms via attack of CoA on the oxoester of the [3H]sT-PhaC chain, leaving the synthase attached to a single HB unit. Comparison of the relative rate constants of thiolysis by CoA and elongation by PhaCCc, and the size of the PHB polymer generated in vivo, suggests a mechanism for chain termination and reinitiation.
Polyhydroxybutyrate (PHB) synthases (PhaCs) catalyze the polymerization of 3-(R)-hydroxybutyryl coenzyme A (HBCoA) to generate PHB. Polyhydroxyalkanoates (PHAs) such as PHB are produced by most microorganisms as an energy reserve in times of nutrient limitation when a carbon source (e.g., a sugar or fatty acid) is available.1−4 As the PHA is synthesized, it is deposited in the cytosol as insoluble inclusions or granules. When the environment becomes conducive to growth, the bacteria degrade the PHA and use the monomer units for biosynthesis and growth. PHAs are of general interest because they are biodegradable polymers from a renewable source. Understanding the mechanism of PHA polymerization, specifically the initiation, elongation, and termination processes, is important to engineering their production in a cost-effective fashion so that they can compete with environmentally unfriendly, oil-based plastics.5−7 In this work, we report our studies of the Caulobacter crescentus class I PHB synthase, PhaCCc, and the new insight into the polymerization process.
Two distinct classes of PhaC that make PHB have been identified and studied in detail. The prototypical class I PhaC from Ralstonia eutropha (PhaCRe) is a homodimer of 65 kDa subunits.8,9 The prototypical class III synthase from Allochromatium vinosum (PhaECAv) is a heterotetramer of two distinct types of subunits: PhaC, the synthase, and PhaE, which is of unknown function but essential for activity.10,11 Studies of these prototypes suggest that PhaCRe and PhaECAv share a common active site housed in a lipase-like domain, containing conserved Cys, His, and Asp residues that are essential for catalysis.12−16 While a number of mechanistic models have been considered, our current working model for polymerization involves both covalent and noncovalent intermediates (Scheme 1). In this model, the His deprotonates the Cys to generate the active site thiolate, which reacts with HBCoA, resulting in acylation of PhaC with hydroxybutyrate (HB). A second HBCoA then binds, and the Asp serves as a general base to activate its hydroxyl group for attack on the HB-PhaC thioester, releasing the growing (HB)n-SCoA chain within the active site. This noncovalent intermediate then rapidly reacylates the active site Cys, and the polymerization continues until the polymer reaches a relatively uniform molecular weight (Mw), which varies by organism, at which point termination occurs by a poorly understood mechanism.
Scheme 1. Working Mechanistic Model for PHB Polymerization.

The initiation and elongation steps of PHB polymerization have been challenging to study with both PhaCRe and PhaECAv. Neither enzyme can be uniformly loaded with HB units even when the HBCoA:PhaC ratio is 1:1 to 5:1, because the elongation rate is much faster than the initiation rate. Thus, a fraction of the enzyme generates a large polymer under these conditions, while most of the synthase remains unmodified.9,17,18 Furthermore, the kinetics of CoA release, used as a measure of polymerization, are distinct between synthases: PhaCRe has a lag phase in activity thought to be caused in part by a requirement for priming and protein dimerization,9 whereas PhaECAv exhibits a burst phase.11 These challenges have precluded the study of initiation and elongation. Therefore, in an effort to mitigate these issues and study the initiation and elongation processes, we designed an artificial primer, [3H]sTCoA [a trimer of HBCoA in which the terminal OH group is replaced with a 3H (Figure 1)]. This primer was shown to acylate the active site Cys of both PhaCRe and PhaECAv with a stoichiometry of 0.5 sT per PhaC.13,14 The [3H]sT attached to PhaC could be chased into PHB. These observations led us to propose half-sites reactivity, with one PHB chain synthesized per dimer of PhaC. From these observations, the expectation was that priming with sTCoA would allow uniform extension of sT-PhaC upon addition of HBCoA. To probe this model, HBCH2CoA [an analogue of HBCoA in which the S of the thioester is replaced with CH2 (Figure 1)] was synthesized and reacted with sT-PhaCRe.19 In principle, if sT-PhaCRe uniformly extends, then this reaction should rapidly generate a saturated tetramer methylene CoA [sT4CH2CoA (Figure 1)], which would be unable to reacylate and thus be isolable. Unexpectedly, the product of this reaction was a saturated trimer methylene CoA intermediate [sTCH2CoA (Figure 1)]. These results suggest that the sT-PhaC with HBCoA bound is in a conformation that can extend its chains rapidly, while with HBCH2CoA (Figure 1) bound, the chain extension rate is greatly reduced and chemistry occurs at the penultimate HB unit leaving an HB-PhaC. The structural basis for these conformational differences with such a modest chemical change (S vs CH2) is unknown.
Figure 1.

Structures of the artificial primer 3H-labeled saturated trimer CoA ([3H]sTCoA), the methylene analogue of HBCoA (HBCH2CoA), the expected elongation product of sT-PhaC reacted with HBCH2CoA (sT4CH2CoA), the observed elongation product of sT-PhaC reacted with HBCH2CoA (sTCH2CoA), and the 3H-labeled saturated dimer CoA ([3H]sDCoA).
The termination process is also of great interest because understanding how PhaCs are capable of generating a high-Mw polymer with low polydispersity is essential to the commercialization of PHAs. Like initiation and elongation, however, termination has also been difficult to study. Several small molecules, including water,20,21 CoA,22 and HB,23 have been proposed to function as chain terminators. However, end group analysis of the native polymer to shed light on the in vivo termination species is not possible because the polymer Mw values are too high. Nonetheless, several experiments have indirectly provided insight into the termination process. The observation by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and autoradiography that PhaECAv-(HB)n (where n = 40–100) is converted in a time-dependent fashion into a PhaECAv-(HB)n species where n = 3–10 suggested that PhaC alone is sufficient to catalyze termination and that termination occurs at an oxoester of the polymer chain to leave a primed synthase.18 Reaction of PhaECAv with the N-acetylcysteamine (NAC) derivative of HB (HB-NAC) produced a polymer with a Mw of 75 kDa, sufficiently small for end group analysis, which revealed that each chain terminated in NAC.24 This result suggested that chain termination could potentially occur in vivo by thiolysis with CoA but does not address the issue of the position of thiolysis or whether termination leaves a primed synthase. Thus, while it is clear that PhaC is sufficient to catalyze termination, the mechanism remains unknown.
In this work, we report the cloning, expression, and isolation of the 73 kDa class I synthase from C. crescentus (PhaCCc) and our studies to understand the polymerization process. PhaCCc is very soluble, in contrast with PhaCRe, and has a comparable turnover number (75 s–1). Kinetic studies with wild-type (wt) and mutant PhaCCcs reveal a protein with properties of both PhaCRe and PhaECAv. Covalent catalysis with the active site cysteine/histidine is required, and the elongation rate is much greater than the initiation rate. However, studies with sTCoA indicate that, in contrast to both PhaCRe and PhaECAv, PhaCCc is acylated with ∼1 sT per PhaC. This observation suggests that the half-sites reactivity model for PhaCs needs to be reconsidered and potentially simplifies mechanistic analysis. We have shown that sT-PhaCCc in the presence of HBCoA is chemically competent to form PHB, and that priming with sTCoA increases the specific activity and appears to increase the uniformity of chain elongation. However, in the absence of HBCoA, the sT moiety of sT-PhaCCc unexpectedly is converted to a saturated dimer CoA [sDCoA (Figure 1)] at a rate of 0.017 s–1, leaving behind HB-PhaC, a reaction reminiscent of the reaction between sT-PhaCRe and HBCH2CoA.19 We propose that this result reports on a potential termination and reinitiation mechanism wherein CoA is the terminator and that a conformational change occurs during elongation to switch from a resting state to an elongation state.
Experimental Procedures
Materials
All chemicals were purchased at the highest purity available from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Primers and plasmids used in this study are listed in Tables S1 and S2 of the Supporting Information, respectively. Oligonucleotide primers were obtained from Integrated DNA Technologies (Coralville, IA). (R/S)-[1-14C]HBCoA was obtained from American Radiolabeled Chemicals, Inc., and diluted with (R)-HBCoA synthesized as previously described.25 sTCoA and [3H]sTCoA were synthesized as previously described.15 Restriction enzymes were purchased from New England Biolabs (Beverly, MA), and PfuII polymerase was purchased from Stratagene (La Jolla, CA). Scintillation fluid was obtained from PerkinElmer (Hopkinton, MA).
Construction of Expression Plasmids and Site-Directed Mutagenesis of phaC
Plasmids pRBphaCCc and pRBΔNphaCCc (Table S2 of the Supporting Information) were constructed from pET28a and encode N-terminally His6-tagged PhaCCc and ΔNPhaCCc, respectively. ΔNPhaCCc is a truncation construct of PhaCCc that is missing the 85 N-terminal amino acids. phaCCc and ΔNphaCCc were amplified from 200 ng of C. crescentus genomic DNA using PfuII Turbo polymerase. For phaCCc, primers phaCfw and phaCrev (Table S1 of the Supporting Information) were used to insert 5′ NheI and 3′ BamHI restriction sites. For ΔNphaCCc, primers ΔNphaCfw and ΔNphaCrev were used to insert the same restriction sites. The amplified gene products were digested with NheI and BamHI and ligated into pET28a digested with the same enzymes to generate pRBphaCCc and pRBΔNphaCCc. The portions of the plasmids containing phaCCc and ΔNphaCCc were sequenced at the Massachusetts Institute of Technology Biopolymers Facility.
The active site mutant plasmids (pRBphaCCc-C406S, -D562N, and -H590Q) were constructed from template pRBphaCCc using PfuII polymerase and primers C406Sfw/C406Srev, D562Nfw/D562Nrev, and H590Qfw/H590Qrev, respectively. Plasmid pRBphaCCc-C406A was constructed using the primer overlap extension method using PfuII polymerase (see the Supporting Information for details).26 Mutations and sequences were confirmed by sequencing at the Massachusetts Institute of Technology Biopolymers Facility.
Expression and Purification of wt and Mutant PhaCs
ΔNPhaCCc, PhaCCc, and mutants of PhaCCc were expressed at 18 °C in Escherichia coli strain BL21(DE3)Gold (Stratagene) harboring the appropriate plasmids. PhaCCcs were purified at 4 °C by standard Ni-NTA affinity chromatography methods (see the Supporting Information). Purified PhaCs were concentrated to 10–20 mg/mL (200–300 μM), exchanged into 20 mM Hepes (pH 7.5) and 200 mM NaCl, and stored at −80 °C until they were used. A typical purification yielded 10 mg of ΔNPhaCCc, or 3 mg of PhaCCc and PhaCCc mutants, per gram of wet cell paste. The concentration of purified PhaCs was determined by A280 (ε = 103270 M–1 cm–1 for ΔNPhaCCc, and ε = 103630 M–1 cm–1 for PhaCCc) or a Bradford assay using BSA as a standard.27
Enzyme Assays
All assays were conducted at 30 °C. A typical assay contained, in a final volume of 170 μL, 20 mM Hepes (pH 7.5), 20 mM NaCl, 0.76–1 mM HBCoA, and 500 nM ΔNPhaCCc, 25 nM wt PhaCCc, or 50–100 μM mutant PhaCs. Reactions were initiated by the addition of enzyme. At various time points, 20 μL aliquots were withdrawn and quenched in 50 μL of ice-cold 10% trichloroacetic acid (TCA). CoA release was determined by the 5,5′-dithiobis(2-nitrobenzoic) acid (DTNB) assay as previously described.11 Specific activity is given in units per milligram, where one unit is 1 μmol of CoA produced per minute. Kinetic data were fit to the Michaelis–Menten equation28 or the Hill equation.29
Analysis of Products of HBCoA Polymerization by SDS–PAGE and Autoradiography
In a final volume of 22 μL, PhaC (4 μM) was first reacted at room temperature for 1 min with 0 or 10 equiv of sTCoA and then chased by 5 or 50 equiv of [1-14C]HBCoA (88000 cpm total per reaction). After 10 s, 10 μL aliquots were withdrawn and quenched in 10 μL of Laemmli buffer (without reducing agent), and the samples were not boiled. PhaCCc standards and the samples (20 μL) were immediately loaded onto two 10% SDS–PAGE gels and run at 150 V on ice for 1 h. One gel was stained for 15 min in fresh Coomassie stain and then destained for 15 min in fast destain. The second gel was rinsed for 3 × 5 min in ddH2O, immediately dried, and then exposed to a low-energy phosphor screen (Molecular Dynamics) for 12 h. The phosphor screen was scanned using the Storm Imaging System and analyzed using ImageQuant TL (Amersham Biosciences).
Gel Permeation Chromatography (GPC) Analysis of PHB Produced by Reaction of PhaCCc and [3H]sT-PhaCCc with HBCoA
The reaction mixture of PhaCCc with 5 or 50 equiv of [1-14C]HBCoA contained, in a final volume of 100 μL, 4 μM PhaCCc and 20 or 200 μM [1-14C]HBCoA (specific activity of 175 cpm/nmol), and the reaction was conducted for 30 min at 30 °C. The reaction mixture of [3H]sT-PhaCCc with HBCoA contained, in a final volume of 12.5 μL, 20 mM Hepes (pH 7.5), 20 mM NaCl, 50 μM PhaCCc, and 500 μM [3H]sTCoA (specific activity of 5700 cpm/nmol). After 10 s, 10 μL of this reaction mixture was added to 990 μL of 20 mM Hepes (pH 7.5) and 20 mM NaCl. A 100 μL aliquot of this reaction mixture was then diluted into 1 mM HBCoA in 1 mL of 20 mM Hepes (pH 7.5) and 20 mM NaCl and incubated for 30 min at 30 °C. A control reaction mixture in a final volume of 100 μL containing 50 nM PhaCCc and 1 mM [1-14C]HBCoA (specific activity of 175 cpm/nmol) was incubated for 30 min at 30 °C. In all cases, when the reaction was complete the mixture was transferred to a borosilicate tube (16 × 125 mm), flash-frozen, and lyophilized. Chloroform (2 mL) and a magnetic stir bar were added, and the tube was capped with a rubber septum that was pierced with a Pasteur pipet that served as a condenser. The samples were refluxed for 48 h at 70 °C, and then the tubes were cooled to room temperature and centrifuged at 4 °C for 10 min at 3000g to pellet the insoluble material. The chloroform was carefully removed and stored at −20 °C until it was analyzed. The remaining insoluble material was refluxed for an additional 8 h in 2 mL of fresh chloroform, and this extract was combined with the first. To remove the remaining insoluble material, the sample was filtered using a glass syringe (0.5 mL) fitted with a 0.4 μm PTFE filter (13 mm) (Pall Life Science, Port Washington, NY). The filter was washed with 3 mL of fresh chloroform, which was added to the filtrate. Pooled samples were concentrated to 250 μL by evaporating chloroform under a stream of air.
For GPC analysis, 100 μL of each sample was injected onto a Plgel Olexis column [two columns in series, 300 mm × 7.5 mm (Varian, Palo Alto, CA)] connected to a Waters 515/2487 high-performance liquid chromatography (HPLC) system equipped with refractive index detection and eluted at a flow rate of 1 mL/min at 30 °C. Fractions (1 mL) were collected and analyzed by scintillation counting. 2-Propanol (retention time of 21 min) was added to each sample prior to GPC analysis and was used to normalize sample retention times. Mw values were determined by comparison to a set of polystyrene standards (Varian) (retention times in parentheses): 3.1 kDa (17.8 min), 10 kDa (17 min), 73 kDa (15.4 min), 205 kDa (14.8 min), 490 kDa (13.9 min), 1800 kDa (12.9 min), and 5000 kDa (12.4 min).
Stoichiometry of Acylation of PhaC with sTCoA Monitoring CoA Release
The reaction was conducted at 30 °C, and the mixture contained, in a final volume of 110 μL, 20 mM Hepes (pH 7.5), 20 mM NaCl, 50 μM PhaCCc, and 500 μM sTCoA. At various time points, 20 μL aliquots were withdrawn and quenched in 50 μL of ice-cold 10% TCA. The samples were analyzed by the DTNB assay as previously described.15
Activity Assay of sT-PhaCCc
In a final volume of 10 μL at 30 °C, PhaCCc (50 μM) was reacted with 500 μM sTCoA. After 10 s, the reaction mixture was diluted with 590 μL of 20 mM Hepes (pH 7.5) and 20 mM NaCl, and 10 μL was withdrawn and added to an assay mixture. The assay mixture contained, in a final volume of 170 μL, 50 nM PhaCCc (acylated with sT), ∼0.5 μM unreacted sTCoA, and 1 mM HBCoA. At various time points, 20 μL aliquots were removed, quenched in 50 μL of 10% TCA, and analyzed by the DTNB assay as described above.
HPLC Analysis of the Products of the Reaction between PhaCCc and [3H]sTCoA
The reaction was conducted at 30 °C, and the mixture contained, in a final volume of 500 μL, 50 μM PhaCCc and 500 μM [3H]sTCoA (specific activity of 147 cpm/nmol) in 20 mM Hepes (pH 7.5) and 20 mM NaCl. Aliquots (50 μL) were withdrawn from 10 s to 5 min and quenched in 20 μL of ice-cold 10% TCA. The samples were centrifuged at 4 °C for 5 min at 20000g to remove precipitated protein. The supernatant was removed; the protein pellets were washed with 50 μL of ice-cold ddH2O, and the wash was combined with the supernatant. The pellets were solubilized in 100 μL of 1% SDS and analyzed by scintillation counting. The combined wash and supernatant samples were adjusted to pH ∼5 by addition of 12 μL of 1 M NaOH. One hundred microliters of each sample was injected onto a nucleoside-nucleotide column (Alltech, 7 μm, 4.6 mm × 250 mm) attached to a Waters 515/2487 HPLC system equipped with a diode array detector. The elution protocol used 20 mM KH2PO4 (pH 4.7) (mobile phase A) and methanol (mobile phase B) and a linear gradient from 5 to 70% mobile phase B from 0 to 60 min, and the eluent was monitored by A260. Fractions were collected and analyzed by scintillation counting and matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (see the Supporting Information).
Results
Class I PhaCCc Has Properties That Are a Hybrid of Those of Class I and III Synthases (PhaCRe and PhaECAv)
Studies of Qi and Rehm using antibodies to PhaCRe revealed a protein with an Mw of ∼75 kDa in crude cell extracts of C. crescentus, distinct from the 65 kDa PhaCCc annotated in the database (accession number NP_420193).30 They identified a second putative start codon, and sequence alignments of this new protein with PhaCRe gave 37% sequence identity; however, the N-terminus of PhaCCc was absent in PhaCRe (Figure S1 of the Supporting Information). This PhaCCc and the annotated 65 kDa form (ΔNPhaCCc) were cloned, expressed, and purified (Tables S3 and S4 and Figure S2 of the Supporting Information). Assays monitoring CoA release revealed a lag phase and a specific activity of 1 unit/mg for ΔNPhaCCc (Figure S3A of the Supporting Information), reminiscent of studies with PhaCRe.9 The 73 kDa PhaCCc, on the other hand, had a specific activity of 50 units/mg and burst kinetics, similar to those of PhaECAv (Figure S3B of the Supporting Information).11 Because of the higher specific activity and the antibody studies,30 the 73 kDa protein became the focus of our studies. The kinetic parameters of this protein were determined, giving a kcat of 75 s–1 and a Km of 0.29 mM (Figure S3C of the Supporting Information).
The active site residues in PhaCRe (Figure S1 of the Supporting Information) align with residues C406, D562, and H590 in PhaCCc, and mutant proteins were made and purified. With PhaCRe, the C319S and C319A mutants were both catalytically inactive,9 while with C149S-PhaECAv, 0.05% of the activity of the wt enzyme was observed.14 C406A-PhaCCc was catalytically inactive, while the C406S mutant had a specific activity of 0.002 unit/mg (0.004% of that of wt) (Figure S3D of the Supporting Information). Mutation of the putative general base catalyst D562 to N (Scheme 1) and H590 to Q gave PhaCCc with 0.006 unit/mg (0.012% of that of wt) and no activity, respectively. PhaCCc is again more similar to PhaECAv than to PhaCRe, and our mutant studies support our model in Scheme 1.
The Initiation Rate Is Much Slower Than the Rate of Elongation in the Polymerization Process
For all PhaCs investigated mechanistically to date, the rate of elongation is much greater than that of initiation, thwarting efforts to understand these processes.9,17,18 To investigate the uniformity of loading of PhaCCc, the protein was incubated with 5 or 50 equiv of [1-14C]HBCoA and analyzed by SDS–PAGE and autoradiography (Figure 2A,B). In both reactions, the synthase remains predominantly unmodified and all of the radioactivity was associated with a species that failed to enter the gel (Figure 2B). The reactions were then repeated, and the PHB was extracted into chloroform for GPC analysis of its Mw. PHB with Mw values of 28 and 64 kDa was formed from the reaction mixtures containing 5 and 50 equiv of [1-14C]HBCoA, respectively. Thus, as in the case of PhaCRe and PhaECAv, the rate of elongation by PhaCCc is much faster than the rate of initiation and a small portion of the enzyme generates a large polymer from the available substrate.
Figure 2.

SDS–PAGE (10%) and autoradiography monitoring the reaction of PhaCCc and sT-PhaCCc with 5 or 50 equiv of [1-14C]HBCoA. (A) Coomassie-stained gel. Lanes 1–3 are as described for panel B. (B) Autoradiography of the gel in panel A. Lanes: M, molecular weight standards; lane 1, PhaC (3 μg); lanes 2 and 3, PhaC (3 μg) reacted with [1-14C]HBCoA at the indicated substrate:enzyme ratio (S:E). The specific activity of [1-14C]HBCoA is 2 × 105 cpm/nmol in lane 2 and 2 × 104 cpm/nmol in lane 3. (C) Coomassie-stained gel. Lanes 1–3 are as described for panel D. (D) Autoradiography of the gel in panel C: M, molecular weight standards; lane 1, PhaC (3 μg); lanes 2 and 3, PhaC (3 μg) reacted with 10 equiv of sTCoA for 1 min and then chased with 50 and 5 equiv of [1-14C]HBCoA, respectively. The specific activity of [1-14C]HBCoA is 2 × 104 cpm/nmol in lane 2 and 2 × 105 cpm/nmol in lane 3. W, wells. GF, gel front.
Priming with sTCoA Increases the Uniformity of Loading of PhaCCc with HBCoA
In an effort to uniformly load PhaCCc so that the elongation process could be studied, PhaCCc was incubated with 10 equiv of sTCoA, and CoA release was monitored. As shown in Figure S4 of the Supporting Information, by 10 s, 0.90 ± 0.07 mol of CoA per mol of PhaCCc was released. This burst was followed by CoA release at a rate of 0.004 s–1, indicating hydrolysis followed by reacylation. The stoichiometry of labeling suggested that [3H]sT-PhaCCc might be isolable if Sephadex G25 chromatography is used. However, these efforts, even at pH 5 where thioesters are stable, resulted in 80–90% loss of the label from the protein. Thus, all subsequent experiments have unreacted sTCoA present.
To determine if the sT of sT-PhaCCc could be elongated into PHB, [3H]sT-PhaCCc was incubated with 20000 equiv of HBCoA for 30 min. A control was run under identical conditions with unprimed PhaCCc and 20000 equiv of [1-14C]HBCoA. The PHB produced in these reactions was extracted into chloroform and its Mw determined by GPC analysis. In the case of the control, PHB had a retention time of 12.9 min, corresponding to an Mw of 680 kDa (Figure S5A of the Supporting Information). With [3H]sT-PhaCCc a broad peak with a retention time of 13.7 min was observed, corresponding to [3H]PHB with an Mw of ∼440 kDa (Figure S5B of the Supporting Information). Quantitation of the radiolabel indicated that 72% of the [3H]sT was chased into the polymer. When the rate of CoA release of sT-PhaCCc reacting with HBCoA was monitored, the specific activity increased 1.7-fold relative to that of the unprimed reaction (Figure S6 of the Supporting Information). These studies together demonstrate the chemical and kinetic competence of the sT species.
To examine whether sT acylation allowed uniform extension of sT-PhaC, 5 and 50 equiv of [1-14C]HBCoA were added and the products examined by SDS–PAGE and autoradiography (Figure 2C,D). The samples loaded onto the gel were not boiled or treated with reductant. Four regions of radioactivity were observed (species I–IV), compared to the single region for the unprimed reaction (Figure 2B). These results are similar to the distribution of radioactive species observed by the same method of analysis when unprimed class III PhaECAv was reacted with 5–45 equiv of [1-14C]HBCoA, an additional similarity between the two synthases.18 By analogy to the unprimed reaction (Figure 2A,B), species I and II may be insoluble high-Mw PHB that fails to enter the separating gel. However, species II appears as two distinct bands, and the lower band migrates into the gel. Our interpretation of our previous results of the reaction of PhaECAv with [1-14C]HBCoA was that species II might be an oligomeric form of the synthase stabilized to electrophoresis by its association with the small PHB polymer. A similar interpretation might be put forward for the results with PhaCCc. SEC studies of PhaCCc reveal that it migrates between dimeric or trimeric forms (not shown); thus, we propose that species II may be a stabilized dimer or trimer [14C]-(HB)n-PhaCCc complex. However, it should be noted that we do not fully understand how (HB)n-PhaC species migrate via SDS–PAGE.
Interestingly, radioactive species designated III and IV that migrate just above and at the position of PhaCCc, respectively, are also observed. As in the unprimed reaction, the majority of the PhaC migrates at the position of the unmodified synthase; however, the autoradiograph reveals associated radioactivity (species IV). This species is likely monomeric PhaCCc covalently attached to small (HB)n oligomers. Species III is also likely to be monomeric PhaCCc attached to (HB)n that is larger than in species IV. Together, these findings suggest that priming with sTCoA does increase the uniformity of loading of PhaCCc such that PhaCCc-(HB)n species (where n is small) are observed.
[3H]sT-PhaC Forms [3H]sDCoA in the Absence of HBCoA
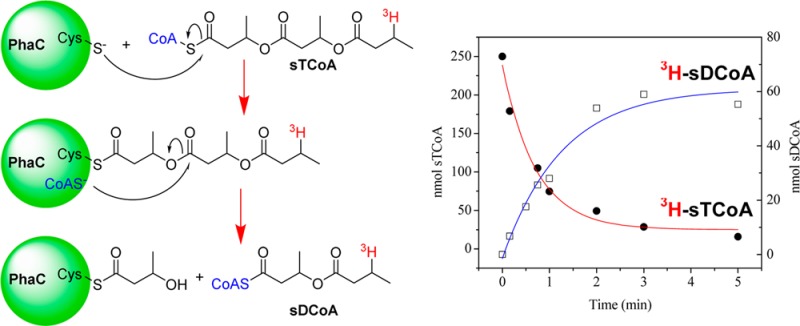
The Sephadex experiments described above demonstrated the lability of the thioester linkage to PhaCCc despite the apparent slow rate of hydrolysis indicated by CoA release subsequent to acylation (Figure S4 of the Supporting Information). To understand the basis of this lability, this process was investigated further by monitoring the reaction of PhaCCc (25 nmol) with [3H]sTCoA (250 nmol). Time points were taken from 10 s to 5 min and the reactions quenched with TCA. The precipitated protein was pelleted, washed, and repelleted, and the combined supernatants were analyzed by HPLC by monitoring A260 and radioactivity (Figure 3 and Figure S7 of the Supporting Information). The labeled peaks in Figure 3A were assigned on the basis of their retention times (tR) relative to authentic standards [CoA (18 min), [3H]sTCoA (47 min), and [3H]sT4CoA (54 min)], except for sDCoA, which was assigned by MALDI-TOF MS, as described subsequently. The time course analysis is shown in panels B and C of Figure 3 and for all species in Figure S7 of the Supporting Information. The protein pellet, analyzed by scintillation counting, contained 0.9 equiv of 3H label per PhaCCc, which decreased with time to 0.6 equiv per PhaCCc (Figure 3B, blue circles).
Figure 3.

Products of the reaction of PhaCCc with [3H]sTCoA. (A) Overlays of HPLC traces of aliquots taken after incubation for 10 s (red), 45 s (green), and 5 min (blue). The peak of radioactivity at 37 min was present in the control. Peaks are labeled as they are described in the text. (B) Amount of CoA (red stars) and [3H]sT associated with PhaC (blue, filled circles) as a function of time. (C) Disappearance of [3H]sTCoA (red, filled circles) and formation of [3H]sDCoA (blue, empty squares) as a function of time.
The HPLC trace in Figure 3A is complex, but the species eluting at tRs of 18, 43, 47, and 54 min are associated with A260 alone or A260 and radioactivity and are kinetically well-behaved. The [3H]sTCoA (tR = 47 min) decreases with a rate constant of 0.023 s–1 (Figure 3C). This rate constant is faster than that of CoA release, which occurs at 0.01 s–1 subsequent to its burst of formation in amounts approximately stoichiometric with PhaCCc (Figure 3B, red stars). Both the rates and amounts of CoA release are similar to the data from the DTNB assays (Figure S4 of the Supporting Information). The loss of [3H]sTCoA measured by scintillation counting does not agree with the changes in A260 (compare the top and bottom traces of Figure 3A). This difference is likely associated with the [3H]sT acid hydrolysis products of [3H]sTCoA that elute as broad peaks with similar tRs.31
The faster loss of starting material relative to CoA appearance suggests that CoA is incorporated into an additional product(s). One of these products (tR = 54 min) has been identified as sT4CoA (see the Supporting Information and Figure S8A for details). Its identification and kinetics of formation are presented in Figure S7 of the Supporting Information, and its production is likely associated with small amounts of contaminating HBCoA, a breakdown product of [3H]sTCoA always generated during its storage.11 The most interesting and major product appearing has a tR of 43 min (Figure 3A) and grows in with a rate constant of 0.017 s–1. The rate constant for CoA release added to that associated with formation of the feature at a tR of 43 min are approximately equal to the rate of [3H]sTCoA disappearance.
To identify this new species, the reaction was run on a larger scale, and the product was isolated by HPLC and analyzed by positive mode MALDI-TOF MS. The species has an ion at m/z 924.76 (Figure S8B of the Supporting Information), consistent with the structure of the sDCoA (Figure 1), which has a calculated m/z of 924.71. The simplest interpretation of this data is that the CoA released on acylation of PhaCCc with [3H]sTCoA partitions between dissociation from the active site and thiolysis of the penultimate oxygen ester of sT-PhaCCc. This reaction is reminiscent of the reaction between sT-PhaCRe and HBCH2CoA (Figure 1) and will be discussed subsequently.19
Discussion
Studies with PhaCCc, in comparison with PhaCRe and PhaECAv, reveal that it has properties that are a hybrid between the properties of these more extensively studied synthases. Like PhaCRe, PhaCCc is a class I synthase composed of a single subunit but has an unusual N-terminal domain that is required for full activity. Kinetically, however, PhaCCc is reminiscent of the class III PhaECAv: it has no lag phase in CoA release during polymerization, and an active site Cys to Ser mutation results in an active enzyme. Finally, PhaCCc is very soluble and well-behaved compared with PhaCRe, which requires the presence of Hecameg, a nonionic detergent, to prevent protein aggregation.9 Our studies have revealed three important findings about PhaCCc, discussed subsequently, that have mechanistic implications.
The first is the observation that PhaCCc can be primed with sTCoA with a stoichiometry of 1 sT per PhaC. In contrast, both PhaCRe and PhaECAv, where the active PhaC unit is proposed to be a dimer,13−15 are primed with a stoichiometry of 0.5 sT per PhaC.13,14 At the time of these experiments, our favored explanation, based on our understanding of fatty acid synthases, was that a Cys from each monomer of PhaC formed a single active site at the protein dimer interface. This active site in turn generated one PHB chain, which transferred back and forth between the Cys during chain elongation.1,2 However, it is difficult to reconcile an active site shared between two PhaC monomers based on the predicted structural similarities between PhaCs and lipases,14 as most lipases are monomers with deeply buried active sites.32,33 Our data showing that PhaCCc can be uniformly labeled with 1 equiv of sTCoA thus provide additional support for our model shown in Scheme 1, assuming all PhaCs use similar mechanistic strategies.1,2
A second finding from our studies is that priming of PhaCCc with sTCoA increases the uniformity of elongation, allowing detection of various (HB)n-PhaCCc species (Figure 2A–D). When 5–50 equiv of [1-14C]HBCoA is added to the unprimed synthase, a small portion of the enzyme initiates and synthesizes the large polymer (see Figure 2A,B), suggesting that the rate of initiation is much slower than the rate of elongation. This same observation has been made with both PhaCRe and PhaECAv in vitro.9,17,18,31 In contrast to the unprimed PhaCCc, when 5 or 50 equiv of [1-14C]HBCoA was added to sT-PhaCCc, a distinct distribution of species is observed by SDS–PAGE and autoradiography (see Figure 2C,D). The distribution is similar to that observed when PhaECAv was incubated with 5–45 equiv of [1-14C]HBCoA.18 The similarity of these species between the class I and class III synthases suggests that they are mechanistically important in PHB formation. We propose that the species migrating at the position of PhaCCc (species IV) represents monomeric PhaCs attached to short (HB)n chains where n is 4–10, by analogy to similar species observed by MS analysis of PhaECAv incubated with sTCoA containing small amounts of contaminating HBCoA.11 Species III migrates more slowly than PhaCCc, and on the basis of its apparent increase of ∼15 kDa relative to that of the unmodified synthase, we propose that it is PhaCCc attached to (HB)n where n is ∼170. Finally, the high-Mw species that is sufficiently soluble to enter the gel (species II) may in fact be oligomeric PhaCCc stabilized through interaction with HB oligomers. However, it should be noted that we do not yet have a good understanding of the structure or oligomeric state of PhaCCc. The various PHB species, observed with both PhaCCc and PhaECAv, may represent distinct stages in polymerization, the levels of which are increased by priming with sTCoA such that they can be detected in these experiments. However, high-Mw species that fail to enter the gel still form in the case of both synthases, suggesting that while a portion of the PhaCCc can uniformly elongate, most of the substrate is still incorporated into the large polymer. This may occur when a small population of PhaC synthesizes PHB of a certain Mw [species III (Figure 2D)], which induces a conformational change to facilitate rapid elongation.
A final interesting result is the unexpected observation that sT-PhaCCc generates sDCoA and presumably HB-PhaCCc as the major products when HBCoA is omitted from the reaction. This reaction likely occurs through thiolysis with CoA at the penultimate oxoester position, rather than at the thioester where chemistry is expected to occur during elongation. Our interpretation of this finding is that sT-PhaCCc, in the absence of HBCoA, is in a conformation that is distinct from the elongation conformation and promotes thiolysis chemistry instead. In the presence of HBCoA, a conformational change occurs and rapid chain elongation proceeds, as demonstrated by SDS–PAGE and autoradiography and GPC studies. However, the omission of HBCoA precludes this rapid elongation and allows us to observe the slower thiolysis reaction (Scheme 2). We propose that this reaction is reporting on a termination mechanism in which CoA is the chain terminator.
Scheme 2. Fates of sT-PhaC in the Presence and Absence of HBCoA.
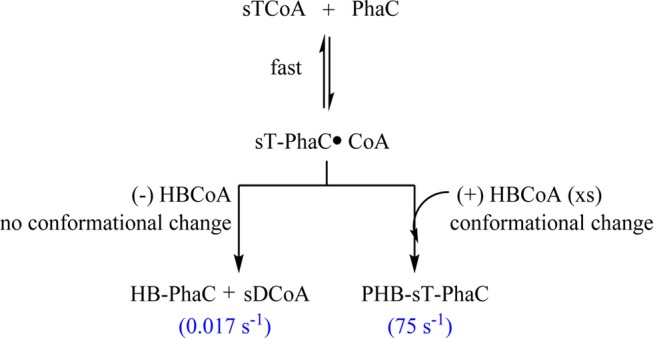
This interpretation is supported by the recent studies with sT-PhaCRe incubated with HBCH2CoA (Figure 1).19 Instead of observing rapid elongation to form sT4CH2CoA, we observed slow formation of sTCH2CoA (Figure 1). Indeed, in the case of both PhaCCc and PhaCRe, the relative rates of turnover versus the very slow rate of formation of the unexpected products from the sT-PhaC can account for the Mw of PHB produced.19 With PhaCCc, if the rate constant for [3H]sDCoA formation (0.017 s–1) is reporting on termination and the turnover number is 75 s–1, then on average, a polymer of ∼4500 HB units (390 kDa) would be synthesized before termination occurs. This Mw is similar to that of PHB isolated from cultures of C. crescentus grown under nitrogen-limited conditions (330–360 kDa) (R. M. Buckley and J. Stubbe, unpublished results). With PhaCRe, the product sTCH2CoA is generated at a rate of 0.01–0.02 s–1 and the turnover number is 200 s–1; given these relative rate constants, one would expect 1–2 × 104 HB units would be polymerized before termination occurred, consistent with PHB with an Mw of 1–2 MDa produced by the organism.19 With both sT-PhaCs, HB-PhaC remains following termination. While the sT-PhaC (which closely resembles a trimer of HB) is not indicative of the priming process that likely occurs in vivo, we believe it reveals two important properties of all synthases: that PhaC is sufficient for termination and that the conformation of the (HB)n-PhaC plays a key role in chain extension versus termination.
Our previous studies with PhaECAv were also interpreted in terms of the synthase’s ability to catalyze termination to leave acylated PhaC. Incubation of PhaECAv with 45 equiv of [1-14C]HBCoA for an extended period of time resulted in conversion of the initially acylated (HB)n-PhaC, where n was estimated to be 40–100, to (HB)n-PhaC, where n = 3–10.18 Additionally, PhaECAv reacted with the poor substrate HB-NAC (HB-N-acetylcysteamine, an HBCoA analogue in which CoA is replaced with NAC) produced ∼75 kDa PHB, with NAC at its terminus, suggesting that this thiol acts as a chain terminator and also partitions between release from the active site (and continued polymerization) and termination.24 Intriguingly, re-examination of the fate of [3H]sT-PhaECAv reveals the formation of a species with an HPLC retention time consistent with that of sDCoA, albeit at a greatly reduced rate relative to that of PhaCCc (Figure S9 of the Supporting Information), suggesting that termination by thiolysis chemistry may be general rather than off-pathway.
The path to engineering recombinant systems for the production of PHAs with high Mw and low polydispersity, with useful properties, requires an understanding of the factors that control these properties in vivo. The results presented for PhaCCc and previously for PhaCRe19,34,35 and PhaECAv18,24,36 demonstrate that PhaCs are sufficient to catalyze termination and that active site conformational states govern the relative rates of elongation and termination. The studies presented here, however, have not addressed the role and importance of additional protein factors, such as the phasin proteins (PhaPs), which are granule-associated proteins involved in PHB production in all organisms.37,38 Early studies demonstrated that expression levels of phasins controlled the amounts of PHB produced in R. eutropha.37,39−41 However, our recent studies suggest the importance of kinetic coupling between the rates of phasin production and the rates of PHB chain elongation in both C. crescentus (R. M. Buckley and J. Stubbe, unpublished results) and R. eutropha (M. Cho and J. Stubbe, unpublished results). Taking into account all of these factors is key to the success of commercialization of PHAs.
Acknowledgments
We thank Prof. Ping Li for synthesizing 3-(R)-HBCoA, sTCoA, and [3H]sTCoA and for helpful discussions. We thank Prof. Michael Laub for providing C. crescentus CB15 for molecular cloning. We thank Dr. Mimi Cho and Dr. Phillip W. Snyder for helpful discussions.
Glossary
Abbreviations
- PHB
polyhydroxybutyrate
- HBCoA
3-(R)-hydroxybutyryl coenzyme A
- PhaCCc
PHB synthase from C. crescentus
- CoA
coenzyme A
- sTCoA
saturated trimer CoA, an analogue of (HB)3CoA in which the terminal hydroxyl group is replaced by H
- HB
hydroxybutyrate
- PHA
polyhydroxyalkanoate
- PhaCRe
PHB synthase from R. eutropha
- PhaECAv
PHB synthase from A. vinosum
- sT-PhaC
PhaC labeled with a terminally saturated (HB)3
- HBCH2CoA
analogue of HBCoA in which the S of the thioester is replaced with a CH2
- sTCH2CoA
analogue of a saturated tetramer CoA in which the S of the thioester is replaced with a CH2
- sTCH2CoA
analogue of sTCoA in which the S of the thioester is replaced with a CH2
- sDCoA
saturated dimer CoA, an analogue of (HB)2CoA in which the terminal OH is replaced by H
- ΔPhaCCc
65 kDa construct of PhaCCc that is missing the 85 N-terminal amino acids
- cv
column volumes
- TCA
trichloroacetic acid
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- GPC
gel permeation chromatography
- Mw
molecular weight
- NB
nutrient broth medium
- PhaP
phasin protein
- HB-NAC
3-(R)-hydroxybutyryl-N-acetylcysteamine.
Supporting Information Available
Plasmids and primers used in this study, protein purification details, and figures referenced in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health Grant GM49171 to J.S.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Stubbe J.; Tian J. (2003) Polyhydroxyalkanoate (PHA) homeostasis: The role of the PHA synthase. Nat. Prod. Rep. 20, 445–457. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Tian J.; He A.; Sinskey A. J.; Lawrence A. G.; Liu P. (2005) Nontemplate-dependent polymerization processes: Polyhydroxyalkanoate synthases as a paradigm. Annu. Rev. Biochem. 74, 433–480. [DOI] [PubMed] [Google Scholar]
- Steinbüchel A.; Valentin H. E. (1995) Diversity of bacterial polyhydroxyalkanoic acids. FEMS Microbiol. Lett. 128, 219–228. [Google Scholar]
- Sudesh K.; Abe H.; Doi Y. (2000) Synthesis, structure and properties of polyhydroxyalkanoates: Biological polyesters. Prog. Polym. Sci. 25, 1503–1555. [Google Scholar]
- Steinbüchel A. (2001) Perspectives for Biotechnological Production and Utilization of Biopolymers: Metabolic Engineering of Polyhydroxyalkanoate Biosynthesis Pathways as a Successful Example. Macromol. Biosci. 1, 1–24. [Google Scholar]
- Keshavarz T.; Roy I. (2010) Polyhydroxyalkanoates: Bioplastics with a green agenda. Curr. Opin. Microbiol. 13, 321–326. [DOI] [PubMed] [Google Scholar]
- Snell K. D.; Peoples O. P. (2009) PHA Bioplastic: A value-added coproduct for biomass biorefineries. Biofuels, Bioprod. Biorefin. 3, 456–467. [Google Scholar]
- Peoples O. P.; Sinskey A. J. (1989) Poly-β-hydroxybutyrate (PHB) biosynthesis in Alcaligenes eutrophus H16. Identification and characterization of the PHB polymerase gene (phbC). J. Biol. Chem. 264, 15298–15303. [PubMed] [Google Scholar]
- Gerngross T. U.; Snell K. D.; Peoples O. P.; Sinskey A. J.; Csuhai E.; Masamune S.; Stubbe J. (1994) Overexpression and purification of the soluble polyhydroxyalkanoate synthase from Alcaligenes eutrophus: Evidence for a required posttranslational modification for catalytic activity. Biochemistry 33, 9311–9320. [DOI] [PubMed] [Google Scholar]
- Liebergesell M.; Sonomoto K.; Madkour M.; Mayer F.; Steinbüchel A. (1994) Purification and characterization of the poly(hydroxyalkanoic acid) synthase from Chromatium vinosum and localization of the enzyme at the surface of poly(hydroxyalkanoic acid) granules. Eur. J. Biochem. 226, 71–80. [DOI] [PubMed] [Google Scholar]
- Müh U.; Sinskey A. J.; Kirby D. P.; Lane W. S.; Stubbe J. (1999) PHA Synthase from Chromatium vinosum: Cysteine 149 is involved in covalent catalysis. Biochemistry 38, 826–837. [DOI] [PubMed] [Google Scholar]
- Liebergesell M.; Steinbüchel A. (1992) Cloning and nucleotide sequences of genes relevant for biosynthesis of poly(3-hydroxybutyric acid) in Chromatium vinosum strain D. Eur. J. Biochem. 209, 135–150. [DOI] [PubMed] [Google Scholar]
- Wodzinska J.; Snell K. D.; Rhomberg A.; Sinskey A. J.; Biemann K.; Stubbe J. (1996) Polyhydroxybutyrate synthase: Evidence for covalent catalysis. J. Am. Chem. Soc. 118, 6319–6320. [Google Scholar]
- Jia Y.; Kappock T. J.; Frick T.; Sinskey A. J.; Stubbe J. (2000) Lipases provide a new mechanistic model for polyhydroxybutyrate (PHB) synthases: Characterization of the functional residues in Chromatium vinosum PHB synthase. Biochemistry 39, 3927–3936. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Yuan W.; Wodzinska J.; Park J.; Sinskey A. J.; Stubbe J. (2001) Mechanistic studies on class I polyhydroxybutyrate (PHB) synthase from Ralstonia eutropha: Class I and III synthases share a similar catalytic mechanism. Biochemistry 40, 1011–1019. [DOI] [PubMed] [Google Scholar]
- Rehm B. H.; Antonio R. V.; Spiekermann P.; Amara A. A.; Steinbüchel A. (2002) Molecular characterization of the poly(3-hydroxybutyrate) (PHB) synthase from Ralstonia eutropha: In vitro evolution, site-specific mutagenesis and development of a PHB synthase protein model. Biochim. Biophys. Acta 1594, 178–190. [DOI] [PubMed] [Google Scholar]
- Tian J.; Sinskey A. J.; Stubbe J. (2005) Detection of intermediates from the polymerization reaction catalyzed by a D302A mutant of class III polyhydroxyalkanoate (PHA) synthase. Biochemistry 44, 1495–1503. [DOI] [PubMed] [Google Scholar]
- Tian J.; Sinskey A. J.; Stubbe J. (2005) Class III polyhydroxybutyrate synthase: Involvement in chain termination and reinitiation. Biochemistry 44, 8369–8377. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Shrestha R.; Buckley R. M.; Jewell J.; Bossmann S. H.; Stubbe J.; Li P. (2014) Mechanistic insight with HBCH2CoA as a probe to polyhydroxybutyrate (PHB) synthases. ACS Chem. Biol. 9, 1773–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y.; Doi Y. (1992) Kinetics and mechanism of synthesis and degradation of poly(3-hydroxybutyrate) in Alcaligenes eutrophus. Macromolecules 25, 2324–2329. [Google Scholar]
- Kusaka S.; Abe H.; Lee S. Y.; Doi Y. (1997) Molecular mass of poly[(R)-3-hydroxybutyric acid] produced in a recombinant Escherichia coli. Appl. Microbiol. Biotechnol. 47, 140–143. [DOI] [PubMed] [Google Scholar]
- Kurja J.; Zirkzee H. F.; de Koning G. M.; Maxwell I. A. (1995) A new kinetic model for the accumulation of poly(3-hydroxybutyrate) in Alcaligenes eutrophus, 1. Granule growth. Macromol. Theory Simul. 4, 839–855. [Google Scholar]
- Madden L. A.; Anderson A. J.; Shah D. T.; Asrar J. (1999) Chain termination in polyhydroxyalkanoate synthesis: Involvement of exogenous hydroxy-compounds as chain transfer agents. Int. J. Biol. Macromol. 25, 43–53. [DOI] [PubMed] [Google Scholar]
- Lawrence A. G.; Choi J.; Rha C.; Stubbe J.; Sinskey A. J. (2005) In vitro analysis of the chain termination reaction in the synthesis of poly-(R)-β-hydroxybutyrate by the class III synthase from Allochromatium vinosum. Biomacromolecules 6, 2113–2119. [DOI] [PubMed] [Google Scholar]
- Yuan W.; Jia Y.; Tian J.; Snell K. D.; Muh U.; Sinskey A. J.; Lambalot R. H.; Walsh C. T.; Stubbe J. (2001) Class I and III polyhydroxyalkanoate synthases from Ralstonia eutropha and Allochromatium vinosum: Characterization and substrate specificity studies. Arch. Biochem. Biophys. 394, 87–98. [DOI] [PubMed] [Google Scholar]
- Heckman K. L.; Pease L. R. (2007) Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. (1976) Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Michaelis L.; Menten M. M. (1913) The kinetics of invertin action. FEBS Lett. 587, 2712–2720. [DOI] [PubMed] [Google Scholar]
- Barcroft J.; Hill A. V. (1910) The nature of oxyhaemoglobin, with a note on its molecular weight. J. Physiol. 39, 411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Q.; Rehm B. A. (2001) Polyhydroxybutyrate biosynthesis in Caulobacter crescentus: Molecular characterization of the polyhydroxybutyrate synthase. Microbiology 147, 3353–3358. [DOI] [PubMed] [Google Scholar]
- Li P.; Chakraborty S.; Stubbe J. (2009) Detection of covalent and noncovalent intermediates in the polymerization reaction catalyzed by a C149S class III polyhydroxybutyrate synthase. Biochemistry 48, 9202–9211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss J.; Fischer M.; Schmid R. D. (1998) Anatomy of lipase binding sites: The scissile fatty acid binding site. Chem. Phys. Lipids 93, 67–80. [DOI] [PubMed] [Google Scholar]
- Lang D.; Hofmann B.; Haalck L.; Hecht H.; Spener F.; Schmid R. D.; Schomburg D. (1996) Crystal structure of a bacterial lipase from Chromobacterium viscosum ATCC 6918 refined at 1.6 Å resolution. J. Mol. Biol. 259, 704–717. [DOI] [PubMed] [Google Scholar]
- Doi Y.; Kawaguchi Y.; Koyama N.; Nakamura S.; Hiramitsu M.; Yoshida Y.; Kimura H. (1992) Synthesis and degradation of polyhydroxyalkanoates in Alcaligenes eutrophus. FEMS Microbiol. Rev. 103, 103–108. [Google Scholar]
- Gerngross T. U.; Martin D. P. (1995) Enzyme-catalyzed synthesis of poly[(R)-(−)-3-hydroxybutyrate]: Formation of macroscopic granules in vitro. Proc. Natl. Acad. Sci. U.S.A. 92, 6279–6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossek R.; Reichelt R.; Steinbüchel A. (1998) In vitro biosynthesis of poly(3-hydroxybutyric acid) by using purified poly(hydroxyalkanoic acid) synthase of Chromatium vinosum. Appl. Microbiol. Biotechnol. 49, 258–266. [DOI] [PubMed] [Google Scholar]
- Wieczorek R.; Pries A.; Steinbüchel A.; Mayer F. (1995) Analysis of a 24-kilodalton protein associated with the polyhydroxyalkanoic acid granules in Alcaligenes eutrophus. J. Bacteriol. 177, 2425–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek R.; Steinbüchel A.; Schmidt B. (1996) Occurrence of polyhydroxyalkanoic acid granule-associated proteins related to the Alcaligenes eutrophus H16 GA24 protein in other bacteria. FEMS Microbiol. Lett. 135, 23–30. [DOI] [PubMed] [Google Scholar]
- Pötter M.; Madkour M. H.; Mayer F.; Steinbüchel A. (2002) Regulation of phasin expression and polyhydroxyalkanoate (PHA) granule formation in Ralstonia eutropha H16. Microbiology 148, 2413–2426. [DOI] [PubMed] [Google Scholar]
- York G. M.; Junker B. H.; Stubbe J. A.; Sinskey A. J. (2001) Accumulation of the PhaP phasin of Ralstonia eutropha is dependent on production of polyhydroxybutyrate in cells. J. Bacteriol. 183, 4217–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York G. M.; Stubbe J.; Sinskey A. J. (2001) New insight into the role of the PhaP phasin of Ralstonia eutropha in promoting synthesis of polyhydroxybutyrate. J. Bacteriol. 183, 2394–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.