

In their recent paper, “Poststress block of kappa opioid receptors rescues long-term potentiation of inhibitory synapses and prevents reinstatement of cocaine seeking,” Polter and colleagues showed that cold-water swim stress exposure blocks LTP of GABAergic synaptic input recorded in ventral tegmental area (VTA) dopamine neurons of male Sprague-Dawley rats (1). The effects of stress exposure on LTPGABA were blocked by pretreatment with the glucocorticoid antagonist RU-486 or the kappa opioid receptor (KOR) antagonist norbinaltorphimine (norBNI). Importantly, norBNI given 4 days after stress exposure could reverse the effects of swim-stress on LTPGABA and block stress-induced reinstatement of cocaine self-administration. Because stress-exposure is such a well-recognized risk factor for the development of compulsive drug abuse and relapse during intervals of abstinence, the demonstration that norBNI given after stress exposure could promote stress-resilience and reverse the adverse effects of prior stress exposure are important preclinical advances in the development of addiction therapeutics. The Polter study also increases our understanding of the complex effects of stress on the reward circuitry controlling addictive drug effects.
Multiple synaptic inputs converge on the VTA dopaminergic neurons to regulate their excitability, and these neurons, in turn, project broadly to critical targets in the brain to coordinate the drug seeking behaviors initiated by addictive drugs (2) (Figure 1). Understanding this circuit and defining the effects of stress-mediators on the functioning of these neurons seems vital in the development of novel treatments for addiction, which has appropriately been described as a ‘stress-surfeit’ disorder by Koob and colleagues (3). The intimate relationship between stress vulnerability and addiction risk was summarized there and in other reviews on this topic, and the reciprocal concepts that stress exposure increases addiction risk and that addictive drug exposure increases stress vulnerability have now been supported by an extensive literature. Breaking this cycle by blocking the actions of the stress-mediators holds great promise, but understanding how these antagonists act on the reward circuit requires the kind of high-resolution circuit analysis described by Polter and colleagues (1).
Figure 1.
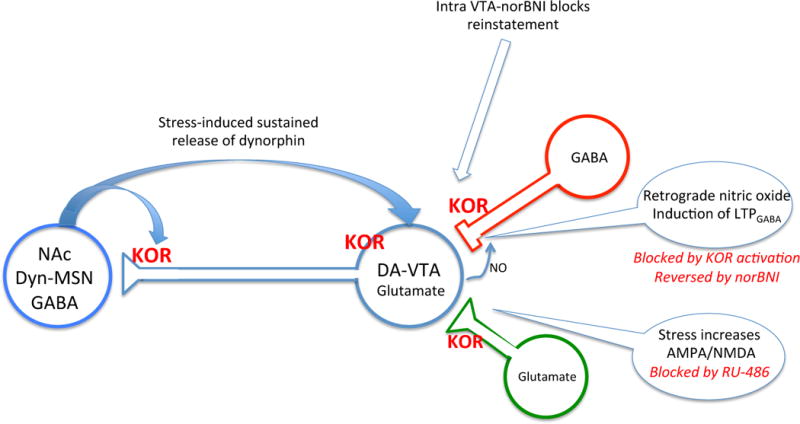
The ventral tegmental area (VTA) is a key player in the brain’s reward system, and the stress-mediators CRF, corticosterone, and dynorphin have dramatic effects on the neurophysiology of this brain region. Some of the principal actions of dynorphin described in the Polter et al study are illustrated in this diagram. The VTA-DA neurons release dopamine and glutamate and project broadly to prefrontal cortex, nucleus accumbens (NAc), hypothalamus, amygdala, lateral habenula, pallidum and bed nucleus of the stria terminalis (BNST). The VTA-DA neuron activity signals ‘incentive salience’ to the NAc, but are not uniquely activated by rewarding stimuli. These neurons are thought to have biophysical characteristic signatures including the expression of the Ih current (but this alone is not considered to be a sufficient identifier). Both excitatory and inhibitory neurons control VTA-DA neuron firing rates. Heterogeneity of the VTA-DA projections and inputs are evident and need further characterization before the functional complexity of this synaptic organization can be understood. Stress mediators have profound and complex effects on the physiology of this system (2), but the key sites of kappa opioid receptor expression are illustrated in this diagram (5). Dynorphin containing afferents to the VTA come from the medium spiny neurons of the NAc (shown), but also from the hypothalamus, amygdala, and BNST (not shown). Kappa receptors are also expressed on multiple elements of this circuit including on both the excitatory and inhibitory inputs as well as on the VTA-DA neurons themselves. Stress-induced activation of dynorphin release is likely to activate all of these components and local norBNI injection will inactivate all of these KORs in ways that are not yet fully understood. The Polter et al study (1) focuses on the dynorphin/kappa regulation of GABAergic input to VTA-DA neurons illustrated in the diagram. Their studies show that KOR activation (pharmacologically or following stress-induced dynorphin release) blocks LTPGABA at this synapse. The source of dynorphin is not yet known, and how dynorphin affects the other sites is not yet clear, but a clear connection to behavior is shown by the effects of norBNI on stress-induced reinstatement of cocaine self-administration reported in this study.
Stress exposure produces adaptive physiological responses vital for the individual’s survival. As such, some mildly stressful experiences can be perceived as ‘exciting’ and positive when controlled by the individual, but persistent uncontrolled stressful experiences are aversive and can cause lasting changes in the reward circuitry (4). Corticotrophin releasing factor (CRF) orchestrates the classically recognized stress response by activating the hypothalamic-pituitary-adrenal glucocorticoid response, but CRF also acts intracerebrally to produce some of the cognitive and emotional responses to stress. These can be adaptive by promoting escape behaviors, but can be maladaptive when promoting mood and cognitive disorders. The mechanisms of these maladaptive responses are being actively studied by many research groups, but a key role of CRF-induced activation of the endogenous dynorphin opioid peptides and their cognate kappa opioid receptors has been implicated as essential mediators of these adverse responses (5). Dynorphins have been shown to encode the dysphoric effects of stress (6); stress-induced release of dynorphins potentiates the rewarding effects of drugs of abuse including cocaine, ethanol, and nicotine (7); and the dynorphins mediate stress-induced reinstatement of drug seeking behaviors (5). Mice lacking the prodynorphin or KOR genes or pretreated with the KOR antagonists norBNI or JDtic demonstrate stress-resilience in assays of anxiety-like, aversion, and reinstatement behaviors (5). This preclinical research suggests that KOR antagonists may have therapeutic potential, and results from early clinical studies support this conception (8).
Nevertheless, understanding of how dynorphin acts in brain to increase addiction and mood disorder risk is still incomplete. Kappa opioid regulation of serotonergic inputs from dorsal raphe to nucleus accumbens (NAc) has been described (9). Dynorphin released during stress exposure within the NAc (presumably by prodynorphin-expressing medium spiny neurons) activates KOR expressed on serotonergic nerve terminals; KOR stimulation activates p38 MAPK to increase cell surface expression of the serotonin transporter SERT; and the resulting transient hyposerotonergic state in the NAc is necessary for stress-induced aversion, social avoidance and reinstatement of extinguished cocaine place preference (9).
The Kauer lab has previously demonstrated that stress can block long-term potentiation of GABAergic inputs onto dopamine neurons (10). They further demonstrated that this effect of stress requires KOR activation, as it is blocked by pretreatment with either the mixed opiate antagonist naloxone or the selective KOR antagonist norBNI. Intriguingly, they also showed that selective injection of norBNI into the VTA attenuates stress-induced reinstatement to cocaine self-administration. This suggests that the potentiation of GABAergic signaling onto dopaminergic neurons may play a key role in reinstatement of cocaine-seeking behavior.
In this issue, Polter et al follow-up on that observation by probing the duration of stress-induced blockade of LTPGABA (1). They find that stress blocks LTPGABA at 1 and 5, but not 10 days after exposure to a single cold water forced swim. They further find that norBNI administered after stress still blocks its effects on GABAergic signaling, whether administered at 2 hr, 24 hr, or 4 days after stress exposure. This is independent of any effect on glucocorticoid signaling, as the authors confirmed results reported previously showing that norBNI pretreatment fails to alter serum corticosterone after stress exposure. Further, although the glucocorticoid receptor antagonist RU-486 can also block the effects of stress on LTPGABA induction, it only does so if administered within one hour of stress exposure. But most dramatically, they demonstrate that after extinction of cocaine self-administration, norBNI can block stress-induced reinstatement of cocaine-seeking even if administered subsequent to stress. These two results combined strongly suggest that in cocaine-experienced animals, a single stress exposure can induce a long-lasting state of dynorphin release and/or KOR activation that can mediate substantial alterations to behavior.
Future studies are needed to further define the key elements of this neural circuit and the sites of stress-induced regulation. The Polter et al study used systemic norBNI administration, and subsequent studies using cell-specific activation and inactivation methods will be revealing. For instance, it has also been demonstrated that KOR activation in the dorsal raphe nucleus is required for stress-induced reinstatement of place preference for cocaine (6). KOR-induced activation of p38 MAPK is also required for CPP reinstatement, an effect that may be mediated specifically by KORs located on serotonergic terminals within the nucleus accumbens (9). If stress acts by inducing a long-lasting state of ongoing dynorphin release, it is also possible that the effect may be mediated by inhibition of dopamine release via action at terminals in the nucleus accumbens, medial prefrontal cortex or even via direct inhibition of dopamine neurons in the VTA.
The development of better treatments for drug use relapse and its exacerbation by stress after prolonged abstinence from drug abuse in vulnerable individuals is urgently needed. Addictive disorders may have well documented pathologies, but the specific role of dynorphin in the human syndromes remains poorly defined. Kappa antagonists developed for human use will hopefully have beneficial effects in stress-vulnerable individuals, but a better understanding of dynorphin’s functions seems essential if we are to properly anticipate the range of beneficial and possibly adverse consequences that a KOR antagonist might clinically produce.
Acknowledgments
CC receives support from the National Institutes of Health grants KO5-DA20570, R21-MH102059, RO1-DA30074, and PO1-DA35764.
Footnotes
The authors report no financial conflicts of interest.
References
- 1.Polter AM, Bishop RA, Briand LA, Graziane NM, Pierce RC, Kauer JA. Poststress Block of Kappa Opioid Receptors Rescues Long-Term Potentiation of Inhibitory Synapses and Prevents Reinstatement of Cocaine Seeking. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.04.019. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polter AM, Kauer JA. Stress and VTA synapses: implications for addiction and depression. Eur J Neurosci. 2014;39:1179–1188. doi: 10.1111/ejn.12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW, Jr, George O. Addiction as a stress surfeit disorder. Neuropharmacology. 2014;76(Pt B):370–382. doi: 10.1016/j.neuropharm.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemos JC, Wanat MJ, Smith JS, Reyes BA, Hollon NG, Van Bockstaele EJ, Chavkin C, Phillips PE. Severe stress switches CRF action in the nucleus accumbens from appetitive to aversive. Nature. 2012;490:402–406. doi: 10.1038/nature11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van’t Veer A, Carlezon WA., Jr Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology (Berl) 2013;229:435–452. doi: 10.1007/s00213-013-3195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carroll FI, Carlezon WA., Jr Development of κ opioid receptor antagonists. J Med Chem. 2013;56:2178–2195. doi: 10.1021/jm301783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C. Selective p38α MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA. Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron. 2013;77:942–954. doi: 10.1016/j.neuron.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]