

Abstract
Bioaccumulative organohalogen chemicals, such as organochlorine (OC) insecticides, have been increasingly associated with disease etiology; however, the mechanistic link between chemical exposure and diseases, such as atherosclerosis, cancer, and diabetes, is complex and poorly defined. Systemic oxidative stress stemming from OC exposure might play a vital role in the development of these pathologies. Monocytes are important surveillance cells of the innate immune system that respond to extracellular signals possessing danger-associated molecular patterns by synthesizing oxyradicals, such as superoxide, for the purpose of combating infectious pathogens. We hypothesized that OC chemicals can be toxic to monocytes because of an inappropriate elevation in superoxide-derived reactive oxygen species (ROS) capable of causing cellular oxidative damage. Reactive oxyradicals are generated in monocytes in large part by NADPH oxidase (Nox). The present study was conducted to examine the ability of two chlorinated cyclodiene compounds, trans-nonachlor and dieldrin, as well as p,p′-DDE, a chlorinated alicyclic metabolite of DDT, to stimulate Nox activity in a human monocytic cell line and to elucidate the mechanisms for this activation. Human THP-1 monocytes treated with either trans-nonachlor or dieldrin (0.1–10 μM in the culture medium) exhibited elevated levels of intracellular ROS, as evidenced by complementary methods, including flow cytometry analysis using the probe DCFH-DA and hydroethidine-based fluorometric and UPLC-MS assays. In addition, the induced reactive oxygen flux caused by trans-nonachlor was also observed in two other cell lines, murine J774 macrophages and human HL-60 cells. The central role of Nox in OC-mediated oxidative stress was demonstrated by the attenuated superoxide production in OC-exposed monocytes treated with the Nox inhibitors diphenyleneiodonium and VAS-2870. Moreover, monocytes challenged with OCs exhibited increased phospho-p47phox levels and enhanced p47phox membrane localization compared to that in vehicle-treated cells. p47phox is a cytosolic regulatory subunit of Nox, and its phosphorylation and translocation to the NOX2 catalytic subunit in membranes is a requisite step for Nox assembly and activation. Dieldrin and trans-nonachlor treatments of monocytes also resulted in marked increases in arachidonic acid (AA) and eicosanoid production, which could be abrogated by the phospholipase A2 (PLA2) inhibitor arachidonoyltrifluoromethyl ketone (ATK) but not by calcium-independent PLA2 inhibitor bromoenol lactone. This suggested that cytosolic PLA2 plays a crucial role in the induction of Nox activity by increasing the intracellular pool of AA that activates protein kinase C, which phosphorylates p47phox. In addition, ATK also blocked OC-induced p47phox serine phosphorylation and attenuated ROS levels, which further supports the notion that the AA pool liberated by cytosolic PLA2 is responsible for Nox activation. Together, the results suggest that trans-nonachlor and dieldrin are capable of increasing intracellular superoxide levels via a Nox-dependent mechanism that relies on elevated intracellular AA levels. These findings are significant because chronic activation of monocytes by environmental toxicants might contribute to pathogenic oxidative stress and inflammation.
Graphical abstract

INTRODUCTION
Epidemiological studies have implicated bioaccumulative organochlorine (OC) pesticide exposure as a risk factor for diseases with a strong inflammatory component, such as atherosclerosis, cancer, and diabetes.1–3 Although the mechanistic relationship between OC chemical exposure and increased disease risk is complex and poorly defined, systemic oxidative stress induced by environmental chemicals is hypothesized to have an important role in disease development.3,4 Elevated levels of reactive oxygen species (ROS), in the context of pathogenesis, increasingly hinge on the inappropriate activation of oxyradical generating enzymes, such as mitochondrial complex III and NADPH oxidase (Nox).5 Nox is a multisubunit holoenzyme, and the only known biochemical function that it has is the biosynthesis of superoxide and/or hydrogen peroxide.6 It is the main ROS-producing enzyme during inflammation. Nox is expressed in many cell types, including, but not limited to, monocytes, macrophages, vascular endothelial cells, and smooth muscle cells. The Nox family consists of several membrane-embedded catalytic subunits (designated Nox1–5 and Dual Oxidase 1/2) that possess a unique tissue distribution and regulation. The principal Nox holoenzyme complex assembled in phagocytes responsible for the respiratory burst is composed of two membrane-bound components, NOX2 and p22phox, in addition to cytosolic regulatory subunits Rac, p47phox, p67phox, and p40phox. In the assembled Nox complex, two electrons are transferred from NADPH to the cofactor flavin adenine dinucleotide via hydride ion. The electrons are then passed sequentially to two heme moieties in the catalytic subunit (NOX2) via one-electron reductions and ultimately to two molecules of oxygen, yielding superoxide radical anion. Superoxide dismutates rapidly to oxygen and hydrogen peroxide, which is capable of crossing membranes. NOX2 is the major catalytic subunit in monocytes, which are important surveillance cells of the innate immune system that respond to extracellular signals possessing danger-associated molecular patterns by synthesizing Nox-derived oxyradicals to combat infectious pathogens. Nox also has important functions in signal transduction and the maintenance of normal physiology.6 However, the inappropriate activation of Nox and resulting increase in oxyradical flux has been implicated in disease development, including atherosclerosis and cardiomyopathy.5,7,8
Pro-oxidative stimuli in the vessel wall include vascular shear stress, oxidized low-density lipoprotein (LDL), isoprostanes, angiotensin II, and interferon γ.9,10 These stressful stimuli act through a variety of signaling networks to stimulate the concerted assembly of regulatory cytosolic Nox subunits with membrane-bound NOX2/p22phox subunits, thereby increasing Nox-dependent ROS levels. In addition to endogenous factors, environmental organohalogen chemicals can accumulate in adipose tissue and lipid-rich atherosclerotic plaques. For example, gas chromatography–mass spectrometry analysis of human aortic plaques detected numerous anthropogenic chemicals, including the organochlorine insecticide metabolite p,p′-DDE.11 Buildup of lipophilic xenobiotics in atheromatous plaques may contribute to atherogenesis because the higher concentrations of these compounds in the lesion as compared to that in the circulation would be expected to increase their bioavailability to cells in the vessel wall intima.
Xenobiotics are known to promote ROS formation in cells. For example, noxious chemicals in cigarette smoke can stimulate Nox-dependent ROS production in endothelial cells and smooth muscle cells.12,13 Furthermore, polychlorinated biphenyls (PCBs) have been shown to activate Nox in rat and human neutrophils,14–16 as did dieldrin and lindane, both chlorinated cyclodiene insecticides.7–9,17 The croton oil component, phorbol myristate acetate, is widely used in experimental settings to stimulate Nox-derived ROS production. In addition, dieldrin, lindane, paraquat, and rotenone also activate microglial Nox, suggesting a role for these chemicals in neurodegenerative disease.8,18–20 Dieldrin was also shown to generate reactive oxygen species as an early event contributing to the apoptosis of dopaminergic PC12 cells.21 Therefore, diverse pesticides appear to have roles in Nox activation. Endogenous chemicals such as lysophosphatidylcholine and arachidonic acid (AA) can also stimulate Nox.6 The mechanisms by which chemical factors activate Nox are incompletely understood, but protein kinase C (PKC) activation is often involved.22 Phospholipase (PL)A2-derived AA is an important second messenger that can activate PKC.23 Furthermore, it was previously shown that exposure of neutrophils to arochlor 1242 and dieldrin enhanced [3H]-AA release in a PLA2-dependent manner.17,23 Similarly, PCBs and polybrominated diphenyl ether (PBDE) flame retardants elicited [3H]-AA release from rat neutrophils24 and cerebellar granule neurons.25 Because PKC is activated following the release of AA from phospholipid reserves,26 these findings suggested a signaling axis initiated by increases in PLA2-dependent AA levels resulting in PKC activation and thereby eliciting Nox-dependent increases in ROS (AA–PKC–Nox–ROS). AA liberated by PLA2 might be the key signaling molecule responsible for the induction of Nox activity following organochlorine exposure.23 AA modulates Nox activity by a complex process that involves regulatory cytosolic subunit translocation and assembly. AA is capable of directly inducing conformational changes in p47phox that uncover Src homology 3 (SH3) domains required for binding to p22phox while also activating protein kinases that are needed for phosphorylating p47phox and p67phox.27–30 In addition, AA can modulate Nox activity by directly binding to the RhoGDP dissociation inhibitor and releasing RacGDP, a small G-protein that augments Nox function when bound to p67phox in its active RacGTP form.31 These processes all appear to act synergistically to activate Nox-catalyzed superoxide biosynthesis.
The goal of the present study was to examine the ability of three bioaccumulative organochlorine compounds, trans-non-achlor, dieldrin, and p,p′-DDE, to induce the production of ROS in cultured human THP-1 monocytes. Specifically, this study evaluated the ability of these compounds to induce NOX2-derived ROS in cultured human monocytes as well as provided evidence for a vital role of PLA2-derived AA in a signaling cascade that yields phospho-p47phox, a biomarker of Nox activation.
EXPERIMENTAL PROCEDURES
Materials
Human THP-1 monocytes, murine J774A.1 macrophages, human HL-60 cells, high-glucose RPMI-1640 (with and without phenol red), Dulbecco’s modified Eagle’s medium (DMEM), Hank’s balanced salt solution (HBSS), gentamicin sulfate solution, and penicillin/streptomycin solution were purchased from American Type Culture Collection (ATCC) (Manassas, VA). Low-endotoxin fetal bovine serum (FBS) and 5-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) were purchased from Invitrogen (Carlsbad, CA). Dieldrin, trans-nonachlor and p,p′-DDE (>98% purity) were from ChemService, Inc. (West Chester, PA). Phosphoserine antibodies and p47phox antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX), and protein disulfide isomerase (PDI) antibody was from Stressgen Biotechnologies (San Diego, CA). Protein A/G-agarose beads and SuperSignal West Pico chemiluminescence reagent were purchased from Thermo Scientific (Waltham, MA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay reagents and the deuterated standards [2H4]-8-iso-PGF2α and [2H8]-AA were purchased from Cayman Chemical Company (Ann Arbor, MI), as was arachidonyl trifluoromethyl ketone (ATK), an inhibitor of cytosolic phospholipase A2 (cPLA2). Bromoenol lactone (BEL), a specific inhibitor of cytosolic calcium-independent phospholipase A2 (iPLA2), was purchased from Sigma (St. Louis, MO). Hydroethidine (HE) was purchased from Invitrogen, and diphenyleneiodonium (DPI), a nonspecific Nox inhibitor, and LDH cytotoxicity assay reagents were from Sigma (St. Louis, MO). VAS-2870, a potent andselective Nox inhibitor, was purchased from Enzo Life Sciences (Farmingdale, NY).
General Cell Culture Conditions
Human THP-1 monocytes (ATCC no. TIB-202) were cultured in RPMI-1640 medium containing 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4500 mg/L glucose, 1500 mg/L sodium bicarbonate, and 50 μg/mL gentamicin and maintained at 37 °C in a humidified atmosphere of 5% CO2 in air. Cells were maintained in suspension at a density below 1 × 106 cells/mL with media changes approximately every 48–72 h.
Murine J774A.1 macrophages (ATCC no. TIB-67) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 4 mM L-glutamine, 1 mM sodium pyruvate, 4500 mg/L glucose, 1500 mg/L sodium bicarbonate, and 100 U/mL penicillin/streptomycin and maintained at 37 °C in a humidified atmosphere of 5% CO2 in air. Cells were subcultured by scraping approximately every 48–72 h. Modified HBSS was used as a treatment medium in some experiments and consisted of 5.33 mM KCl, 0.44 mM KH2PO4, 4.17 mM NaHCO3, 137.93 mM NaCl, 0.34 mM Na2HPO4, 5.56 mM D-glucose, 25 mM HEPES, and 0.1 mM diethylene triaminepentaacetic acid (DTPA).
THP-1 Monocytes Treated with Toxicants: DCFH-DA ROS Assay
Intracellular production of ROS in THP-1 monocytes following OC treatment was determined by measuring DCF-derived fluorescence by flow cytometry. THP-1 monocytes (2 × 106 cells/sample) were pretreated for 1 h with 50 μM DCFH-DA, followed by 1–10 μM trans-nonachlor, 10 μM dieldrin, 10 μM p,p′-DDE, or ethanol vehicle (0.1% v/v) in serum-free RPMI-1640 at 37 °C. Following incubation at 37 °C with chemicals (2–16 h), the treated cells were washed and resuspended in PBS. The cells were analyzed by flow cytometry using a BD Biosciences FACSCalibur system measuring DCF fluorescence in the FL1 channel (530/30). Data were analyzed using CellQuest Pro software, and statistical significance was assessed by one-way ANOVA with Tukey’s Studentized range test for multiple comparisons using PROC GLM in SAS 9.3.
THP-1 Monocytes Treated with Toxicants: Hydroethidine ROS Assay
Production of ROS by THP-1 monocytes following OC treatment was determined with the ROS probe HE using both fluorometric and mass spectrometry-based methods.32,33 For each approach, THP-1 monocytes (2 × 106 cells/sample) were treated with either ethanol vehicle (0.1% v/v) or trans-nonachlor (0.1–10 μM) for 2 h in the presence or absence of 50 μM DPI, 0.1–10 μM VAS-2870, or 20 μM ATK, in either serum-free/phenol red-free RPMI-1640 medium (fluorometric assay) or modified HBSS (mass spectrometry assay) at 37 °C. For the fluorometric kinetic analysis,32 HE was added to cells to a final concentration of 10 μM in the culture medium following 1 h of toxicant exposure. The fluorescence derived from 2-hydroxyethidium (2-OH-Et+) (the specific product of the reaction of superoxide with HE) and nonspecific HE oxidation products (e.g., ethidium) was monitored for an additional hour (λexcit, 490 nm; λemis, 565 nm) on a Molecular Devices SpectraMax M5 fluorescence plate reader set at 37 °C.34 Readings were taken every 5 min and plotted against time. Because of spectral overlap of 2-OH-Et+ and ethidium, one cannot separate the fluorescent signals derived from the specific and nonspecific oxidation products of HE by this approach. For mass spectrometry analysis,33 cells were incubated with toxicants for 2 h in modified HBSS at 37 °C in silinized glass tubes prior to addition of 20 μM HE. After a 15 min incubation with HE at 37 °C, the cells were pelleted by centrifugation (400g for 5 min) and washed with PBS. Cells were lysed by brief sonication in 200 μL of 1:1 (v/v) methanol/water with 0.1% formic acid containing an internal standard (atrazine, 1 μM final concentration) followed by centrifugation (16 100g for 3 min). (It was predetermined that the lysing protocol (sonication) did not generate 2-OH-Et+ and ethidium as artifacts.) The resulting supernatant was loaded into LC vials and analyzed by UPLC/ESI-MS using a Waters Acquity ultra-performance liquid chromatograph coupled to a Thermo TSQ Quantum Access MAX triple quadrupole mass spectrometer. Analytes were injected onto a Mac-Mod Analytical ACE C18-column (3 μm, 100 × 2.1 mm) containing a guard column and eluted with the following linear gradient solvent system: 0 min (85% A, 15% B), 0.5 min (85% A, 15% B), 15 min (50% A, 50% B), 17 min (5% A, 95% B), 17.5 min (5% A, 95% B), and 19 min (85% A, 15% B). Solvent A was 0.1% acetic acid in water, and solvent B was 0.1% acetic acid in methanol; flow rate, 0.3 mL/min. 2-OH-Et+ was quantified via single ion monitoring (SIM) of the m/z 330 ion, and Et+, by monitoring the m/z 314 ion. Alternatively, HE and its oxidation products were separated on a Phenomenex C6-phenyl column (3 μm, 50 × 2.1 mm) and detected by single reaction monitoring (SRM) using the following transitions: m/z 330 > 255 (2-OH-Et+), m/z 314 > 284 (Et+), m/z 313 > 298 (Et+-Et+), and m/z 316 > 210 (HE).35 Positive control reactions included a cell-free xanthine/xanthine oxidase system that was incubated with 10 μM HE for 10 min, followed by a similar work up procedure. Statistical significance was assessed by one-way ANOVA with Tukey’s Studentized range test for multiple comparisons using PROC GLM in SAS 9.3.
Arachidonic Acid and Eicosanoid Liberation
The levels of AA and AA-derived prostanoids liberated into cell culture media were determined to evaluate phospholipase A2 (PLA2)/cyclooxygenase activity after OC treatment of cells. THP-1 monocytes (2 × 106 cells/sample) were treated with dieldrin (10 μM), trans-nonachlor (0.01–10 μM), or vehicle (0.1% v/v ethanol) in RPMI-1640 medium containing 0.5% v/v FBS with and without pretreatment with 0.1–20 μM arachidonoyl trifluoromethylketone (ATK), an inhibitor of cPLA2, or 0.1–10 μM bromoenol lactone (BEL), a specific inhibitor of calcium-independent phospholipase A2 (iPLA2). After 2, 4, 8, or 12 h incubation at 37 °C, lipid mediators in cell culture media were extracted into ethyl acetate containing 0.1% v/v acetic acid, followed by organic solvent evaporation under N2 and resuspension of residues in 100 μL of 1:1 v/v methanol/H2O. AA and prostanoid (PGE2 and thromboxane B2) concentrations were quantified by UPLC/ESI-MS/MS using a Waters Acquity ultra-performance liquid chromatograph coupled to a Thermo TSQ Quantum Access MAX triple quadrupole mass spectrometer. Chromatography was performed on a Waters BEH C18 2.1 × 50 mm column, as described previously,36 with each analyte detected via single reaction monitoring (SRM) using the following transitions: m/z 303 > 259 (arachidonic acid), m/z 351 > 271 (PGE2), and m/z 369 > 169 (thromboxane B2). The internal standards [2H4]-8-iso-PGF2α and [2H8]-AA were monitored by the transitions m/z 357 > 197 and m/z 311 > 267, respectively. Chromatographic separation of AA and the PLA2 inhibitor ATK required 86:14 (v/v) acetonitrile/methanol containing 0.1% v/v acetic acid as the strong elution solvent instead of 100% methanol. Statistical significance was determined by one-way ANOVA with Tukey’s Studentized range test for multiple comparisons using PROC GLM in SAS 9.3.
Serine Phosphorylation of p47phox
Phosphoserine post-translational modifications of p47phox were detected by immunoblotting to assess the activation state of Nox following OC treatments of cells. Human THP-1 monocytes (2 × 106 cells/sample) were treated with trans-nonachlor (1–10 μM), AA (50 μM), or vehicle (0.1% v/v ethanol) in RPMI-1640 containing 0.5% v/v FBS with and without 20 μM ATK pretreatment. After 2, 4, 8, or 12 h incubation at 37 °C, cells were lysed in RIPA buffer containing protease inhibitors and immunoprecipitated by incubation with rabbit anti-human p47phox IgG overnight at 4 °C prior to capture on Thermo protein A/G agarose beads in accordance with the manufacturer’s protocol for indirect immunoprecipitation. After collecting and washing the beads, the immunoprecipitated proteins (30 μg of protein per lane) were separated by SDS-PAGE (10%) prior to semidry transfer onto PVDF membranes (20 V for 30 min). Membranes were blocked in 1% BSA in Tris-buffered saline with Tween-20 (TBST: 10 mM Tris, 150 mM NaCl, 0.1% Tween-20) at room temperature for 2 h. Phosphoserine modification was detected by western blot analysis (Santa Cruz SC-81514, 1:300 dilution in 0.1% w/v BSA solution). The membrane was subsequently stripped, blocked for 2 h in 5% w/v nonfat milk, and reprobed for p47phox (Santa Cruz SC-14015, 1:500 in 1% w/v nonfat milk). After washing, the blots were visualized via enhanced chemiluminescence (Thermo SuperSignal West Pico chemiluminescence reagent), and films were scanned. Densiometry analysis was performed using ImageJ v1.49a (NIH), and statistical significance was determined by one-way ANOVA with Tukey’s Studentized range test for multiple comparisons using PROC GLM in SAS 9.3.
Subcellular Fractionation: Membrane Translocation of p47phox
Membrane translocation of p47phox following OC treatment was assessed as a complementary means of Nox activation. THP-1 monocytes (2 × 106 cells/sample) were treated with trans-nonachlor (1–10 μM), AA (50 μM), or vehicle (0.1% v/v ethanol) in serum-free RPMI-1640. After 4 h incubation at 37 °C, cells were lysed in buffer containing 0.1 M Tris-HCl, 0.1 M KCl, 1.0 mM EDTA, 20 μM BHT, and 1× Promega protease inhibitor cocktail (PIC). Crude membrane (cytoplasmic and ER) fraction was prepared using sequential ultracentrifugation and wash steps (100 000g, 4 °C, 105 min total) and resuspended in lysis buffer containing PIC and 1% v/v Triton X-100. The membrane fraction (30 μg of protein per lane) was separated by SDS-PAGE (10% gel) prior to semidry transfer (20 V for 30 min) onto PVDF membranes. Membranes were blocked in 1% w/v BSA in Tris-buffered saline with Tween-20 (TBST: 10 mM Tris, 150 mM NaCl, 0.1% Tween-20) at room temperature for 2 h. p47phox was detected by western blot analysis (Santa Cruz SC-14015, 1:500 with 0.1% w/v BSA). The membrane was subsequently stripped, blocked for 2 h in 5% w/v nonfat milk, and reprobed for protein disulfide isomerase (PDI) for loading control (Stressgen SPA-890, 1:4000 with 0.1% w/v BSA). Following final washes, blots were visualized via enhanced chemiluminescence (Thermo SuperSignal West Pico chemiluminescence reagent), and films were scanned. Densiometry analysis was performed using ImageJ v1.49a (NIH), and statistical significance was determined by one-way ANOVA with Tukey’s Studentized range test for multiple comparisons using PROC GLM in SAS 9.3.
RESULTS
ROS Induced by OC Exposure: DCFH-DA Assay
The maximum concentrations of dieldrin, trans-nonachlor, and p,p′-DDE were selected as the highest concentration that did not cause marked cytotoxicity in human THP-1 monocytes (<10%), as measured by either the LDH assay or MTT assay (for trans-nonachlor, see Figure S1A,B). All experiments in this study used toxicant concentrations ≤ 10 μM.
To determine whether OC chemicals affected the production of superoxide and other ROS in cultured human monocytes, we used a flow cytometry-based approach to analyze changes in DCF-derived fluorescent signals indicative of ROS production. THP-1 monocytes preloaded with DCFH-DA were exposed to varying concentrations of dieldrin, trans-nonachlor, or p,p′-DDE for up to 16 h. Monocytes treated with 10 μM trans-nonachlor exhibited increased DCF-derived fluorescence, indicative of elevated ROS levels compared to that in vehicle-treated monocytes after 2, 4, 8, and 16 h (Figure 1A, top panel), whereas treatment with 1 and 2.5 μM trans-nonachlor did not produce an increase in DCF-derived fluorescence (data not shown). For the 10 μM trans-nonachlor treatment, the DCF-derived mean fluorescence intensity increased by 30% relative to that from vehicle-treated cells at 2 h (Figure 1B,C), whereas treatment with exogenous AA (10 μM) for 2 h caused a 60% increase in DCF-derived mean fluorescence intensity (Figure 1C). On the other hand, treatment with 10 μM dieldrin elevated ROS levels at 2 and 4 h compared to that in vehicle-treated cells, but not at later time points (Figure 1A, bottom panel). The apparent loss of DCF-derived fluorescence over time likely resulted from the gradual leakage of the fluorescent probe from the cells or its metabolism during the course of these experiments. THP-1 monocytes treated with p,p′-DDE (10 μM) did not exhibit elevated ROS production at any time point (data not shown); thus, we did not look at this compound further. Because trans-nonachlor gave a more robust and sustained oxidative stress response than either dieldrin or p,p′-DDE, we focused on this compound in the majority of the experiments reported here.
Figure 1.
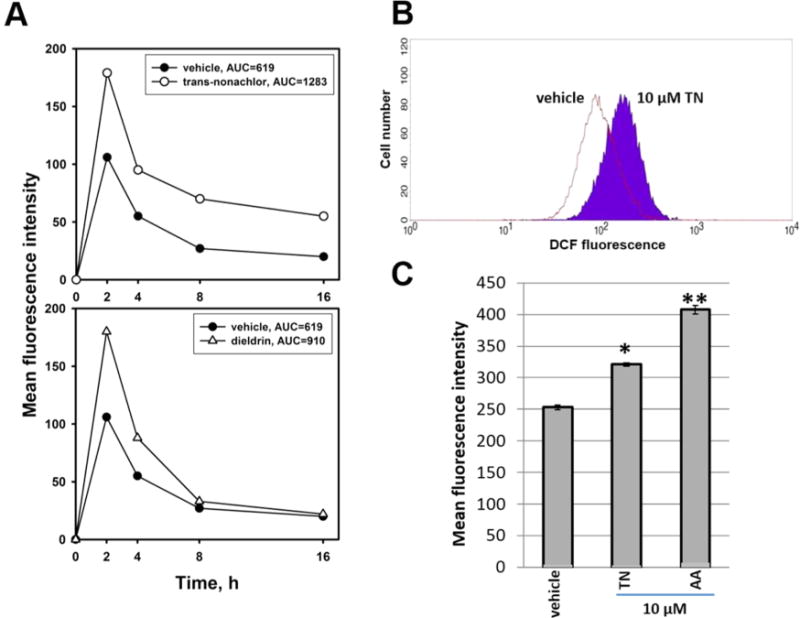
Enhanced production of ROS of uncertain identity in THP-1 monocytes following treatment with OC chemicals or exogenous AA: DCFH-DA fluorescence assay. (A) Time course of DCF-derived mean fluorescence intensity in monocytes exposed to trans-nonachlor (top panel) and dieldrin (bottom panel) for up to 16 h. Data represent mean values of duplicate measurements. THP-1 monocytes were preloaded with DCFH-DA (50 μM) for 30 min and then treated with 10 μM OC for the indicated amount of time. The vehicle was ethanol (0.1% v/v). Area under curve (AUC) values indicates the time-integrated fluorescence intensity. (B) Representative histogram of DCF-derived fluorescence in vehicle-treated and 10 μM trans-nonachlor (TN)-treated monocytes. The abscissa represents fluorescence intensity, and ordinate represents cell number. (C) Mean fluorescence intensity following treatment of monocytes with vehicle (ethanol, 0.1% v/v), 10 μM TN, or 10 μM arachidonic acid (AA) for 2 h. Data represents the mean ± SD of three experiments. *, p < 0.05; **, p < 0.01; one-way ANOVA with Tukey’s Studentized range test.
ROS Induced by OC Exposure: Hydroethidine Assay
A complementary approach to determine whether OC exposure increases ROS and superoxide production in cultured monocytes involved the ROS probe hydroethidine (HE). HE-derived oxidation products, such as 2-OH-Et+ and ethidium, exhibit red fluorescence, whereas HE does not.33 Moreover, HE can react with superoxide, yielding the fluorescent compound 2-hydroxyethidium (2-OH-Et+), a specific product derived from the superoxide/HE reaction (Figure 3A). Thus, the rate of HE oxidation in intact living THP-1 monocytes treated with OCs can be used as an index of oxyradical flux and be monitored in a plate reader by fluorometry (Figure 2A,C). It is important to emphasize here that the increased fluorescence caused by HE oxidation is not strictly due to presence of 2-OH-Et+ because other nonspecific HE oxidation products might be formed as well. Thus, data obtained using a plate reader fluorometric assay with the HE probe measures ROS of uncertain identity.37 The fluorometric kinetic assay revealed that monocytes treated with 10 μM trans-nonachlor exhibited an elevated rate of HE oxidation (∼25–50%) when compared to that in vehicle-treated cells, suggestive of enhanced superoxide biosynthesis (Figure 2B,D). The rate of HE oxidation in cells treated with trans-nonachlor and Nox inhibitor (DPI or VAS-2870), which have different mechanisms of inhibition, was significantly attenuated relative to that in trans-nonachlor only treated cells (Figure 2B,D); rates of HE oxidation were reduced 2.5- and 7.8-fold for DPI and VAS-2870, respectively. It was also noted that DPI and VAS-2870 could reduce the constitutive (basal) Nox activity in control (vehicle-treated) THP-1 cells (data not shown).
Figure 3.

Determination of superoxide levels following trans-nonachlor treatment. (A) HPLC-MS analysis of 2-hydroxyethidium (2-OH-Et+, m/z 330), the superoxide-dependent oxidation product of HE, in a xanthine/xanthine oxidase (XO) cell-free system. (B) 2-OH-Et+ production in THP-1 monocytes treated with 10 μM TN in the presence or absence of 50 μM DPI, measured by UPLC-MS analysis. DPI suppressed superoxide production in TN-exposed monocytes. (C) UPLC-MS/MS chromatograms of 2-OH-Et+ (m/z 330 > 255) following treatments with vehicle (a), 5 μM TN (b), and 5 μM TN + 2.5 μM VAS-2870 (c). (D) Profiles of 2-OH-Et+, Et+, and Et+-Et+ following treatment of THP-1 cells with vehicle or indicated amount of trans-nonachlor. The relative amounts of each analyte were normalized to the level detected in vehicle-treated cells. (E) Relative levels of 2-OH-Et+ detected in THP-1 cells treated with 5 μM trans-nonachlor in the absence or presence of indicated amount of VAS-2870. The difference in elution time for 2-OH-Et+ peaks in panels (A–C) reflects that the HPLC-MS system was used in (A) with a Thermo C18-column (100 × 2.1 mm), whereas UPLC-MS system was used with a Mac-Mod ACE C18-column (100 × 2.1 mm) (B) or Phenomenex C6 phenyl column (50 × 2.1 mm) (C–E). Data represents the mean ± SD of at least three experiments. *, p < 0.05 relative to TN alone (B); *, p < 0.05 relative to vehicle control (D, E); †, p < 0.05 relative to 5 μM TN without the inhibitor (E).
Figure 2.

Enhanced production of ROS of uncertain identity in THP-1 monocytes following treatment with trans-nonachlor: hydroethidine fluorescence assay. Rate of hydroethidine (HE) oxidation in THP-1 monocytes treated with 10 μM TN in the presence or absence of Nox inhibitors (A) DPI (50 μM) and (C) VAS-2870 (10 μM) was determined by a fluorometric kinetic assay (λexcit, 490 nm; λemis, 565 nm). Production of HE-derived oxidation products in intact cells was monitored for 33–60 min in a fluorescence plate reader. (B, D) Treatment with 10 μM TN caused a significant increase in the rate of HE oxidation (slopes of curves shown in A and C) compared to that in vehicle (ethanol)-treated cells, which could be attenuated by concomitant treatment with either 50 μM DPI (B) or 10 μM VAS-2870 (D). Data represent the mean ± SD (or individual data points) of three experiments. *, p < 0.05; one-way ANOVA with Tukey’s Studentized range test. RFU, relative fluorescence units; TN, trans-nonachlor.
In addition, UPLC-MS analysis of 2-OH-Et+ (m/z 330), the superoxide-specific oxidation product of HE (Figure 3A), following xenobiotic treatments supported the finding that DPI could suppress superoxide production in trans-nonachlor–exposed monocytes (Figure 3B). The concentration of DPI used (50 μM) and its duration of treatment (2 h) was determined to be not cytotoxic to THP-1 cells on the basis of the LDH assay (Figure S1C). Moreover, following treatment of cells with increasing amounts of trans-nonachlor, the profile of HE oxidation products (2-OH-Et+, Et+, and Et+-Et+) determined by UPLC-MS/MS indicated that significantly elevated levels of 2-OH-Et+ were generated by 1–10 μM trans-nonachlor but not 0.1 μM trans-nonachlor (Figure 3C,D). The increased levels of 2-OH-Et+ induced by trans-nonachlor at 1, 5, and 10 μM were statistically greater than the level in the vehicle control, but they were statistically indistinguishable from one another (Figure 3D). On the other hand, Et+ levels were only slightly elevated following treatment with 10 μM trans-nonachlor, whereas Et+-Et+ levels were unchanged at all concentrations of trans-nonachlor (Figures 3D and S2). Et+ and Et+-Et+ are nonspecific oxidation products that can be generated by nonenzymatic reactions of the HE probe with oxidants, such as peroxynitrite, and by enzymatic reactions with NADP+-dependent dehydrogenases or peroxidases. Importantly, the trans-nonachlor-induced 2-OH-Et+ levels were attenuated in a concentration-dependent manner by the Nox inhibitor VAS-2870 (Figure 3C,E). Even 1 μM VAS-2870 was shown to significantly reduce 2-OH-Et+ levels (Figure 3E).
The ability of trans-nonachlor to induce reactive oxygen flux was further confirmed in a murine macrophage cell line, J774a.1, in which 10 μM trans-nonachlor treatment caused a significant increase (more than 2-fold) in the rate of HE oxidation compared to that in vehicle-treated cells, as measured by fluorometric kinetic assay (Figure 4A). Furthermore, the human HL-60 cell line, which express Nox2, was also sensitive to trans-nonachlor-induced reactive oxygen flux (Figure 4B). Additionally, the rate of HE oxidation induced by trans-nonachlor in the HL-60 cells was significantly attenuated by the Nox-specific inhibitor VAS-2870 (Figures 4B and S3).
Figure 4.
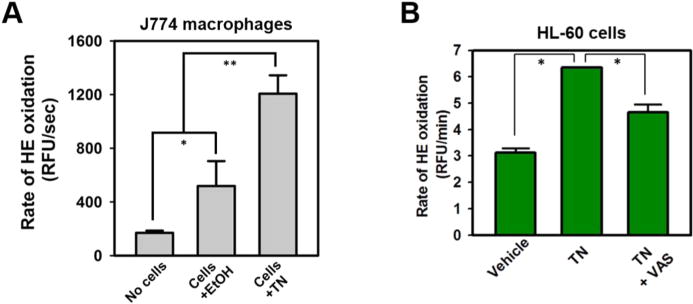
trans-Nonachlor-induced production of ROS of uncertain identity in J774 macrophaghes and HL-60 cells. (A) Murine J774a.1 macrophages and (B) human HL-60 cells treated with 10 μM TN exhibited a significant increase in the rate of HE oxidation compared to that in vehicle (ethanol)-treated cells, as measured by the fluorometric kinetic assay, further demonstrating the ability of TN to induce reactive oxygen flux in phagocytes. Data represents the mean ± SD of two to three experiments. *, p < 0.05; **, p < 0.01; one-way ANOVA with Tukey’s Studentized range test. RFU, relative fluorescence units; TN, trans-nonachlor; VAS, VAS-2870.
Arachidonic Acid and Eicosanoid Liberation Caused by OC Treatments
Liberation of AA and AA-derived eicosanoids into the cell culture medium was quantified by UPLC-MS/MS and used to assess PLA2 activity, which liberates fatty acids from the sn-2 position of phospholipids (Figure 5A). THP-1 monocytes treated for 12 h with either 10 μM trans-nonachlor or 10 μM dieldrin exhibited a 30-fold increase in AA levels in the cell culture medium compared to that in the vehicle control (Figure 5B). Furthermore, monocytes treated for 12 h with increasing concentrations of trans-nonachlor exhibited a dose-dependent increase in AA levels (Figure 5C), with an apparent threshold for this effect observed between 0.1 and 1 μM. Treatment of cells for 2 to 12 h with 1 μM trans-nonachlor induced significant AA liberation into the cell culture media compared to that from the vehicle control at all time points examined (Figure 5D), indicating that activation of PLA2 was a relatively early and persistent event following OC exposure.
Figure 5.
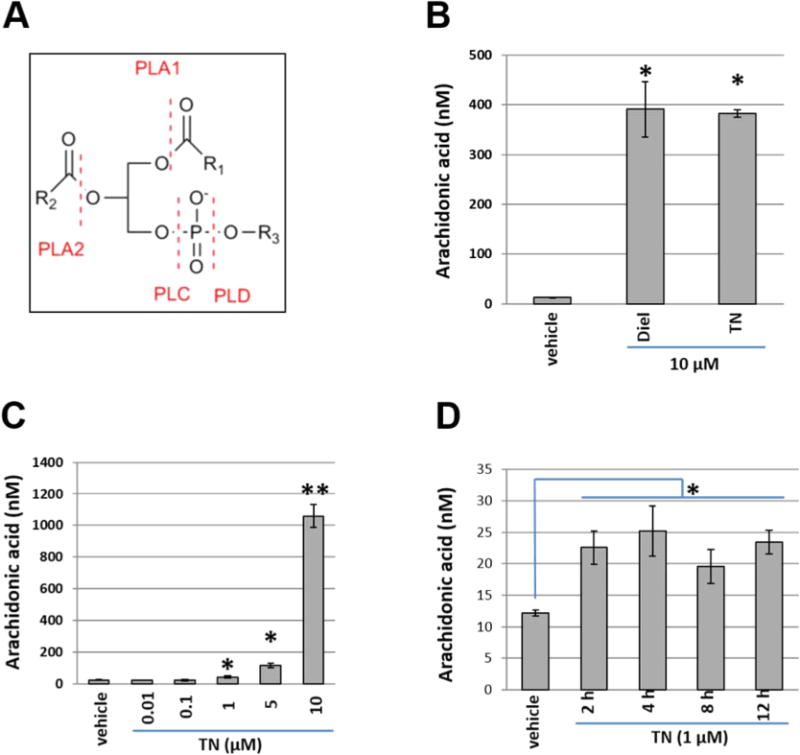
OC chemical treatment stimulates arachidonic acid (AA) liberation. (A) Release of AA from phospholipids into the cell culture medium was quantified by UPLC-MS/MS to evaluate phospholipase A2 (PLA2) activity following OC treatment. Generic phospholipid structure is shown with sites modified by phospholipases indicated. R1 and R2 represent acyl groups; R3 represents the polar headgroup. (B) AA release from THP-1 monocytes treated for 12 h with either vehicle (ethanol), 10 μM dieldrin (Diel), or 10 μM TN. (C) Dose–response curve of monocytes treated for 12 h with increasing concentrations of TN. A dose-dependent increase in AA levels was observed, with an apparent threshold for this effect observed between 0.1 and 1 μM. (D) Time course of AA release from monocytes treated for 2 to 12 h with 1 μM TN. Elevated AA liberation compared to that from the vehicle control was evident at all time points. Data in panels B–D represent the mean ± SD of two to three experiments. *, p < 0.05; **, p < 0.01; one-way ANOVA with Tukey’s Studentized range test. Vehicle, ethanol (0.1% v/v); TN, trans-nonachlor.
In light of the enhanced production of AA caused by OC treatments, we examined whether eicosanoid levels were also elevated due to the greater availability of AA substrate to the cyclooxygenase protein. As shown in Figure 6A–C, prostaglandin E2 and thromboxane B2 levels in the culture medium were significantly elevated by either 10 μM dieldrin or 10 μM trans-nonachlor treatments after 12 h. Furthermore, pretreatment of monocytes with ATK, a potent inhibitor of PLA2, prior to exposure to either trans-nonachlor or dieldrin effectively blocked the production of AA-derived prostaglandin E2 (Figure 6A,B) and thromboxane B2 (Figure 6C). In-source fragmentation of ATK during UPLC-MS/MS analysis prevented quantitation of AA released from ATK-pretreated cells; however, the eicosanoids were a good surrogate for AA formation and provided information on inflammatory lipid mediators released during OC chemical exposure. The technical issues surrounding in-source fragmentation could be alleviated by chromatographically separating ATK and AA using a shallower elution gradient and substituting 84:16 (v/v) acetonitrile/methanol for 100% methanol as the strong elution solvent. This enabled the impact of increasing concentrations of ATK on cellular AA production to be determined (Figure 6D). Furthermore, it was previously reported that ATK could inhibit COX1/2 activity,38 further justifying the need to separate ATK from AA in order to establish that organochlorine chemicals could stimulate AA release. Figure 6D shows that ATK could indeed attenuate the trans-nonachlor-induced release of AA in a concentration-dependent manner. On the other hand, the iPLA2-specific inhibitor BEL did not significantly attenuate trans-nonachlor-induced AA production (Figure 6E). These results suggested that cPLA2, and not iPLA2, was primarily responsible for the OC chemical-induced release of AA.
Figure 6.

Increased bioactive lipid liberation following OC chemical treatment. Eicosanoid concentrations in culture medium were assessed by UPLC-MS/MS analysis. (A) UPLC-MS/MS chromatograms of prostaglandin E2 (PGE2; m/z 351 > 271) following treatments with vehicle (a), 10 μM TN (b), and 10 μM TN + 20 μM ATK (c). (B) Prostaglandin E2 and (C) thromboxane B2 levels in the culture medium were significantly elevated following either 10 μM dieldrin or 10 μM TN treatments. Pretreatment with 20 μM ATK prior to OC exposure effectively blocked the production of AA-derived eicosanoids. Data represents the mean ± SD of two separate experiments. *, p < 0.05; one-way ANOVA with Tukey’s Studentized range test. Vehicle, ethanol (0.1% v/v); TN, trans-nonachlor. Arachidonic acid levels in culture medium following treatment with 5 μM trans-nonachlor in the absence or presence of indicated amounts of ATK (D) or BEL (E). Data represents the mean ± SD of three separate experiments. *, p < 0.05 relative to vehicle control; †, p < 0.05 relative to 5 μM TN without the inhibitor.
p47phox Serine Phosphorylation and Membrane Translocation
NOX2 is the major catalytic subunit of Nox in monocytes/macrophages, but to become active, it requires the phosphorylation and subsequent translocation of cytosolic p47phox to membrane bound NOX2 for full activity.5 Therefore, to assess activation of Nox following OC treatment, serine phosphorylation of p47phox and its translocation to the cell membrane fraction were determined. Treatment of cells for 12 h with trans-nonachlor induced serine phosphorylation of p47phox subunit (Figure 7A). Furthermore, treatment of THP-1 monocytes with 1 μM trans-nonachlor resulted in increased p47phox serine phosphorylation compared to that in the vehicle control at 2, 4, 8, and 12 h (Figure 7B). Treatments of THP-1 monocytes with 1, 2.5, 5, and 10 μM trans-nonachlor for 12 h resulted in markedly increased p47phox phosphorylation relative to that in the vehicle control (Figure 8). Importantly, pretreatment of monocytes with ATK prior to trans-nonachlor exposure blocked the induction of p47phox serine phosphorylation (Figure 8). Treatment with 10 and 20 μM dieldrin also yielded a dose-dependent increase in p47phox phosphorylation compared to that in the vehicle control (data not shown). With regard to p47phox membrane translocation, treatment of THP-1 monocytes with 1–10 μM trans-nonachlor for 4 h resulted in a dose-dependent increase in the p47phox content in membrane fraction immunoblots compared to that in the vehicle control (Figure 9). The localization of p47phox in the membrane fraction is evidenced by the detection of protein disulfide isomerase (PDI) by immunoblotting (Figure 9). PDI is localized in the endoplasmic reticulum. The membrane fraction immunoblot following AA treatment is shown because it is known to activate p47phox phosphorylation and stimulate translocation.29 The concentration of AA used (50 μM) is not unwarranted because during inflammation the levels of free AA in cells can reach high micromolar amounts.39 Levels between 10 and 100 μM AA are observed in a (patho)-physiological setting. For example, it has been shown that free AA in leukocytes can reach ranges of 15–150 μM following activation.40 Moreover, concentrations of AA that affect Nox begin around 5–10 μM, and maximal responses occurs with 50–100 μM.39
Figure 7.

OC chemical treatment stimulates p47phox serine phosphorylation. (A) Western blot analysis of p47phox serine phosphorylation in THP-1 monocytes treated with indicated amounts of trans-nonachlor for 12 h. (B) Western blot analysis of p47phox phosphorylation in THP-1 monocytes treated with 1 μM TN for 2 to 12 h. p47phox serine phosphorylation was increased in monocytes treated with 1 μM TN compared to that with vehicle (ethanol, 0.1% v/v) at 2, 4, 8, and 12 h, which is indicated by the values beneath the blot. TN, trans-nonachlor; IP, immunoprecipitation.
Figure 8.

OC-stimulated p47phox serine phosphorylation: concentration–response. Western blot analysis of p47phox phosphorylation in THP-1 monocytes treated with vehicle (ethanol, 0.1% v/v) or the indicated amounts of TN for 12 h. Ordinate depicts phospho-p47phox band density normalized to p47phox. Treatment with 1, 2.5, 5, and 10 μM TN resulted in markedly increased p47phox phosphorylation relative to that in the vehicle control, whereas treatment with 20 μM ATK prior to TN exposure blocked the induction of p47phox serine phosphorylation at all concentrations. Data represents the mean ± SD of duplicate experiments. *, p < 0.05; **, p < 0.01; one-way ANOVA with Tukey’s Studentized range test. TN, trans-nonachlor; IP, immunoprecipitation.
Figure 9.
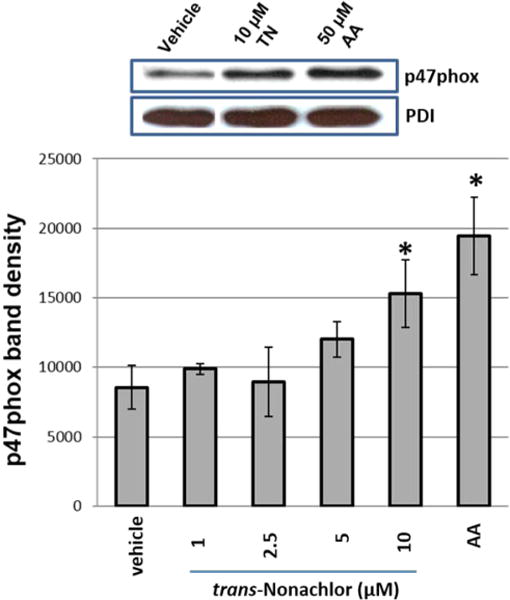
Enhanced p47phox membrane translocation following OC exposure. Western blot analysis of THP-1 cell membranes after cell treatments with TN (1–10 μM), AA (50 μM), or vehicle (0.1% v/v ethanol) for 4 h. Arachidonic acid is a known inducer of p47phox translocation and served as a positive control. Treatment with 10 μM TN resulted in a significant increase in the p47phox content of the membrane fraction compared to that in the vehicle control (ethanol, 0.1% v/v), indicative of activation of the NAPDH oxidase enzyme complex. Data represents the mean ± SD of duplicate experiments. *, p < 0.05; one-way ANOVA with Tukey’s Studentized range test. TN, trans-nonachlor; PDI, protein disulfide isomerase.
Attenuated Rate of OC-Mediated Oxidative Stress by PLA2 Inhibitor ATK
Finally, because ATK can block PLA2-catalyzed AA release, eicosanoid formation, and p47phox phosphorylation via PLA2 inhibition, we reasoned that pretreatment of THP-1 monocytes with ATK would reduce the amount of Nox-dependent ROS induced by trans-nonachlor. As shown in Figure 10, the rate of HE oxidation following trans-nonachlor treatment was attenuated 23% by pretreatment of cells with ATK, suggesting that PLA2 activation was, in part, responsible for the elevated Nox-dependent ROS levels caused by trans-nonachlor exposure.
Figure 10.

Production of ROS of uncertain identity in THP-1 monocytes following trans-nonachlor exposure is attenuated by PLA2 inhibitor ATK. (A) THP-1 monocytes were pretreated with 20 μM ATK for 1 h, followed by treatment with 10 μM trans-nonachlor for 2 h. One hour after the cells were treated with trans-nonachlor, HE was added and the extent of HE oxidation was monitored for an additional 60 min by a fluorometric kinetic assay (λexcit, 490 nm; λemis, 565 nm). (B) Treatment with 10 μM TN caused a significant increase in the rate of HE oxidation (slopes of curves shown in A) compared to that in vehicle (ethanol)-treated cells, which could be attenuated by 20 μM ATK. Data represents the mean ± SD of three experiments. *, p < 0.05; one-way ANOVA with Tukey’s test. RFU, relative fluorescence units; TN, trans-nonachlor.
DISCUSSION
Due to their persistent nature and numerous adverse ecological effects, the use of bioaccumulative OC insecticides was discontinued several decades ago in many parts of the world. Nevertheless, these so-called “legacy” compounds and their metabolites are still present in tissues of food animals and plants worldwide in amounts that lead to significant human exposure. For example, organochlorine body burden data from the 2003–2004 National Health and Nutrition Examination Survey revealed that detectable levels of trans-nonachlor and p,p′-DDE were present in 92.6 and 99.7% of the study population, respectively.41 Although human adipose tissue concentrations of these compounds vary considerably based on geography and diet, several recent studies place average adipose levels for North American and Western European populations in the range of 40–70 μg/kg lipid for trans-nonachlor and 630–695 μg/kg lipid for p,p′-DDE.42–45 The recent characterization of dieldrin, trans-nonachlor, p,p′-DDE, and certain PCBs as risk factors linked to the development of metabolic syndrome and/or cardiovascular disease in humans suggests a need for further investigation into the mechanisms that underpin the toxicity of these chemicals.1,2,46,47 For example, summed plasma levels of OC chemicals, including trans-nonachlor and p,p′-DDE, were associated with the levels of oxidized LDL, a marker of oxidative stress, in a population-based Swedish cohort.4 In addition, plasma levels of p,p′-DDE were higher in type 2 diabetics than in normal patients in a cross-sectional study of two U.S. Air Force medical facilities.48 The purpose of this study was to explore one such potential mechanism by characterizing the ability of bioaccumulative organochlorine insecticides to stimulate ROS production in human monocytes and to examine the signaling mechanisms involved. In cells, superoxide flux is defined by the difference between its rates of production and degradation. Both are highly dynamic processes and subject to competitive pathways, thus quantifying superoxide levels is a difficult proposition. We elected to use the general nonspecific ROS probe DCFH-DA to quantify reactive oxygen stress in monocytes exposed to OCs because of its relative ease of use and robustness;49 however, a probe that is more specific for superoxide, HE, was also used in two different formats, a fluorometric-based assay and a mass spectrometric-based assay.50
We focused on the effects of trans-nonachlor, dieldrin, and p,p′-DDE in human monocytes because these compounds are detectable in the serum of the U.S. population and are associated with metabolic and cardiovascular disease risk.1,2,41,46,47 Monocytes were studied due to their central roles in immune surveillance and inflammation as well as in the pathogenesis of atherosclerosis. Exposure to chlorinated cyclodiene insecticides has been reported to produce a wide variety of immunotoxic effects,51 including significant disruption of monocyte and macrophage maturation and function52,53 and, in the case of dieldrin, the activation of oxidative burst in neutrophils via increased intracellular AA levels produced by PLA2.17,23 We hypothesized that a similar mechanism involving the inappropriate activation of oxyradical producing enzymes through OC stimulation of PLA2 activity might operate in monocytes. It was found that micromolar concentrations of two chlorinated cyclodiene compounds, dieldrin and trans-nonachlor (a component of the chlordane insecticide mixture), elevated ROS levels in THP-1 monocytes, but p,p′-DDE, a chlorinated alicyclic metabolite of DDT, did not. trans-Nonachlor appeared to be a more potent inducer of oxidative stress than dieldrin, on the basis of DCF-derived fluorescence [area under the curve (AUC), Figure 1A]. Moreover, the use of the superoxide-selective probe HE and UPLC-MS/MS assay to measure 2-OH-Et+ demonstrated that trans-nonachlor could stimulate superoxide production (Figure 3). Further support that trans-nonachlor could activate Nox activity was demonstrated by increased serine phosphorylation of the regulatory p47phox subunit and suggested that elevated Nox activity was responsible for the increase in superoxide. It was noted that a monotonic dose–response in 2-OH-Et+ levels was not observed between 1 and 10 μM trans-nonachlor (Figure 3D). This suggested that levels of superoxide did not strictly correlate with levels of phosphorylated p47phox, which, as assessed by western blot, appeared to be higher at 2.5 μM compared to that at 10 μM (Figure 8). We speculate that this apparent inconsistency might result from efficient scavenging of superoxide by cellular antioxidant enzymes once Nox is activated, such as superoxide dismutase, thereby preventing a further buildup of superoxide levels at concentrations of trans-nonachlor ≥ 1 μM. This could result from the induction of the cellular Nrf2 antioxidant response. Alternatively, feedback mechanisms that result in reductions of phosphorylated p47phox at higher concentrations of trans-nonachlor might also be operating.
Further support for trans-nonachlor-stimulated Nox activation was shown by the phosphorylation and subsequent translocation of p47phox to cell membranes (Figures 8 and 9), the attenuated rates of HE oxidation by Nox inhibitors (Figures 2–4), and cPLA2 inhibitor (Figure 10). These findings provide direct evidence for the ability of chlorinated cyclodienes to activate Nox and shows, for the first time, that p47phox can be phosphorylated following trans-nonachlor exposure. Interestingly, both DPI and VAS-2870 attenuated the rates of HE oxidation to levels below that in the vehicle control, which indicates the extent of constitutive (basal) Nox-mediated superoxide production in non-OC stimulated (control) THP-1 monocytes. Indeed, the specific Nox inhibitor VAS-2870 nearly abrogated Nox activity in vehicle (ethanol)-treated cells (data not shown). Furthermore, the substantial increases in AA levels and its oxygenated metabolites (eicosanoids) following OC exposure, and the marked attenuation of arachidonic acid and eicosanoid production by ATK, a potent inhibitor of cytosolic PLA2,38 suggested that cPLA2 has a crucial upstream role in the induction of Nox activity (Figure 11). This conclusion is supported by the fact that the calcium-independent PLA2 inhibitor BEL did not significantly influence AA production (Figure 6D,E). Because pharmacological inhibitors, such as ATK, are not completely selective, it is difficult to completely distinguish between cPLA2 and calcium-independent PLA2 (iPLA2) enzymes. Nevertheless, it should be noted that Group IVA cPLA2 enzymes have a known preference over iPLA2 for arachidonic acid at the sn-2 position of phospholipids.54 Furthermore, ATK has a preference for inhibiting cPLA2 over iPLA2: ∼150-fold on the basis of IC50 values for the pure enzymes.55 Thus, the cPLA2 enzyme is most likely the PLA2 isoform activated by the OC chemical, especially since the iPLA2 inhibitor did not impact AA production. In addition to the provision of increased amounts of substrate (AA) to cyclooxygenase protein following OC chemical treatment, the increased eicosanoid levels could also be due to induction of COX2 levels via OC chemical-mediated Nox-dependent increases in ROS. Precedence for such a mechanism was recently shown when 2-aminobiphenyl was found to induce COX2 in a bladder cancer cell line by a Nox- and MAPK-dependent mechanism.56 Additionally, the attenuated OC-induced p47phox serine phosphorylation and ROS levels in ATK-pretreated monocytes (Figures 8 and 10) also provided compelling support for an important role of PLA2-derived AA in Nox activation.
Figure 11.
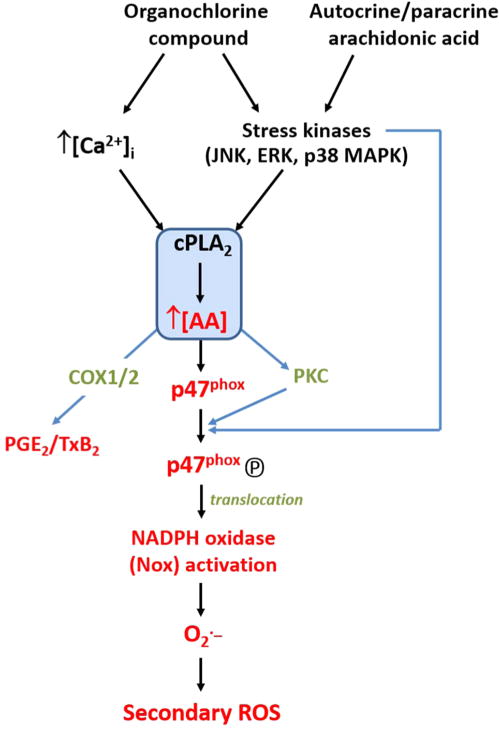
Proposed scheme for cPLA2/AA-mediated and Nox-derived superoxide in monocytes following treatment with organochlorine chemicals or extracellular arachidonic acid. Molecules denoted in red indicate those that were assayed in the current study. Calcium ions and stress kinases are known to activate cytosolic PLA2. AA, arachidonic acid; COX1/2, cyclooxygenase 1 and 2; ERK, extracellular-signal regulated kinases; JNK, c-jun N-terminal kinases; MAPK, mitogen-activated protein kinases; PKC, protein kinase C; cPLA2, cytosolic phospholipase A2.
Due to their hydrophobic nature, the distribution/disposition of trans-nonachlor and dieldrin in vivo is likely to be similar. However, they are slightly different chemically, with dieldrin containing an epoxide moiety. Yet, this epoxide functionality is apparently unreactive toward model nucleophiles.57 In addition, the cellular localization of OC chemicals is primarily confined to lipid membranes.57 Although the neuronal targets responsible for the insecticidal activity of OC pesticides are known to be ion channels, the specific non-neuronal targets responsible for mammalian cell toxicity are undefined.57 Multiple triggers of Nox activation exist, and it is likely that OCs act on upstream signaling molecules of Nox, including PLA2/AA and PKC. Members of the PKC family are the primary catalysts of serine phosphorylation of p47phox during Nox activation, with PKC α, βII, δ, and ζ being the individual isoforms responsible.29,30 All three PKC subfamilies are represented in this process, including conventional calcium-dependent PKC isoforms, such as α and βII, as well as the calcium-independent novel and atypical isoforms (δ and ζ, respectively). This indicates that calcium is not the sole signaling molecule responsible for stimulating p47phox phosphorylation by PKC following OC stimulation. Importantly, AA has been shown to stimulate the membrane translocation and activation of all the above PKC isoforms.27,28,58,59 The reduction in OC-induced phosphoserine-p47phox levels following PLA2 inhibition by ATK (Figure 8) suggested that AA is primarily responsible for PKC activation in this signaling pathway. In addition to the stimulation of p47phox phosphorylation through PKC activation, AA promotes RacGDP/GTP exchange and induces conformational changes in p47phox that reveal SH3 domains needed for interaction with membrane-embedded p22phox during Nox complex formation.29,31 AA has several attributes of a second messenger molecule that is responsible, in part, for OC-mediated Nox activation, although signal transduction pathways emanating from activated stress kinases that bypass PLA2 might also be involved (Figure 11).
Taken together, the results of this study suggest that trans-nonachlor and dieldrin are capable of increasing intracellular superoxide levels through a Nox-dependent release of ROS that relies on an upstream PLA2/AA signaling node, which might be significant in the etiology of atherosclerosis development and other diseases exhibiting an inflammatory component. These findings might offer a molecular mechanism (Figure 11) to explain, in part, the epidemiological evidence implicating trans-nonachlor and related bioaccumulative OC compounds as risk factors for metabolic and cardiovascular diseases. Furthermore, it provides a framework for future studies that investigate how triggering molecules, such as xenobiotics and endogenous toxins, activate downstream signaling pathways that stimulate the PLA2/AA/Nox axis.
Supplementary Material
Acknowledgments
The MSU College of Veterinary Medicine Flow Cytometry Core facility is acknowledged for generous use of the BD Biosciences FACSCalibur system. Dr. Edward Sharman, University of California, Irvine, is gratefully acknowledged for reviewing the manuscript.
Funding: This study was supported by NIH 1R15ES015348-02 (M.K.R.). A.T.M. is supported by predoctoral fellowship NIH F31HL122082-01A1.
ABBREVIATIONS
- AA
arachidonic acid
- BEL
bromoenol lactone
- DCFH-DA
5-carboxy-2′,7′-dichlorodihydrofluorescein diacetate
- Nox
NADPH oxidase
- PKC
protein kinase C
- PLA2
phospholipase A2
- PGE2
prostaglandin E2
- ROS
reactive oxygen species
- TN
trans-nonachlor
- TxB2
thromboxane B2
- ATK
arachidonoyl trifluoromethylketone
- DPI
diphenyleneiodonium
Footnotes
Supporting Information
Assessment of trans-nonachlor- and DPI-induced cytotoxicity in THP-1 cells; LC-MS chromatograms of THP-1 cell extracts obtained following treatment with vehicle or trans-nonachlor; and rate of HE oxidation in human HL-60 cells following treatment with trans-nonachlor. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Lee DH, Steffes MW, Sjodin A, Jones RS, Needham LL, Jacobs DR., Jr Low dose of some persistent organic pollutants predicts type 2 diabetes: a nested case-control study. Environ Health Perspect. 2010;118:1235–1242. doi: 10.1289/ehp.0901480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Min JY, Cho JS, Lee KJ, Park JB, Park SG, Kim JY, Min KB. Potential role for organochlorine pesticides in the prevalence of peripheral arterial diseases in obese persons: results from the National Health and Nutrition Examination Survey 1999–2004. Atherosclerosis. 2011;218:200–206. doi: 10.1016/j.atherosclerosis.2011.04.044. [DOI] [PubMed] [Google Scholar]
- 3.Alavanja MC, Ross MK, Bonner MR. Increased cancer burden among pesticide applicators and others due to pesticide exposure. CA–Cancer J Clin. 2013;63:120–142. doi: 10.3322/caac.21170. [DOI] [PubMed] [Google Scholar]
- 4.Kumar J, Monica Lind P, Salihovic S, van Bavel B, Lind L, Ingelsson E. Influence of persistent organic pollutants on oxidative stress in population-based samples. Chemosphere. 2014;114:303–309. doi: 10.1016/j.chemosphere.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 7.Gomez C, Bandez MJ, Navarro A. Pesticides and impairment of mitochondrial function in relation with the parkinsonian syndrome. Front Biosci. 2007;12:1079–1093. doi: 10.2741/2128. [DOI] [PubMed] [Google Scholar]
- 8.Mao H, Liu B. Synergistic microglial reactive oxygen species generation induced by pesticides lindane and dieldrin. NeuroReport. 2008;19:1317–1320. doi: 10.1097/WNR.0b013e32830b3677. [DOI] [PubMed] [Google Scholar]
- 9.Abid MR, Spokes KC, Shih SC, Aird WC. NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J Biol Chem. 2007;282:35373–35385. doi: 10.1074/jbc.M702175200. [DOI] [PubMed] [Google Scholar]
- 10.Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, Boullier A, Gonen A, Diehl CJ, Que X, Montano E, Shaw PX, Tsimikas S, Binder CJ, Witztum JL. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248. doi: 10.1161/CIRCRESAHA.110.223875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrario JB, DeLeon IR, Tracy RE. Evidence for toxic anthropogenic chemicals in human thrombogenic coronary plaques. Arch Environ Contam Toxicol. 1985;14:529–534. doi: 10.1007/BF01055381. [DOI] [PubMed] [Google Scholar]
- 12.Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, Ferdinandy P, Wolin MS, Rivera A, Ungvari Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. Am J Physiol: Heart Circ Physiol. 2007;292:H130–H139. doi: 10.1152/ajpheart.00599.2006. [DOI] [PubMed] [Google Scholar]
- 13.Motley ED, Kabir SM, Gardner CD, Eguchi K, Frank GD, Kuroki T, Ohba M, Yamakawa T, Eguchi S. Lysophosphatidylcholine inhibits insulin-induced Akt activation through protein kinase C-alpha in vascular smooth muscle cells. Hypertension. 2002;39:508–512. doi: 10.1161/hy02t2.102907. [DOI] [PubMed] [Google Scholar]
- 14.Tithof PK, Schiamberg E, Peters-Golden M, Ganey PE. Phospholipase A2 is involved in the mechanism of activation of neutrophils by polychlorinated biphenyls. Environ Health Perspect. 1996;104:52–58. doi: 10.1289/ehp.9610452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown AP, Olivero-Verbel J, Holdan WL, Ganey PE. Neutrophil activation by polychlorinated biphenyls: structure-activity relationship. Toxicol Sci. 1998;46:308–316. doi: 10.1006/toxs.1998.2551. [DOI] [PubMed] [Google Scholar]
- 16.Myhre O, Mariussen E, Reistad T, Voie OA, Aarnes H, Fonnum F. Effects of polychlorinated biphenyls on the neutrophil NADPH oxidase system. Toxicol Lett. 2009;187:144–148. doi: 10.1016/j.toxlet.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Tithof PK, Olivero J, Ruehle K, Ganey PE. Activation of neutrophil calcium-dependent and -independent phospholipases A2 by organochlorine compounds. Toxicol Sci. 2000;53:40–47. doi: 10.1093/toxsci/53.1.40. [DOI] [PubMed] [Google Scholar]
- 18.Mao H, Fang X, Floyd KM, Polcz JE, Zhang P, Liu B. Induction of microglial reactive oxygen species production by the organochlorinated pesticide dieldrin. Brain Res. 2007;1186:267–274. doi: 10.1016/j.brainres.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Mao YR, Jiang L, Duan YL, An LJ, Jiang B. Efficacy of catalpol as protectant against oxidative stress and mitochondrial dysfunction on rotenone-induced toxicity in mice brain. Environ Toxicol Pharmacol. 2007;23:314–318. doi: 10.1016/j.etap.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Taetzsch T, Block ML. Pesticides, microglial NOX2, and Parkinson’s disease. J Biochem Mol Toxicol. 2013;27:137–149. doi: 10.1002/jbt.21464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptopic cell death in dopaminergic cells. Free Radical Biol Med. 2001;31:1473–1485. doi: 10.1016/s0891-5849(01)00726-2. [DOI] [PubMed] [Google Scholar]
- 22.Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 23.Tithof PK, Peters-Golden M, Ganey PE. Distinct phospholipases A2 regulate the release of arachidonic acid for eicosanoid production and superoxide anion generation in neutrophils. J Immunol. 1998;160:953–960. [PubMed] [Google Scholar]
- 24.Brown AP, Ganey PE. Neutrophil degranulation and superoxide production induced by polychlorinated biphenyls are calcium dependent. Toxicol Appl Pharmacol. 1995;131:198–205. doi: 10.1006/taap.1995.1062. [DOI] [PubMed] [Google Scholar]
- 25.Kodavanti PR, Derr-Yellin EC. Differential effects of polybrominated diphenyl ethers and polychlorinated biphenyls on [3H]arachidonic acid release in rat cerebellar granule neurons. Toxicol Sci. 2002;68:451–457. doi: 10.1093/toxsci/68.2.451. [DOI] [PubMed] [Google Scholar]
- 26.Cathcart MK. Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis. Arterioscler, Thromb, Vasc Biol. 2004;24:23–28. doi: 10.1161/01.ATV.0000097769.47306.12. [DOI] [PubMed] [Google Scholar]
- 27.Murakami K, Routtenberg A. Direct activation of purified protein kinase C by unsaturated fatty acids (oleate and arachidonate) in the absence of phospholipids and Ca2+ FEBS Lett. 1985;192:189–193. doi: 10.1016/0014-5793(85)80105-8. [DOI] [PubMed] [Google Scholar]
- 28.Lester DS, Collin C, Etcheberrigaray R, Alkon DL. Arachidonic acid and diacylglycerol act synergistically to activate protein kinase C in vitro and in vivo. Biochem Biophys Res Commun. 1991;179:1522–1528. doi: 10.1016/0006-291x(91)91745-x. [DOI] [PubMed] [Google Scholar]
- 29.Shiose A, Sumimoto H. Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J Biol Chem. 2000;275:13793–13801. doi: 10.1074/jbc.275.18.13793. [DOI] [PubMed] [Google Scholar]
- 30.Fontayne A, Dang PM, Gougerot-Pocidalo MA, El-Benna J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41:7743–7750. doi: 10.1021/bi011953s. [DOI] [PubMed] [Google Scholar]
- 31.Miyano K, Sumimoto H. Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie. 2007;89:1133–1144. doi: 10.1016/j.biochi.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci USA. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zielonka J, Hardy M, Kalyanaraman B. HPLC study of oxidation products of hydroethidine in chemical and biological systems: ramifications in superoxide measurements. Free Radical Biol Med. 2009;46:329–338. doi: 10.1016/j.freeradbiomed.2008.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zielonka J, Zielonka M, Sikora A, Adamus J, Joseph J, Hardy M, Ouari O, Dranka BP, Kalyanaraman B. Global profiling of reactive oxygen and nitrogen species in biological systems: high-throughput real-time analyses. J Biol Chem. 2012;287:2984–2995. doi: 10.1074/jbc.M111.309062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zielonka J, Cheng G, Zielonka M, Ganesh T, Sun A, Joseph J, Michalski R, O’Brien WJ, Lambeth JD, Kalyanaraman B. High-throughput assays for superoxide and hydrogen peroxide: design of a screening workflow to identify inhibitors of NADPH oxidases. J Biol Chem. 2014;289:16176–16189. doi: 10.1074/jbc.M114.548693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang R, Borazjani A, Matthews AT, Mangum LC, Edelmann MJ, Ross MK. Identification of palmitoyl protein thioesterase 1 in human THP1 monocytes and macrophages and characterization of unique biochemical activities for this enzyme. Biochemistry. 2013;52:7559–7574. doi: 10.1021/bi401138s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forman HJ, Augusto O, Brigelius-Flohe R, Dennery PA, Kalyanaraman B, Ischiropoulos H, Mann GE, Radi R, Roberts LJ, II, Vina J, Davies KJ. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radical Biol Med. 2015;78:233–235. doi: 10.1016/j.freeradbiomed.2014.10.504. [DOI] [PubMed] [Google Scholar]
- 38.Leis HJ, Windischhofer W. Inhibition of cyclooxygenases 1 and 2 by the phospholipase-blocker, arachidonyl trifluoromethyl ketone. Br J Pharmacol. 2008;155:731–737. doi: 10.1038/bjp.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brash AR. Arachidonic acid as a bioactive molecule. J Clin Invest. 2001;107:1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramanadham S, Gross R, Turk J. Arachidonic acid induces an increase in the cytosolic calcium concentration in single pancreatic islet beta cells. Biochem Biophys Res Commun. 1992;184:647–653. doi: 10.1016/0006-291x(92)90638-2. [DOI] [PubMed] [Google Scholar]
- 41.Fourth National Report on Human Exposure to Environmental Chemicals. Centers for Disease Control and Prevention; Atlanta, GA: 2009. [Google Scholar]
- 42.Aronson KJ, Miller AB, Woolcott CG, Sterns EE, McCready DR, Lickley LA, Fish EB, Hiraki GY, Holloway C, Ross T, Hanna WM, SenGupta SK, Weber JP. Breast adipose tissue concentrations of polychlorinated biphenyls and other organochlorines and breast cancer risk. Cancer Epidemiol, Biomarkers Prev. 2000;9:55–63. [PubMed] [Google Scholar]
- 43.Zheng T, Holford TR, Tessari J, Mayne ST, Zahm SH, Owens PH, Zhang B, Ward B, Carter D, Zhang Y, Zhang W, Dubrow R, Boyle P. Oxychlordane and trans-nonachlor in breast adipose tissue and risk of female breast cancer. J Epidemiol Biostat. 2000;5:153–160. [PubMed] [Google Scholar]
- 44.Raaschou-Nielsen O, Pavuk M, Leblanc A, Dumas P, Philippe Weber J, Olsen A, Tjonneland A, Overvad K, Olsen JH. Adipose organochlorine concentrations and risk of breast cancer among postmenopausal Danish women. Cancer Epidemiol, Biomarkers Prev. 2005;14:67–74. [PubMed] [Google Scholar]
- 45.Muscatqq JE, Britton JA, Djordjevic MV, Citron ML, Kemeny M, Busch-Devereaux E, Pittman B, Stellman SD. Adipose concentrations of organochlorine compounds and breast cancer recurrence in Long Island, New York. Cancer Epidemiol, Biomarkers Prev. 2003;12:1474–1478. [PubMed] [Google Scholar]
- 46.Lind PM, Riserus U, Salihovic S, Bavel B, Lind L. An environmental wide association study (EWAS) approach to the metabolic syndrome. Environ Int. 2013;55:1–8. doi: 10.1016/j.envint.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 47.Lee DH, Porta M, Jacobs DR, Jr, Vandenberg LN. Chlorinated persistent organic pollutants, obesity, and type 2 diabetes. Endocr Rev. 2014;35:557–601. doi: 10.1210/er.2013-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eden PR, Meek EC, Wills RW, Olsen EV, Crow JA, Chambers JE. Association of type 2 diabetes mellitus with plasma organochlorine compound concentrations. J Exposure Sci Environ Epidemiol. 2014 doi: 10.1038/jes.2014.69. [DOI] [PubMed] [Google Scholar]
- 49.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radical Biol Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 50.Kalyanaraman B, Dranka BP, Hardy M, Michalski R, Zielonka J. HPLC-based monitoring of products formed from hydroethidine-based fluorogenic probes—the ultimate approach for intra- and extracellular superoxide detection. Biochim Biophys Acta. 2014;1840:739–744. doi: 10.1016/j.bbagen.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tryphonas H, Bondy G, Hodgen M, Coady L, Parenteau M, Armstrong C, Hayward S, Liston V. Effects of cis-nonachlor, trans-nonachlor and chlordane on the immune system of Sprague–Dawley rats following a 28-day oral (gavage) treatment. Food Chem Toxicol. 2003;41:107–118. doi: 10.1016/s0278-6915(02)00184-9. [DOI] [PubMed] [Google Scholar]
- 52.Theus SA, Tabor DR, Soderberg LS, Barnett JB. Macrophage tumoricidal mechanisms are selectively altered by prenatal chlordane exposure. Agents Actions. 1992;37:140–146. doi: 10.1007/BF01987903. [DOI] [PubMed] [Google Scholar]
- 53.Theus SA, Lau KA, Tabor DR, Soderberg LS, Barnett JB. In vivo prenatal chlordane exposure induces development of endogenous inflammatory macrophages. J Leukocyte Biol. 1992;51:366–372. doi: 10.1002/jlb.51.4.366. [DOI] [PubMed] [Google Scholar]
- 54.Lucas KK, Dennis EA. Distinguishing phospholipase A2 types in biological samples by employing group-specific assays in the presence of inhibitors. Prostaglandins Other Lipid Mediators. 2005;77:235–248. doi: 10.1016/j.prostaglandins.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 55.Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 56.Chen CC, Cheng YY, Chen SC, Tuan YF, Chen YJ, Chen CY, Chen LC. Cyclooxygenase-2 expression is up-regulated by 2-aminobiphenyl in a ROS and MAPK-dependent signaling pathway in a bladder cancer cell line. Chem Res Toxicol. 2012;25:695–705. doi: 10.1021/tx2004689. [DOI] [PubMed] [Google Scholar]
- 57.Allen EM, Florang VR, Davenport LL, Jinsmaa Y, Doorn JA. Cellular localization of dieldrin and structure–activity relationship of dieldrin analogues in dopaminergic cells. Chem Res Toxicol. 2013;26:1043–1054. doi: 10.1021/tx300458b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muller G, Ayoub M, Storz P, Rennecke J, Fabbro D, Pfizenmaier K. PKC zeta is a molecular switch in signal transduction of TNF-alpha, bifunctionally regulated by ceramide and arachidonic acid. EMBO J. 1995;14:1961–1969. doi: 10.1002/j.1460-2075.1995.tb07188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Flaherty JT, Chadwell BA, Kearns MW, Sergeant S, Daniel LW. Protein kinases C translocation responses to low concentrations of arachidonic acid. J Biol Chem. 2001;276:24743–24750. doi: 10.1074/jbc.M101093200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.