

Abstract
A persistent mystery about the ataxias has been why mutations in genes – many of which are expressed widely in the brain – primarily cause ataxia, and not, for example, epilepsy or dementia. Why should a poly-glutamine stretch in the TATA-binding protein (that is important in all cells) particularly disrupt cerebellar coordination? We propose that advances in the genetics of cerebellar ataxias suggest a rational hypothesis for how so many different genes lead to predominantly cerebellar defects. We argue that the unifying feature of many genes involved in cerebellar ataxias is their impact on the signaling protein ITPR1 (inositiol 1,4,5-triphosphate receptor type 1), that underlies coincidence detection in Purkinje cells and could play an important role in cerebellar coordination.
A brief review of cerebellar ataxias and the excitatory wiring of Purkinje cells (PCs)
Human cerebellar ataxias are caused by mutations in a wide variety of genes that at first glance appear to have little in common – except that their disruption leads to uncoordinated movements. We propose that many of these genes are linked by their interaction with one receptor, the inositol 1,4,5-triphosphate receptor type 1 (or ITPR1) encoded by the ITPR1 gene [gene reference 147265 at Online Mendelian Inheritance in Man (OMIM); http://www.ncbi.nlm.nih.gov/omim/] that is disrupted in several mouse and human ataxic phenotypes. We begin by reviewing the main pathological features of the human cerebellar ataxias, and the minimal cerebellar circuit where ITPR1-dependent signaling plays a central role in coincidence detection. Finally we summarize why the unusual function of ITPR1 in PCs can explain how purely cerebellar disorders can arise from the disruption of widely-expressed genes for several well-characterized human genetic ataxias.
Expanded polyglutamine repeats are the commonest cause of autosomal dominant spinocerebellar ataxia (SCA). Polyglutamine repeats are responsible for around 40% of cases, and the disorders are categorized as SCA1–3, 6–8, 17 or dentatorubral-pallidoluysian atrophy (DRPLA) according to the gene where the expansion is located [1]. The other, less common, types of SCA are caused by mutations in specific genes associated with ataxia (Table 1). The key neuropathological feature of the SCAs associated with polyglutamine expansions is the deposition of neuronal intranuclear or cytoplasmic inclusions. These novel pathological features have a characteristic distribution and type for each type of expansion: SCA1, 7, and 8 have nuclear deposits, SCA2 and 6 cytoplasmic inclusions, and SCA3 and DRPLA have widespread distribution of both types of inclusions [2]. There have been few reports of the neuropathology of the SCAs that are caused by point mutations in distinct genes, and the neuropathology tends to be cerebellar atrophy, and particularly involving PC loss [3,4].
Table 1. Human ataxias not associated with CAG repeats.
| Ataxia | Gene | Protein | Phenotype | Mutation | ITPR1 | Protein function | OMIM |
|---|---|---|---|---|---|---|---|
| SCA15, SCA16 | ITPR1 | ITPR1 | Pure | Deletion | Yes | ITPR1 is an intracellular calcium-release channel gated by IP3 | 606658 |
| SCA14 | PRKCG | PKCγ | Pure | Missense | M | PKCγ is a kinase expressed in cerebellum | 605361 |
| QG ataxia | CA8 | Carbonic anhydrase 8 | Pure | Missense | Yes | CA8 is an ITPR1 antagonist; quadrupedal gait ataxia | 613227 |
| SCA5 | SPTBN2 | Spectrin β | Pure | Deletion, missense | M | Spectrin β is an EAAT4 (glutamate transporter) and GluRδ2 anchor | 600224 |
| EA6 | SLC1A3 | EAAT1 | Episodic, fairly pure | Missense | M | EAAT1 is a glutamate transporter | 612656 |
| EA2 (SCA6) | CACNA1A | Cav2.1 | Episodic and pure | Nonsense, missense, CAG repeat | Yes | Cav2.1 is the main P-type calcium channel in PCs. Nonsense/missense mutations cause episodic ataxia type 2; expansion of a CAG repeat causes SCA6, a pure ataxia | 108500 183086 |
| EA5 | CACNB4 | CavB4 | Episodic Pure | Nonsense, missense | Yes | CavB4 is an accessory subunit that regulates P-type channels encoded by Cav2.1 | 601949 |
| EA1 | KCNA1 | Kv1.1 | Episodic Pure | Missense | M | Kv1.1 contributes to rheobase and neurotransmitter release of at terminals | 160120 |
| SCA13 | KCNC3 | Kv3.3 | Ataxia + | Missense | M | Kv3 channels contribute to repolarization of dendritic Ca2+ spikes in PCs | 605259 |
| SCA27 | FGF14 | Pure | Missense, nonsense | No | FGF14, intracelluar fibroblast growth factor plays a role in localising Na(v) channels. | 609307 | |
| SCA10 | ATXN10 | Ataxin-10 | Ataxia +, also pure | (ATTCT)n intron | ?? | Some present with non-consensus repeat units; Ataxin-10 function unknown | 603516 |
| SCA11 | TTBK2 | Pure | Frameshift | ?? | TTBK2 function unknown | 604432 | |
| SCA4 | PLEKHG4 | Puratrophin-1 | Pure | Substitution 5′UTR | No | Puratrophin-1 function is largely unknown; could be involved in Golgi cytoskeleton (associated with ataxia in Japanese families) | 117210 |
| SCA8 | ATXN8OS (KLHL1) | Ataxia + | (CTG)n 3′UTR | M | Repeats located in the 3′UTR. Endogenous antisense RNA overlaps the KLHL1 gene. KLHL1 is an actin-organizing protein | 608769 |
EA, episodic ataxia; QG, quadrupal gait; ITPR1, likelihood of interaction with ITPR1-dependent signaling; SCA, Spinocerebellar ataxia. m, maybe; ??, protein function unknown.
Major advancements in two fields, genetics and physiology, have paved the way for a mechanistic explanation of ataxias. On the genetic front, a large number of genes have now been linked to human cerebellar ataxias (Table 1 and Ref. [5]). In terms of cellular physiology, a clear understanding of the modulation of ITPR-dependent signaling and its contribution to plasticity in PCs (Figure 1; for recent reviews see Refs [6,7]) reveals how the two major excitatory inputs to PCs converge to regulate ITPR1 activity. We will focus exclusively on two forms of excitatory input to PCs: parallel fibers (PFs) and climbing fibers (CFs; Figure 1). We believe the interaction between these two inputs to PCs could be sufficient to explain the mechanism of many cerebellar ataxias in humans.
Figure 1.
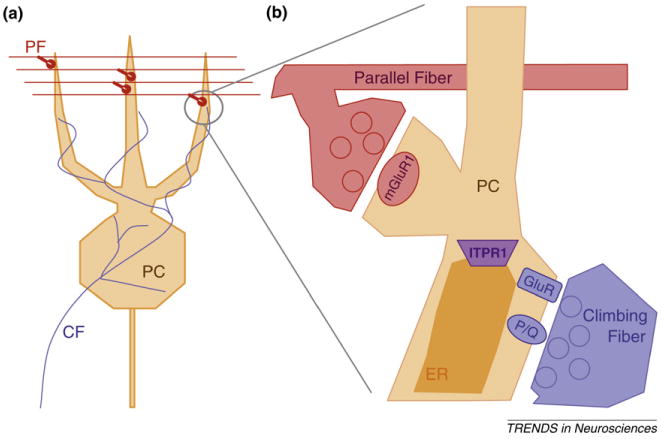
The excitatory input to Purkinje cells (PCs). (a) PCs receive two fundamentally different forms of excitatory input. Parallel fibers (PF, red) arise from abundant granule cells in the cerebellum and provide many small inputs to PCs. Climbing fibers (CF, blue) arise from the inferior olive and each PC receives a single, powerful input from a CF. (b) Parallel fibers tend to activate metabotropic glutamate receptors, particularly mGluR1, that produces IP3 and activates endoplasmic reticulum (ER) IP3Rs (purple) encoded by ITPR1. Climbing fibers activate AMPA receptors (GluR, blue) that depolarize the cell enough to allow P-type calcium channels (P/Q) to open and allow calcium to enter the PC. Climbing fiber synapses are largely on the soma of PCs, for illustration purposes they are shown adjacent to PF synapses here.
PCs, that represent the sole output of the cerebellar cortex, are thought to fire regularly in vivo, but their firing rate is modulated by the activity of PFs. An individual PC receives ∼ 100,000 PF inputs in its distal dendrites, and these inputs represent a trickle of excitatory activity, individually not strong enough to drive the firing of the PC. In contrast, each PC receives a massive synaptic input from a single climbing fiber – one of the strongest excitatory synapses in the brain [7] – that triggers a robust complex spike in the PC and large calcium transients throughout the PC's dendritic tree [6,7]. The interaction between these two pathways drives an unusual form of heterosynaptic plasticity in PCs that relies on ITPR1-dependent signaling.
This article does not aim to address whether cerebellar plasticity underlies motor learning – a topic that has been robustly debated elsewhere [6–11] – our objective is simply to highlight the tendency for human mutations linked to ataxia to disrupt genes that converge upon ITPR1, a function that plays a central role in PC plasticity and synaptic wiring. We suggest that, because these mutations manifest as movement disorders in patients, it is likely that the genes affected and the processes they converge upon are important for motor coordination.
ITPR1 underlies coincidence detection in PCs
In classic hippocampal long term potentiation (LTP), NMDA receptors serve as coincidence detectors that link strong depolarization and presynaptic activity to provide the high levels of calcium required to activate kinases that phosphorylate GluR1-containing AMPA receptors, sending them into the synaptic membrane. In hippocampal long term depression (LTD), by contrast, a low trickle of calcium through modestly active NMDARs activates phosphatases that dephosphorylate GluR1-AMPARs, causing them to be internalized.
Mature PCs in the cerebellum, however, seem not to rely heavily on NMDA receptors, and therefore they cannot use the properties of these receptors to link post-synaptic depolarization to calcium influx. In rats NMDARs are absent from cerebellar PCs [12], whereas in mice there is a small NMDA component to CF-evoked synaptic currents [13,14]. Instead of NMDARs, PCs rely on the neuronal inositol tri-phosphate receptor (type 1; ITPR1) to link different levels of synaptic activity to LTD or LTP. Most neurons have calcium-containing branches of endoplasmic reticulum (ER) in their dendrites that release calcium from intracellular stores in response to a sufficiently strong calcium influx. In pyramidal cells of the hippocampus, and most other neuronal types, these post-synaptic loops of ER use ryanodine receptors to release calcium from the stores. Instead of ryanodine receptors, PCs have dense concentrations of ITPR1 on their post-synaptic ER stores [15], and they express a slightly different splice form than other CNS regions [16]. It is the interaction between calcium and IP3 (inositol tri-phospate) on ITPR1 that underlies coincidence detection in PCs [17]. A final difference between mature PCs and hippocampal neurons is that PCs express mainly GluR2-containing AMPA receptors that, unlike GluR1 receptors, are internalized when phosphorylated [18,19].
As summarized in Figure 2, the properties of ITPR1 have been shown to underlie reversible bi-directional plasticity in PCs [20,21]. Based on the presence of ITPR1, the lack of NMDARs, and the dominance of GluR2 AMPA receptors, Jőrntell and Hansel described the reversed calcium dependence of ITPR1-dependent PC plasticity [6]. In the case of human ataxias, genetic deficiencies that disrupt ITPR1-dependent coincidence detection can be linked to changes in motor coordination – providing a rare opportunity to translate from molecule to movement [6,7].
Figure 2.
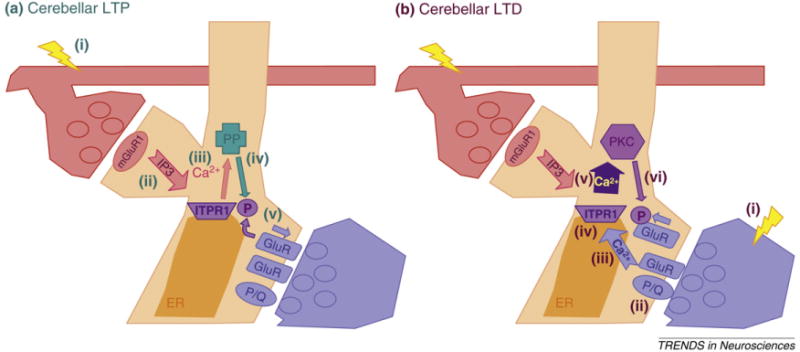
ITPR1-dependent signaling in cerebellar PCs. (a) Cerebellar LTP. (i) Parallel fiber synapses activate mGluR1 (pink ball) in PC spines. (ii) mGluR1 activation triggers an increase in IP3. (iii) IP3 binds the IP3 receptor (encoded by ITPR1) on the ER within the PC spine, and leads to a local release of calcium from intracellular stores. (iv) Volleys of parallel fiber activation (in the absence of climbing fiber activation) lead to a steady, relatively low level calcium signal that binds to and activates protein phosphatases (PP). (v) Phosphatases, then dephosphorylate GluR2-containing AMPA receptors, and stabilize them in the membrane, leading to LTP of the active parallel fiber synapses. (b) Cerebellar LTD. (i) When a climbing fiber is stimulated it activates Ca2+-impermeable GluR2-containing AMPA receptors. (ii) Depolarization mediated by the activated GluR2-AMPA receptors opens voltage-gated P-type calcium channels CaV2.1. Individual climbing fiber activation leads to a long depolarization spread throughout the PC dendrites, and a calcium-dependent dendrite action potential. (iii) ITPR1 binds calcium that enters during the massive CF-mediated depolarization. Calcium works as an allosteric enhancer of IP3R gating. (iv) When IP3 produced by parallel fiber-mGluR1 activation binds to ITPR1 that is primed with bound-Ca2+ (from climbing fibers) ITPR1 produces a much larger (supra-linear) calcium signal. (v) The increased calcium is sufficient to activate PKC. (vi) Phosphorylation of GluR2 triggers internalization, leading to LTD.
ITPR1-dependent signaling in ataxias
The centre of the storm: ITPR1
Genetics, both experiments of nature and deliberate manipulations in mice [22,23], confirm the importance of ITPR1 for motor coordination in mammals, including humans. For the parallel fiber pathway (Figure 2a), auto-antibodies targeting mGluR1 (encoded by GRM1; OMIM 604473), the receptor that triggers inositol triphosphate (IP3) production upon parallel fiber stimulation, are associated with severe paraneoplastic cerebellar ataxia [24,25]. Moreover, mice with deliberate or spontaneous disruption of mGluR1 are profoundly ataxic [26] and have no cerebellar LTD [27]. Downstream deletions in the ITPR1 gene have been shown to underlie spinocerebellar ataxia 15 in people [3], and mice with disruption of the ITPR1 locus are ataxic [22].
For the climbing fiber pathway (Figure 2b), mutations that disrupt P/Q-type calcium channels cause episodic ataxia type 2 in humans (EA2; OMIM 108500) and multiple forms of ataxia in mice [28]. Mutations in AMPA receptors lead to widespread neurological problems in mice, including epilepsy, collapsed dendrites and altered exploratory and motor behavior (summarized on OMIM 138247; GLUR2). This is consistent with the importance of these subunits throughout the nervous system. However, more subtle models that disrupt phosphorylation of GluR2 [19] or its interactions with the scaffolding protein PICK1 [29] disrupt cerebellar plasticity, but do not produce robust ataxia, suggesting that whereas direct disruption of ITPR1 can cause ataxia, downstream block of cerebellar plasticity might not (Box 1).
Box 1. Outstanding Questions.
Does long-term loss of ITPR1 activity drive dendritic collapse and PC death? It is possible to uncouple LTD from ataxia – blocking ITPR1 with T-588 blocks LTD but leaves motor learning intact [58]. Also some mouse models with selective disruption of LTD do not have obvious behavioral correlates (reviewed in Ref. [8]). These mice have very little PC death, suggesting that if loss of ITPR1 activity does lead to pruning and death, it is not via phosphorylation or internalization of GluR2. Thus, is ataxia in humans due to the disruption of ITPR1-dependent signaling, or does disruption of ITPR1 lead to pruning of dendrites and death of PCs, that then leads to ataxia?
Is there a normal role for genes containing CAG repeats? The so-called language gene, FOXP2, is a CAG-containing gene that is expressed in PCs but is so far not associated with cerebellar ataxia. Mutations affecting the CAG-containing FOXP2 transcript (although not the CAG repeat itself) lead to profound ataxia and loss of vocalizations in mice [93]. The role of FoxP2 in cerebellar development, although clearly important, remains unknown, but CAG repeats are clearly not all-or-nothing in their impact on ITPR1. There could be a ‘normal’ role for the interaction between polyglutamine stretches and ITPR1 in PCs.
Do the polyglutamine tracts from all CAG-containing transcripts get to the cytosol where they might interact with ITPR1? Whereas several of these tracts clearly do reach the dendrites of PCs in mouse models [41,42], further models will be needed to test the hypothesis that this is a ubiquitous mechanism for CAG repeat-associated human ataxias.
Given that CA8 inhibits ITPR1, whereas CAG repeats appear to activate ITPR1, do these two pathways (and the pathologies associated with them) interact?
Why are some ataxias episodic? A common theme is that mutations in ion channels lead to episodic disorders (epilepsy and migraine as well as EAs) where periods of dysfunction are interspersed with long stretches of normal behavior. The mechanism underlying this feature of channelopathies is unknown, although it appears to be conserved in the cerebellum in EA1 and EA2.
In a seminal study, Wang et al. [2000] demonstrated that the timing of calcium entry (such as that expected from activation of the climbing fiber pathway) had the most robust supra-linear enhancement of an ITPR1 signal if it occurred about 100 ms after the arrival of IP3 from mGluRs triggered by parallel fiber activation [30]. The time window is similar to the amount of time IP3 remains bound to the ITPR1 [31], and suggests that ITPR1 acts as a coincidence detector dependent on precise timing of IP3 and calcium influx.
Robust activation of ITPR1 is thought to activate PKC. The PKCγ subunit is highly and specifically expressed in PCs, and blocking PKCγ can block cerebellar LTD [32,33]. Missense mutations in PKCγ, particularly those associated with gain-of-function [34,35], cause SCA14 [36,37]. Thus it is possible that uncoupling the coincidence-detection role of ITPR1 from PKCγ activation is sufficient to derange motor control and lead to ataxia. However, it is important to note that whereas disruption of PKCγ can lead to ataxia, its role could be restricted to early in development [38], with mature LTD and AMPA trafficking relying on PKCα [39].
How do polyglutamine repeats lead to ataxia?
Several recent papers suggest that polyglutamine repeats can bind directly to and interfere with ITPR1 (reviewed in Ref. [40]). Ataxin-2 and Ataxin-3, both engineered to have polyglutamine repeats that would cause ataxia in humans, have recently been shown to bind to and activate ITPR1 and disrupt calcium signaling in PCs in mice [41,42]. This raises the possibility that it is not the toxicity of the polyglutamine repeat itself that is disrupting PC function, but the effects of the polyglutamine repeats on ITPR1 that are central to synthesizing input from parallel and climbing fibers that lead to failed motor control [43]. These results will need to be extended to other CAG repeat genes that are linked to ataxia (PPP2R2B, SCA12 [44]; KLHL1, SCA8 [45]).
SCA6 represents a crossover of two pathways. The CAG repeat is in the CACNA1A gene that encodes P/Q-type calcium channels, and P-type channels are required for coordinating AMPA activation by climbing fibers with calcium influx and modulation of IP3 binding to ITPR1 [46,47,11]. A recent paper suggests that the SCA6 expansion represents a gain-of-function of the P-type Ca2+ channels [48], suggesting that this expansion potentially has a double effect – both via polyglutamine binding to ITPR1 and through altered calcium entry via the P-type channel.
Not all CAG repeats cause cerebellar ataxia. Among genes containing CAG repeats, 8 are strongly associated with SCAs. Eleven CAG-repeat-containing genes are not strongly associated with SCAs (although HTT/Huntingtin is clearly associated with movement disorders). If polyglutamine repeats are non-specific activators of ITPR1 that disrupt its signaling and lead to muddled control of movements, then why do only some genes containing CAG repeats lead to cerebellar ataxia? One interesting case is the small conductance Ca2+-activated potassium channel SK3 encoded by KCNC3. SK channels are known to be crucial for regulating the firing of PCs, but PCs do not express SK3 – they only express SK2 (KCNC2 [49]), thereby suggesting an explanation for the specificity of CAG repeats – only those genes expressed in PCs can generate polyglutamine repeats to activate their ITPR1s. Thus, to return to the ubiquitously-expressed TATA-binding protein (TBP) in which polyglutamine repeats cause SCA17, this ataxia could have nothing to do with the normal function of TBP in initiating transcription in the nucleus, but could instead simply be a result of accumulation of polyglutamine tracts binding to and disrupting ITPR-dependent coincidence detection in PCs (Box 1).
A newly described mutation in carbonic anhydrase related protein 8 (CA8) causes quadruple gait ataxia [50]. Previous work also implicates auto-antibodies against CA8 in paraneoplastic cerebellar degeneration (PCD) [51]. In contrast to CAG repeats that activate ITPR1, CA8 appears to be an ITPR1 antagonist that inhibits IP3 binding, and loss of CA8 is associated with aberrant PF–PC contacts, including multiple parallel fibers contacting single PC spines [52]. This sort of disruption of the PF– PC–CF synaptic architecture is a theme in spontaneous ataxic mutations in mice (reviewed in Ref. [7]). Notably, PC dendrites are dramatically shrunken in mice harboring the classic Lurcher mutation (a gain of malfunction affecting Grid2, that turns a glutamate receptor-like protein into a leaky channel), that is now thought to disrupt pre-and post-synaptic targeting between PFs and PCs [7,53].
Calcium homeostasis, ITPR1-dependent signaling, and ataxia
A complementary, more general hypothesis was recently the subject of a review about the mechanism underlying ataxia. The hypothesis holds that loss of control of calcium buffering leads to Purkinje dysfunction [5]. Clearly calcium buffering plays an important role. PCs have two main calcium buffers (calbindin and parvalbumin) and deletion of either leads to mild, but distinct ataxic phenotypes in mice (reviewed in Ref. [54]).
Whereas no gene mutations affecting calcium-buffering proteins (CALB1, or PVALB; OMIM 114050, 168890), have yet been reported in human ataxias, it has been shown that changing calcium buffering substantially alters the severity of ataxic models. Deleting calbindin significantly accelerates the phenotype of mice carrying CAG repeats in ataxin1 [55], that is associated with the fairly pure cerebellar ataxia, SCA1, in humans (ATXN1; OMIM 601556). Moreover, it has been shown that one of the first indications of polyglutamine toxicity in SCA1 mice is downregulation of the expression of genes encoding calcium buffering proteins [56], suggesting one reason why the pathology of these disorders could be delayed. Early on, calcium buffering might obscure the deficit in ITPR1 signaling, but in time this buffering capability appears to be lost (Box 1). One significant observation is that the loss of each of these calcium buffers also leads to defects in spine geometry in the PF–PC synapses [57], but is it calcium buffering or disruption of synaptic wiring that leads to ataxia?
Plasticity, pruning or death?
As we have mentioned, ITPR1-dependent coincidence detection appears to be necessary for several forms of cerebellar plasticity. However, it is not true that disrupting cerebellar plasticity is sufficient to cause ataxia. Several mice with profound disruption of cerebellar plasticity have little or no detectable movement disorders [29,32,58–60]. Thus the defect in ITPR1-signaling could lead to ataxia through a mechanism that is upstream of CF–PC plasticity. Post-mortem examinations of patients with ataxia suggest the deficits could be larger than subtle changes in plasticity might justify.
A prominent feature in SCA cerebella is the loss of PCs (reviewed in Refs [1,61]). PCs are particularly sensitive to many insults, but the exact mechanisms leading to their death remain unclear (reviewed in Ref. [62]). In some circumstances PCs seem to be preserved, such as in the Tau-related dementias. However, the pruning of PC synapses is a common theme in ataxias. It is the most notable feature in Lurcher mice [7,53], and is prominent in Cacna1a ataxic mice [7], and in a quadruple gait ataxia model [52]. In mice with mutations affecting ITPR1 the onset of severe ataxia coincides with PF–PC synaptogenesis [22,23,3], and IP3 signaling is required to maintain PF–PC synapses via retrograde signaling with brain-derived neurotrophic factor (BDNF) [63].
In contrast to mutations that lead to PC pruning or death, acute pharmacological block of ITPR1 with T-588 is neuroprotective for PCs and, although it blocks LTD [64], T-588 does not alter two types of motor learning [58]. The question remains, does the long-term loss of ITPR1-dependent signaling lead to pruning and death of PCs in patients with ataxia, and is ataxia itself delayed in onset until PCs are lost, or until their PF synapses are pruned?
Cerebellar ataxias from ITPR1-dependent functions of widely-expressed genes
Whereas the importance of ITPR1-signalling in PCs could explain the cerebellar pathology associated with polyglutamine repeats, there are several other widely-expressed genes that also can have predominantly cerebellar defects when they are disrupted, but that do not contain polyglutamine repeats. For three different groups of these genes we propose mechanisms that suggest how a link to ITPR1-dependent signaling could lead to their specific cerebellar pathology.
Mutations that disrupt glutamatergic signaling at PCs
Ikeda and colleagues identified the SPTBN2 gene as the cause of SCA5 [65], suggesting that the β3-spectrin domain of this gene, that is an important anchor for glutamate transporters (particularly EAAT4) that help to clear glutamate from synapses, particularly disrupts PC function. Whereas EAAT4 itself has not been implicated in human ataxias, mutations affecting EAAT1, another glutamate transporter (SLC1A3; OMIM 600111), cause episodic ataxia type 6, and mutations in a transporter that is used to pump glutamate into vesicles (SLC17A5; OMIM 604322) cause Salla disease that is characterized by cerebellar ataxia.
As with many SCAs, the disruption of this pathway is associated with PC death. In humans, the expression levels of the glial glutamate transporter (GLAST) and EAAT4 are correlated with neonatal ischemic PC death, and high levels of transporters tend to protect PCs [66]. It is plausible that loss of efficient glutamate transport disrupts the temporal specificity of ITPR1-dependent signaling, and it is this disruption that leads to a disproportionately cerebellar pathology. Indeed, blocking glutamate transporters has been shown to increase mGluR signaling, to enhance ITPR1-dependent cerebellar LTD [67], and even to induce LTD in conditions where it normally would not be triggered [68]. The interactions could be local – PCs with higher levels of EAAT4 expression in GLAST mice have reduced LTD [69]. As with the ITPR1 blocker T-588, transient block of transporters appears both protective and able to increase ITPR1-dependent LTD (possibly by increasing mGluR signaling), but congenital disruption of these proteins leads to aberrant wiring and ataxia.
Mutations in P/Q-type calcium channels and episodic ataxia
CACNA1A, the gene that encodes P/Q-type calcium channels is associated with both cerebellar and cortical disorders (migraine) in patients (OMIM 601011). As mentioned above, a polyglutamine expansion in this gene causes SCA6, whereas a growing number of loss-of-function mutations, including deletions and duplications [70,71] lead to episodic ataxia type 2 (EA2). Mutations in P/Q type channels should affect synaptic transmission throughout the CNS; however in humans haploinsufficiency of the gene is usually associated with pure cerebellar episodic ataxia. This argues that, whereas the CNS-wide role of these channels in transmitter release can be compensated in haploinsufficient individuals, the specific role these channels play in the cerebellum remains vulnerable. The unusual task these channels have in PCs, linking depolarization to modification of ITPR1-dependent signaling, could be the function that is disrupted by haploinsufficiency. In fact, PCs have one of the highest concentration of P-type channels of any neuron (the channels were originally named for their density in these cells [72]). A failure to adequately modify ITPR1-dependent signaling would not be predicted to have major effects elsewhere in the CNS where ITPR1 is used at a much lower density, or where ER stores are accessed via ryanodine receptors and NMDARs provide calcium influx.
Potassium channels and PC excitability
Two different potassium channel genes are implicated in human ataxia – mutations, typically dominant loss-of-function mutations in shaker voltage-gated potassium channels (Kv1.1, encoded by KCNA1) cause episodic ataxia type 1 (EA1), and mutations in the Shaw channels (Kv3.3 encoded by KCNC3) cause spinocerebellar ataxia type 13 (SCA13). Both channels are widespread in the brain [73].
Kv1.1 appears to be necessary for preventing aberrant spontaneous firing of the climbing fibers that activate the AMPA-P-type path to ITPR1 [74,75], and Kv1 channels – particularly those paired with Kv1.2 – are important for preventing the dendritic tree of PCs from depolarizing enough to produce random calcium spikes [76]. Importantly, all EA1 mutations to date are dominant-negative [77]. Because functional Kv1 channels require the co-assembly of four subunits to form a functional channel [78], mutant subunits can disrupt the function of subunits from the unaffected allele, or from other Kv1 channels, leading to fewer available channels than if the gene encoding Kv1.1 was simply deleted. Consistent with this, mice lacking KCNA1 have epilepsy, but not overt ataxia [79], but mice with knock-ins corresponding to known EA1 mutations in humans are homozygous-lethal [80]. It is possible that, in PCs of individuals with EA1 mutations, the loss of Kv1.2 function (disrupted by assembly with mutant Kv1.1 subunits) leads to aberrant depolarization of the PC dendritic tree and possibly lowered threshold for spiking [76]. We recently demonstrated that individual neurons expressing a Kv1.1 mutation associated with severe, drug-resistant EA1 lowered rheobase (the amount of current needed to trigger a spike) and increased transmitter release in autaptic hippocampal neurons, whereas a mutation associated with EA1 complicated by seizures altered only release probability [81]. It is thus possible that increased release probability from the CFs contributes to ataxia in EA1. It is also clear that different mutations within the same channel can selectively alter distinct aspects of neuronal function.
Kv3 channels share a similar four subunit composition with Kv1 channels [78], however Kv3 channels have high thresholds for activation and inactivate quickly – they help promote rapid firing [82]. Kv3 channels seem to be predominantly functioning in the soma of PCs, and are important for regulating their output [83,84]. In response to climbing fiber stimulation, PCs normally fire a stereotypic complex spike. Block of the rapid-inactivating Kv3 channels prevents the generation of repetitive spikelets that form a complex spike [85]. Consistent with their specific role in PCs, targeted restoration of Kv3.3 in PCs of knockout mice restored both the complex spikes and motor function [86]. Thus Kv3.3 appears to be necessary for triggering the complex spike that exemplifies the PC response to CF stimulation, and our model suggests these complex spikes are necessary to preserve ITPR1-dependent signaling.
Recently, mutations in KCNJ10 (encoding an inwardly-rectifying potassium channel) have been shown to be linked to a complex syndrome associated with epilepsy, deafness and electrolyte imbalance, as well as ataxia [87,88]. However, in contrast to the cerebellar-specific functions targeted by mutations affecting Kv1.1, Kv3.3 or P-type calcium channels, the mutations in KCNJ10 channels clearly disrupt functions that are not restricted to the cerebellum, because the patients manifest with many symptoms that are not cerebellar. Consequently, disorders associated with KCNJ10 do not fit with the model of a cerebellar-restricted deficit associated with a widely expressed gene.
Some caveats to the model
We do not propose that ITPR1-dependent signaling explains all ataxias, but rather that a large subset of genetic changes underlying human ataxias could disrupt the signaling of these receptors.
Currently (January 2010) some 50 genes in OMIM refer to ‘cerebellar ataxia’. Of these, our ITPR1-signaling model incorporates the seven associated with CAG repeats, and most of the SCAs and EAs not caused by repeats (Table 1). However there are several genes linked to cerebellar ataxia that do not target ITPR1-dependent signaling. Six different mitochondrial genes are implicated in ataxia, and more than ten mutations in ‘housekeeping’ genes involved in DNA repair, RNA processing or protein folding lead to cerebellar ataxias, that in some cases (TDP1, a topoisomerase; and SYNE1, a nuclear envelope protein) lead to a fairly pure cerebellar phenotype. Why loss of what are prima fasci ubiquitously important proteins would cause ataxia (and not, say, death) is still mysterious, but could simply reflect the metabolic challenges PCs face with such large volumes and extensive dendritic arbors.
The model also leaves several important points unresolved, including the latency of the disease, the episodic nature of some of the disorders, and whether cerebellar plasticity itself plays an important role in human ataxias (Box 1).
Some predictions of the model
Mutations in TTBK2 have recently been shown to underlie SCA11 [89], but the function of this gene is currently unknown. The model we propose here predicts that animal models of SCA11 will have disrupted ITPR1-dependent signaling (that could manifest as disrupted timing in cerebellar LTD, or lost PF–PC synapses). Investigating the mechanism of these deficits could reveal the function of the TTBK2 gene product in healthy PCs.
There are several human genetic ataxias that are not yet linked to specific genes. In the cases where the loci have been narrowed to a reasonably small number of genes, it could be worth investigating local genes for those with possible roles in modifying ITPR1-dependent signaling.
Those human ataxias caused by alterations in genes clearly implicated in ITPR1-dependent signaling tend to be associated with a more pure phenotype (e.g. CACNA1A, ITPR1, PKCG, CACN4B; Table 1), whereas most of those associated with polyglutamine repeats have mixed phenotypes (‘Ataxia +’ in Table 1). An exception is FGF14, that although it gives a pure ataxic phenotype, is not a gene that has been clearly associated with ITPR1. It has recently been shown that the FGF14 protein regulates neuronal sodium channels [90]. An implication of our model is that FGF14 also plays an important, but as yet unknown, role in modifying ITPR1.
Finally, increased cerebellar LTD, that relies on ITPR1, has recently been associated with increased motor learning. Two papers have reported on mice with enhanced cerebellar LTD that also showed enhanced motor learning or optokinetic responses [91,92]. These papers did not explore the link between the altered genes (class I MHC molecules [91]; Delphilin [92]) and ITPR1-dependent signaling. Our model suggests that both molecules could modify ITPR1, possibly by reducing its activity as with the ITPR1 blocker T-588 that also increases cerebellar LTD and appears protective.
Basic cerebellar synaptic physiology has offered valuable insights into the genetics of human ataxias. It is now possible that the genetics of ataxias, through careful studies of mouse models with ataxic genes, will lead to new insights into the mechanisms of ITPR1-dependent signaling in cerebellar PCs
Acknowledgments
This work was undertaken at University College London Hospital (UCLH) and University College London (UCL) with support from the Department of Health's National Institute for Health Research Biomedical Research Centers funding scheme. Work in our laboratories is sponsored by the MRC, Ataxia UK, and the Wellcome Trust. S.S. holds a fellowship from the Worshipful Company of Pewterers. This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services; project Z01 AG000958-06.
References
- 1.Manto M. The wide spectrum of spinocerebellar ataxias (SCAs) Cerebellum. 2005;4:2–6. doi: 10.1080/14734220510007914. [DOI] [PubMed] [Google Scholar]
- 2.Yamada M, et al. CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:71–86. doi: 10.1007/s00401-007-0287-5. [DOI] [PubMed] [Google Scholar]
- 3.van de Leemput J, et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007;3:e108. doi: 10.1371/journal.pgen.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson J, et al. Clinical and genetic analysis of spinocerebellar ataxia type 11. Cerebellum. 2008;7:159–164. doi: 10.1007/s12311-008-0022-3. [DOI] [PubMed] [Google Scholar]
- 5.Carlson KM, et al. Emerging pathogenic pathways in the spinocerebellar ataxias. Curr Opin Genet Dev. 2009;19:247–253. doi: 10.1016/j.gde.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jԁrntell H, Hansel C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber–Purkinje cell synapses. Neuron. 2006;52:227–238. doi: 10.1016/j.neuron.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe M. Molecular mechanisms governing competitive synaptic wiring in cerebellar Purkinje cells. Tohoku J Exp Med. 2008;214:175–190. doi: 10.1620/tjem.214.175. [DOI] [PubMed] [Google Scholar]
- 8.Evans GJO. Synaptic signalling in cerebellar plasticity. Biol Cell. 2007;99:363–378. doi: 10.1042/BC20070010. [DOI] [PubMed] [Google Scholar]
- 9.Porrill J, Dean P. Cerebellar motor learning: when is cortical plasticity not enough? PLoS Comput Biol. 2007;3:1935–1950. doi: 10.1371/journal.pcbi.0030197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manto M. The cerebellum, cerebellar disorders, and cerebellar research – two centuries of discoveries. Cerebellum. 2008;7:505–516. doi: 10.1007/s12311-008-0063-7. [DOI] [PubMed] [Google Scholar]
- 11.Higley MJ, Sabatini BL. Calcium signaling in dendrites and spines: practical and functional considerations. Neuron. 2008;59:902–913. doi: 10.1016/j.neuron.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 12.Farrant M, Cull-Candy SG. Excitatory amino acid receptor-channels in Purkinje cells in thin cerebellar slices. Proc Biol Sci. 1991;244:179–184. doi: 10.1098/rspb.1991.0067. [DOI] [PubMed] [Google Scholar]
- 13.Renzi M, et al. Climbing-fibre activation of NMDA receptors in Purkinje cells of adult mice. J Physiol (Lond) 2007;585:91–101. doi: 10.1113/jphysiol.2007.141531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piochon C, et al. NMDA receptor contribution to the climbing fiber response in the adult mouse Purkinje cell. J Neurosci. 2007;27:10797–10809. doi: 10.1523/JNEUROSCI.2422-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bardo S, et al. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol Sci. 2006;27:78–84. doi: 10.1016/j.tips.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Nucifora FC, et al. Molecular cloning of a cDNA for the human inositol 1,4,5-trisphosphate receptor type 1, and the identification of a third alternatively spliced variant. Brain Res Mol Brain Res. 1995;32:291–296. doi: 10.1016/0169-328x(95)00089-b. [DOI] [PubMed] [Google Scholar]
- 17.Inoue T, et al. Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci. 1998;18:5366–5373. doi: 10.1523/JNEUROSCI.18-14-05366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung HJ, et al. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung HJ, et al. Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science. 2003;300:1751–1755. doi: 10.1126/science.1082915. [DOI] [PubMed] [Google Scholar]
- 20.Coesmans M, et al. Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron. 2004;44:691–700. doi: 10.1016/j.neuron.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 21.Lev-Ram V, et al. Reversing cerebellar long-term depression. Proc Natl Acad Sci U S A. 2003;100:15989–15993. doi: 10.1073/pnas.2636935100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsumoto M, et al. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- 23.Street VA, et al. The type 1 inositol 1,4,5-trisphosphate receptor gene is altered in the opisthotonos mouse. J Neurosci. 1997;17:635–645. doi: 10.1523/JNEUROSCI.17-02-00635.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sillevis Smitt P, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med. 2000;342:21–27. doi: 10.1056/NEJM200001063420104. [DOI] [PubMed] [Google Scholar]
- 25.Coesmans M, et al. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann Neurol. 2003;53:325–336. doi: 10.1002/ana.10451. [DOI] [PubMed] [Google Scholar]
- 26.Conquet F, et al. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- 27.Aiba A, et al. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- 28.Fletcher CF, et al. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 29.Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, et al. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49(6):845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 30.Wang SS, et al. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3:1266–1273. doi: 10.1038/81792. [DOI] [PubMed] [Google Scholar]
- 31.Sarkisov DV, et al. Expression of protein kinase C inhibitor blocks cerebellar long-term depression without affecting Purkinje cell excitability in alert mice. J Neurosci. 2001;21:5813–5823. doi: 10.1523/JNEUROSCI.21-15-05813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goossens J, Daniel H, Rancillac A, van der Steen J, Oberdick J, Crépel F, et al. Expression of protein kinase C inhibitor blocks cerebellar long-term depression without affecting Purkinje cell excitability in alert mice. J Neurosci. 2001;21(15):5813–5823. doi: 10.1523/JNEUROSCI.21-15-05813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kano M, et al. Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell. 1995;83:1223–1231. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 34.Verbeek DS, et al. Protein kinase C gamma mutations in spinocerebellar ataxia 14 increase kinase activity and alter membrane targeting. Brain. 2005;128:436–442. doi: 10.1093/brain/awh378. [DOI] [PubMed] [Google Scholar]
- 35.Adachi N, et al. Enzymological analysis of mutant protein kinase Cgamma causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J Biol Chem. 2008;283:19854–19863. doi: 10.1074/jbc.M801492200. [DOI] [PubMed] [Google Scholar]
- 36.Chen D, et al. Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet. 2003;72:839–849. doi: 10.1086/373883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yabe I, et al. Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma. Arch Neurol. 2003;60:1749–1751. doi: 10.1001/archneur.60.12.1749. [DOI] [PubMed] [Google Scholar]
- 38.Patten SA, et al. A unique PDZ ligand in PKCalpha confers induction of cerebellar long-term synaptic depression. Neuron. 2004;44:585–594. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 39.Leitges M, Kovac J, Plomann M, Linden DJ. A unique PDZ ligand in PKCalpha confers induction of cerebellar long-term synaptic depression. Neuron. 2004;44(4):585–594. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 40.Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci. 2008;28:12713–12724. doi: 10.1523/JNEUROSCI.3909-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 2. J Neurosci. 2009;29:9148–9162. doi: 10.1523/JNEUROSCI.0660-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oue M, et al. Characterization of mutant mice that express polyglutamine in cerebellar Purkinje cells. Brain Res. 2009;1255:9–17. doi: 10.1016/j.brainres.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 44.Holmes SE, et al. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat Genet. 1999;23:391–392. doi: 10.1038/70493. [DOI] [PubMed] [Google Scholar]
- 45.Koob MD, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8) Nat Genet. 1999;21:379–384. doi: 10.1038/7710. [DOI] [PubMed] [Google Scholar]
- 46.Gruol DL, et al. Ca2+ signaling pathways linked to glutamate receptor activation in the somatic and dendritic regions of cultured cerebellar purkinje neurons. J Neurophysiol. 1996;76:3325–3340. doi: 10.1152/jn.1996.76.5.3325. [DOI] [PubMed] [Google Scholar]
- 47.Okubo Y, et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc Natl Acad Sci U S A. 2008;105:11987–11992. doi: 10.1073/pnas.0804350105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watase K, Barrett CF, Miyazaki T, Ishiguro T, Ishikawa K, Hu Y, et al. Spinocerebellar ataxia type 6 knockin mice develop a progressive neuronal dysfunction with age-dependent accumulation of mutant CaV2.1 channels. Proc Natl Acad Sci U S A. 2008;105(33):11987–11992. doi: 10.1073/pnas.0804350105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cingolani LA, et al. Developmental regulation of small-conductance Ca2+-activated K+ channel expression and function in rat Purkinje neurons. J Neurosci. 2002;22:4456–4467. doi: 10.1523/JNEUROSCI.22-11-04456.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Türkmen S, et al. CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet. 2009;5:e1000487. doi: 10.1371/journal.pgen.1000487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bataller L, et al. Carbonic anhydrase-related protein VIII: autoantigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2004;56:575–579. doi: 10.1002/ana.20238. [DOI] [PubMed] [Google Scholar]
- 52.Hirasawa M, et al. Carbonic anhydrase related protein 8 mutation results in aberrant synaptic morphology and excitatory synaptic function in the Cerebellum. Mol Cell Neurosci. 2007;35:161–170. doi: 10.1016/j.mcn.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ito-Ishida A, et al. Cbln1 regulates rapid formation and maintenance of excitatory synapses in mature cerebellar Purkinje cells in vitro and in vivo. J Neurosci. 2008;28:5920–5930. doi: 10.1523/JNEUROSCI.1030-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwaller B, et al. ‘New’ functions for ‘old’ proteins: the role of the calcium-binding proteins calbindin D-28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum. 2002;1:241–258. doi: 10.1080/147342202320883551. [DOI] [PubMed] [Google Scholar]
- 55.Vig PJ, et al. Calcium homeostasis and spinocerebellar ataxia-1 (SCA-1) Brain Res Bull. 2001;56:221–225. doi: 10.1016/s0361-9230(01)00595-0. [DOI] [PubMed] [Google Scholar]
- 56.Lin X, et al. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt H, et al. Spino-dendritic cross-talk in rodent Purkinje neurons mediated by endogenous Ca2+-binding proteins. J Physiol (Lond) 2007;581:619–629. doi: 10.1113/jphysiol.2007.127860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Welsh JP, et al. Normal motor learning during pharmacological prevention of Purkinje cell long-term depression. Proc Natl Acad Sci U S A. 2005;102:17166–17171. doi: 10.1073/pnas.0508191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boyden ES, et al. Selective engagement of plasticity mechanisms for motor memory storage. Neuron. 2006;51:823–834. doi: 10.1016/j.neuron.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 60.Feil R, et al. Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. J Cell Biol. 2003;163:295–302. doi: 10.1083/jcb.200306148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koeppen AH. The pathogenesis of spinocerebellar ataxia. Cerebellum. 2005;4:62–73. doi: 10.1080/14734220510007950. [DOI] [PubMed] [Google Scholar]
- 62.Potts MB, et al. Models of traumatic cerebellar injury. Cerebellum. 2009;8:211–221. doi: 10.1007/s12311-009-0114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Furutani K, et al. Postsynaptic inositol 1,4,5-trisphosphate signaling maintains presynaptic function of parallel fiber-Purkinje cell synapses via BDNF. Proc Natl Acad Sci U S A. 2006;103:8528–8533. doi: 10.1073/pnas.0600497103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kimura T, et al. Purkinje cell long-term depression is prevented by T-588, a neuroprotective compound that reduces cytosolic calcium release from intracellular stores. Proc Natl Acad Sci U S A. 2005;102:17160–17165. doi: 10.1073/pnas.0508190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ikeda Y, et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38:184–190. doi: 10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- 66.Inage YW, et al. Expression of two glutamate transporters, GLAST and EAAT4, in the human cerebellum: their correlation in development and neonatal hypoxic-ischemic damage. J Neuropathol Exp Neurol. 1998;57:554–562. doi: 10.1097/00005072-199806000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Brasnjo G, Otis TS. Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron. 2001;31:607–616. doi: 10.1016/s0896-6273(01)00377-4. [DOI] [PubMed] [Google Scholar]
- 68.Su L, Shen Y. Blockade of glutamate transporters facilitates cerebellar synaptic long-term depression. Neuroreport. 2009;20:502–507. doi: 10.1097/WNR.0b013e328328f397. [DOI] [PubMed] [Google Scholar]
- 69.Wadiche JI, Jahr CE. Patterned expression of Purkinje cell glutamate transporters controls synaptic plasticity. Nat Neurosci. 2005;8:1329–1334. doi: 10.1038/nn1539. [DOI] [PubMed] [Google Scholar]
- 70.Riant F, et al. Large CACNA1A deletion in a family with episodic ataxia type 2. Arch Neurol. 2008;65:817–820. doi: 10.1001/archneur.65.6.817. [DOI] [PubMed] [Google Scholar]
- 71.Labrum RW, et al. Large scale calcium channel gene rearrangements in episodic ataxia and hemiplegic migraine: implications for diagnostic testing. J Med Genet. 2009;46:786–791. doi: 10.1136/jmg.2009.067967. [DOI] [PubMed] [Google Scholar]
- 72.Llinás R, et al. Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison. Proc Natl Acad Sci U S A. 1989;86:1689–1693. doi: 10.1073/pnas.86.5.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vacher H, et al. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88:1407–1447. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haghdoust H, et al. Physiological role of dendrotoxin-sensitive K+ channels in the rat cerebellar Purkinje neurons. Physiol Res. 2007;56:807–813. doi: 10.33549/physiolres.931041. [DOI] [PubMed] [Google Scholar]
- 75.McKay BE, et al. Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons. J Neurosci. 2005;25:1481–1492. doi: 10.1523/JNEUROSCI.3523-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khavandgar S, et al. Kv1 channels selectively prevent dendritic hyperexcitability in rat Purkinje cells. J Physiol (Lond) 2005;569:545–557. doi: 10.1113/jphysiol.2005.098053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rajakulendran S, et al. Episodic ataxia type 1: a neuronal potassium channelopathy. Neurotherapeutics. 2007;4:258–266. doi: 10.1016/j.nurt.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 78.Armstrong CM. Voltage-gated K channels. Sci STKE. 2003;2003:re10. doi: 10.1126/stke.2003.188.re10. [DOI] [PubMed] [Google Scholar]
- 79.Smart SL, et al. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- 80.Herson PS, et al. A mouse model of episodic ataxia type-1. Nat Neurosci. 2003;6:378–383. doi: 10.1038/nn1025. [DOI] [PubMed] [Google Scholar]
- 81.Heeroma JH, et al. Episodic ataxia type 1 mutations differentially affect neuronal excitability and transmitter release. Dis Model Mech. 2009;2:612–619. doi: 10.1242/dmm.003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rudy B, et al. Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. J Neurosci. 2008;28:1291–1300. doi: 10.1523/JNEUROSCI.4358-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McKay BE, Turner RW. Kv3 K+ channels enable burst output in rat cerebellar Purkinje cells. Eur J Neurosci. 2004;20(3):729–739. doi: 10.1111/j.1460-9568.2004.03539.x. [DOI] [PubMed] [Google Scholar]
- 84.Joho RH, Hurlock EC. The role of Kv3-type potassium channels in cerebellar physiology and behavior. Cerebellum. 2009;8(3):323–333. doi: 10.1007/s12311-009-0098-4. [DOI] [PubMed] [Google Scholar]
- 85.Zagha E, Lang EJ, Rudy B. Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. J Neurosci. 2008;28(6):1291–1300. doi: 10.1523/JNEUROSCI.4358-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hurlock EC, et al. Purkinje-cell-restricted restoration of Kv3.3 function restores complex spikes and rescues motor coordination in Kcnc3 mutants. J Neurosci. 2008;28:4640–4648. doi: 10.1523/JNEUROSCI.5486-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bockenhauer D, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scholl UI, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Houlden H, et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet. 2007;39:1434–1436. doi: 10.1038/ng.2007.43. [DOI] [PubMed] [Google Scholar]
- 90.Lou J, et al. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J Physiol (Lond) 2005;569:179–193. doi: 10.1113/jphysiol.2005.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McConnell MJ, et al. H2-K(b) and H2-D(b) regulate cerebellar long-term depression and limit motor learning. Proc Natl Acad Sci U S A. 2009;106:6784–6789. doi: 10.1073/pnas.0902018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takeuchi T, et al. Enhancement of both long-term depression induction and optokinetic response adaptation in mice lacking delphilin. PLoS ONE. 2008;3:e2297. doi: 10.1371/journal.pone.0002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fujita E, et al. Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells. Proc Natl Acad Sci U S A. 2008;105:3117–3122. doi: 10.1073/pnas.0712298105. [DOI] [PMC free article] [PubMed] [Google Scholar]