

Abstract
Purpose
Diabetes mellitus (DM) is a metabolic disorder caused by an absolute or relative deficiency of insulin, a debilitating and costly disease with multiple serious complications. Lower urinary tract (LUT) complications are among the most common complications of DM. The most common and bothersome LUT complication of DM is diabetic cystopathy, or diabetic bladder dysfunction (DBD). We reviewed the current translational knowledge of DBD.
Materials and Methods
We performed a search of the English literature through PUBMED. The key words used were “diabetes” and “bladder dysfunction” or “cystopathy”. Our data and perspective are provided for consideration of future direction of research.
Results
Despite traditional recognition of DBD, as a voiding problem, characterized by poor emptying and overflow incontinence, recent clinical and experimental evidence indicate a presence of storage problems such as urgency, and urge incontinence in DM. Recent experimental evidence from studies of DBD on small animal models of DM, indicate the presence of a temporal effect on DBD: Early phase of DM causes compensated bladder function; and late phase of DM causes decompensated bladder function. The ‘temporal theory’ could plausibly provide the scientific road map for correlation between clinical and experimental findings as well as identification of role of mechanisms such as polyuria, hyperglycemia, oxidative stress, autonomic neuropathy and decompensation of contractile apparatus of the bladder in creation of clinical and experimental manifestations of the DBD.
Conclusions
DBD includes time-dependent manifestations of storage and emptying problems. Identification of mechanistic pathways would lead us to identification of therapeutic intervention.
Keywords: Diabetes, Bladder Dysfunction, Cystopathy, Complications
INTRODUCTION
About one of every 14 Americans, including almost one of every seven African-Americans and one of every five seniors (≥65 years old), has DM, with roughly 30% of those undiagnosed. Type 1 DM accounts for 5-10% of all diagnosed cases. About one of every 500 children and adolescents has type 1 DM 1. The incidence of type 2 DM, accounts for 90-95% of cases, increased by 33% between 1990 and 1998 and by 75% among those 30 to 39 years of age 2. The U.S. Centers for Disease Control and Prevention estimated in 2005 that 20.8 million people in the U.S. (7% of the population) have DM. The total medical and indirect costs of DM and its complications were estimated to be $132 billion in the U.S. in 2005, accounting for about 10% of total health care costs 1. Continuation of this trend is expected due to the continuing rise in obesity, a major risk factor for type 2 DM. Diabetics live decades with the disease and are susceptible to numerous burdensome and costly complications. It is indeed the complications of DM that render it a debilitating and devastating disease. Lower urinary tract (LUT) complications are among the most common complications of DM. The most common and bothersome LUT complication of DM is diabetic cystopathy, or diabetic bladder dysfunction (DBD) 3. We reviewed the current translational knowledge of DBD.
DIABETIC BLADDER DYSFUNCTION
LUT complications are found in more than 80% of individuals diagnosed with DM, a higher rate than that of widely recognized complications such as neuropathy and nephropathy, which affect less than 60% and 50% of patients, respectively 4. The most common and bothersome LUT complication of DM is DBD. Although DBD is not life threatening, it affects quality of life substantially. Yet, little is known about the natural history and pathophysiology of DBD. The paucity of knowledge has been a barrier to developing the best methods of prevention and treatment.
DBD: storage or voiding problem?
The bladder has two major and distinct functions: urine storage and urine disposal. A simplified categorization of bladder dysfunction into problems of storage or voiding has been widely accepted 5. Urodynamic studies (UDS) are often used to provide more information on the storage or voiding nature of the bladder dysfunction, as outlined in Table 1. DBD has been described traditionally as a triad of decreased sensation, increased capacity and poor emptying, but many inconsistencies with those “classic” findings have been found. In most of the asymptomatic diabetic patients they studied, Ueda et al. found increased bladder volume at first sensation to void and a decrease in detrusor contractility, with resultant increased post void residual urine volume, but they also found a 25% incidence of detrusor overactivity 6. A review by Kaplan and coworkers of urodynamic findings in 182 diabetic patients revealed 55% with detrusor overactivity but only 23% with impaired contractility, with 10% of patients areflexic and 11% “indeterminate” 7. The mixed clinical picture of DBD has also been revealed in recent large-scale studies, in which DM was associated with a 40-80% increased risk of urge incontinence and a 30-80% increased risk for overflow incontinence in controlled multivariate analyses 8. So, it is now clear that DBD manifestations are a combination of storage and voiding bladder problems.
Table 1.
Types of Bladder Dysfunction
| Storage problems | Voiding problems | |
|---|---|---|
| Symptoms | urgency, urge incontinence | hesitancy, slow urine stream |
| Urodynamics results | sensory urgency, detrusor overactivity | slow flow, high detrusor pressure, post-void residual |
Temporal theory of DBD: A potential unifying mechanism of the pathogenesis of DBD
In examination of natural history of DBD, we have observed that morphological and functional manifestations of DBD in STZ-induced DM are time-dependent. Bladder hypertrophy and remodeling, increased contractility and associated neurogenic changes occur soon after the onset of DM 9,10,11, while the drop of peak voiding pressure in the cystometric measure, develop only at a later stage of DM 12,13. The time-dependent alterations of DBD served as the basis for our ‘temporal hypothesis’ of DBD with mixed clinical manifestations, in which we propose that DM causes the bladder to undergo two phases of alterations via two main mechanisms (Table 2): In the early phase, hyperglycemia-induced osmotic polyuria is the main mechanistic factor that causes compensatory bladder hypertrophy and associated myogenic and neurogenic alterations. In the later phase, accumulation of oxidative stress products during prolonged hyperglycemia causes decompensation of the bladder tissues and function. This temporal hypothesis of the pathophysiology of DBD provides a potentially unifying theory under which the complex interaction between seemingly confusing bladder dysfunction can be explained. Further, it provides a scientific road map under which the timing and specific roles of various components such as detrusor, urothelium, autonomic nerves and urethra can be explored.
Table 2.
Proposed natural history of progression of diabetic bladder dysfunction

|
PATHOGENESIS
Pathogenesis of DBD could be related to hyperglycemia-induced polyuria and oxidative stress as discussed below:
Polyuria and the early phase of DBD
Unlike most other organs affected by DM, the bladder faces not only hyperglycemia, but also an exceptionally high volume of urine output. In experimental models, sucrose-induced diuresis causes rapid and substantial bladder hypertrophy and increased bladder contractility, capacity and compliance that are similar to those changes observed in diabetic rats 10,14. Those similarities suggest that bladder hypertrophy in diabetic animals may result from a physical adaptation to increased urine production. On the other hand, bladder hypertrophy may also initiate the process of increased oxidative stress 15. Further separation of role of hyper-osmol polyuria from a normal-osmol polyuria in the mediation of bladder remodeling requires future studies in which separation of role of osmolality vs. increased flow or stretch on sensory elements of the urothelium should be explored.
Prolonged hyperglycemia, oxidative stress and late phase DBD
Accumulation of oxidative stress products within most types of cell is a prominent feature of prolonged hyperglycemia 16. Oxidative stress in diabetes could originate from a variety of mechanisms, which include: oxygen radical production from auto-oxidations of glucose, glycated proteins, stimulations of cytochrome P450–like activity, alterations of NADPH/NADP ratio by excess glucose going through the polyol pathway, increased production of super oxide dismutase and increased production of lipid peroxidation 16,17,18. Dr. Brownlee has promoted a unifying mechanism that links together all of the seemingly unconnected hyperglycemia-induced pathways, all of which stem from increased mitochondrial production of reactive oxygen species (ROS), primarily superoxide 18. The excess ROS cause, in turn, DNA strand breaks, activation of poly (ADP-ribose) polymerase (PARP) and inhibition of glyceraldehyde-3 phosphate dehydrogenase (GAPDH), culminating in activation of the four damaging pathways 18.
While there are numerous studies on the role of oxidative stress on the pathogenesis of diabetic complications in the eye, nervous system, kidney and cardiovascular system, studies on the direct effect of oxidative stress in the urologic complications has not yet been investigated in detail. A few studies on its role in erectile dysfunction 11,19 and on cystopathy 20,21 have indicated the importance of oxidative stress in the pathogenesis of urologic diabetic complications. Using a rabbit model of alloxan-induced diabetes, we showed that the decrease in the contractility of the detrusor smooth muscle (DSM) is associated with increased lipid peroxidation products (Figure 1) and overexpression of aldose reductase 21. The increased expressions of aldose reductase favor the cycling of elevated glucose through the polyol pathway and produced increased levels of sorbitol 21. We also have evidence that the exposure of human bladder smooth muscle cells to high glucose also increases the expression of aldose reductase in these cells (unpublished data). Exposure of these cells grown in high glucose to the aldose reductase inhibitor Zopolrestat reverses the overexpression of aldose reductase, which supports the conclusion from our studies on intact muscle from diabetic rabbits that the overexpression of aldose reductase and its over-activity might contribute to the rise in redox and lipid peroxidation (unpublished data).
Figure 1. Increase in Lipid Peroxidation.
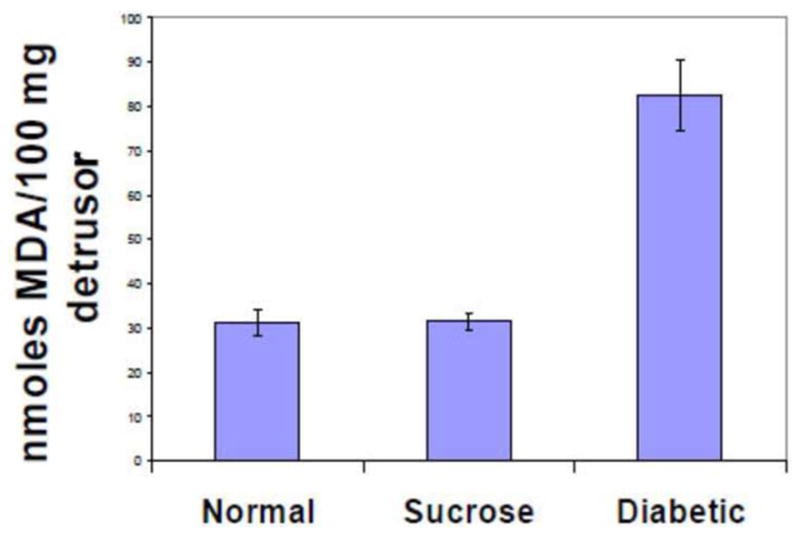
Products in Detrusor MDA levels are given for detrusor from normal (N=11), sucrose fed controls (N=13) and diabetic rabbits (N=5). MDA levels were significantly higher in detrusor muscle from diabetic rabbits compared to normal and sucrose feed controls. Data are given as mean±S.E. *, p<0.05 compared to normal sucrose.
Mitochondria are the major source of superoxide, peroxynitrite and hydroxyl radicals in all types of cells 22. Our preliminary data show that treatment of high glucose increases the mitochondrial membrane potential and ROS in cultured human bladder smooth muscle cells (hBSMC) (Figure 2), in agreement with published reports showing that the mitochondrial dysfunction is a key mechanistic step in diabetes complications 23. Mitochondrial dysfunction reduces the production of ATP, affecting the ability of cross-bridges to cycle during force generation.
Figure 2. Mitochondrial ROS production.
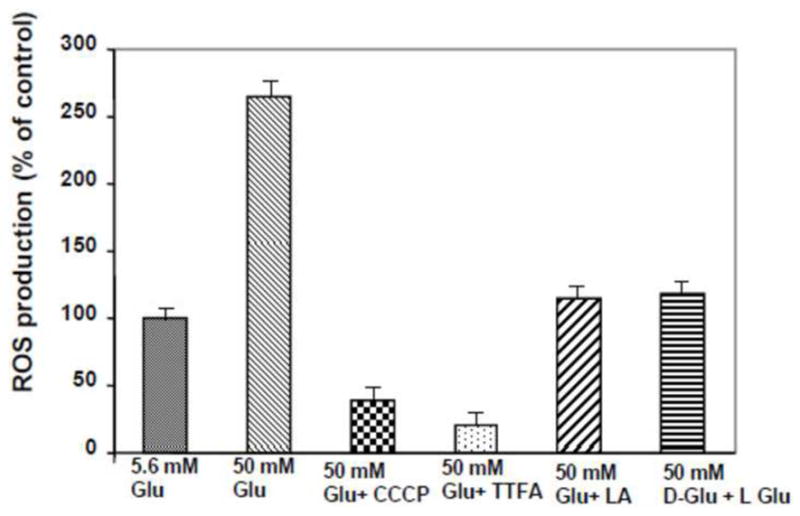
Incubation of human BSM cells with high glucose increases ROS generation and is prevented by the antioxidants alpha lipoic acid. Human BSM cells were grown with low (6mM) and high (50 mM) glucose plus 10 uM TTFA, 1uM CCCP, 200 uM of alpha lipoic acid and 50 mM of L-glucose for 48 hrs. The mitochondrial fractions were assayed for the ROS production. The ROS concentration was determined from a standard curve of H2O2 (95-100 umol/l) and was expressed as a percentage of ROS incubated at 6 mmol/l glucose.
Endogenous antioxidants are able to destroy the ROS and create a balance between antioxidant and free radicals in a normal situation; however, in diabetes the antioxidant defense system is deficient due to the high level of oxidative stress. Intake of antioxidants, such as vitamin E 24 and α-lipoic acid 25, which functions as a cofactor in multi-enzyme complexes, have been successfully used to reverse the oxidative stress produced by hyperglycemia in human diabetics and in STZ-induced diabetic animal models. Oral treatment of α-lipoic acid (600 mg per day orally for five weeks) improved neuropathic deficits in diabetic patients with distal symmetric polyneuropathy in a recent clinical trial 25. It has not been reported whether urinary bladder function improved in these patients. In our preliminary studies on the effects of antioxidants on oxidative-stress induced by high glucose in cultured hBSMC, we are able to decrease the production of lipid peroxidation (Figure 3).
Figure 3. Lipid peroxidation induced by high glucose.
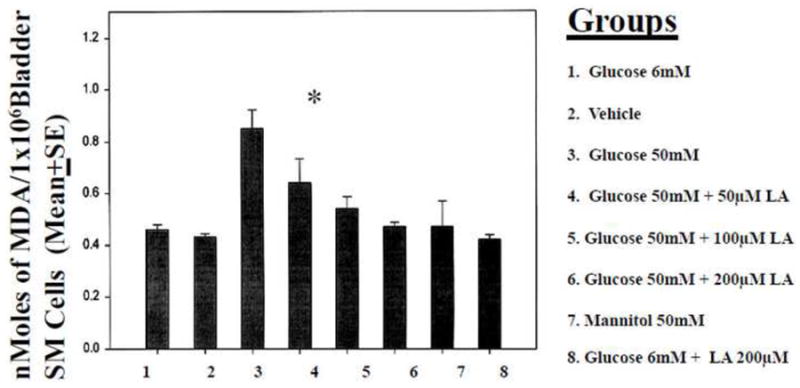
Increase in lipid peroxidation products in BSM cells treated with high glucose (50 mM). Mannitol 50 mM treated group served as control for osmotic shock. High glucose (50 mM) induced lipid peroxidation, was inhibited by lipoic acid treatment (4&5). Normal (6 mM) glucose treatment and mannitol treatment did not induce any changes in lipid peroxidation. *p<0.05 compared to Glucose 6 mM.
PATHOPHYSIOLOGY
Pathophysiology of DBD is multifactorial as it involves alterations in detrusor, nerve, urothelium and urethra
The traditional views recognized autonomic neuropathy as the sole pathophysiological cause of DBD 26. That view would consider decreased sensation of the bladder as the primary event, with patients being unaware of bladder filling and lacking desire to empty, is presumed to result from autonomic neuropathy, and it results in high post void residual and overflow incontinence. Details of how autonomic neuropathy, or loss of sensation leads to the mixed clinical manifestations of DBD are unknown. Evolution of that view is represented by the view held by most contemporary investigators who agree that the pathophysiology of DBD is multifactorial, including disturbances of the bladder detrusor, urethra, autonomic nerves, and perhaps the urothelium 27. We and others have observed that, upon induction of DM in rodents by destruction of the pancreatic β-cells with STZ, the bladder and urethra undergo morphometric and functional changes in both myogenic and neurogenic components 9-13,28-30. Other study has demonstrated the potentially obstructive effects of urethral sphincteric mechanisms in DBD 31.
Myogenic changes in diabetes
In vivo and in vitro experimental studies on DSM from animal models of DM do give evidence for myogenic changes. Earlier studies on the effects of diabetes on detrusor contractility show both decreased 32 and increased 33 force production in rat DSM strips. We investigated the effects of a long-term diabetic state on DSM contractility and associated oxidative stress changes 21. Contractility of the DSM was decreased in response to stimulation by KCl and carbachol, and the decrease was associated with not only the duration of the hyperglycemic state but also the level of hyperglycemia (Figure 4). Changes in muscarinic receptor population have also been linked to altered contractility 34. Unlike the changes in the DSM from an obstructed bladder, we found, both using STZ-induced rat diabetic model and alloxan-induced rabbit model, that there is no change in the myosin isoform composition in the DSM from diabetic animals 35. Recent physiologic and biochemical studies on the DSM from our group 36,37, as well as others 13,38 show a distinct deficit in the regulation of contraction in the DSM in diabetes.
Figure 4. Force generation by detrusor muscle strips.
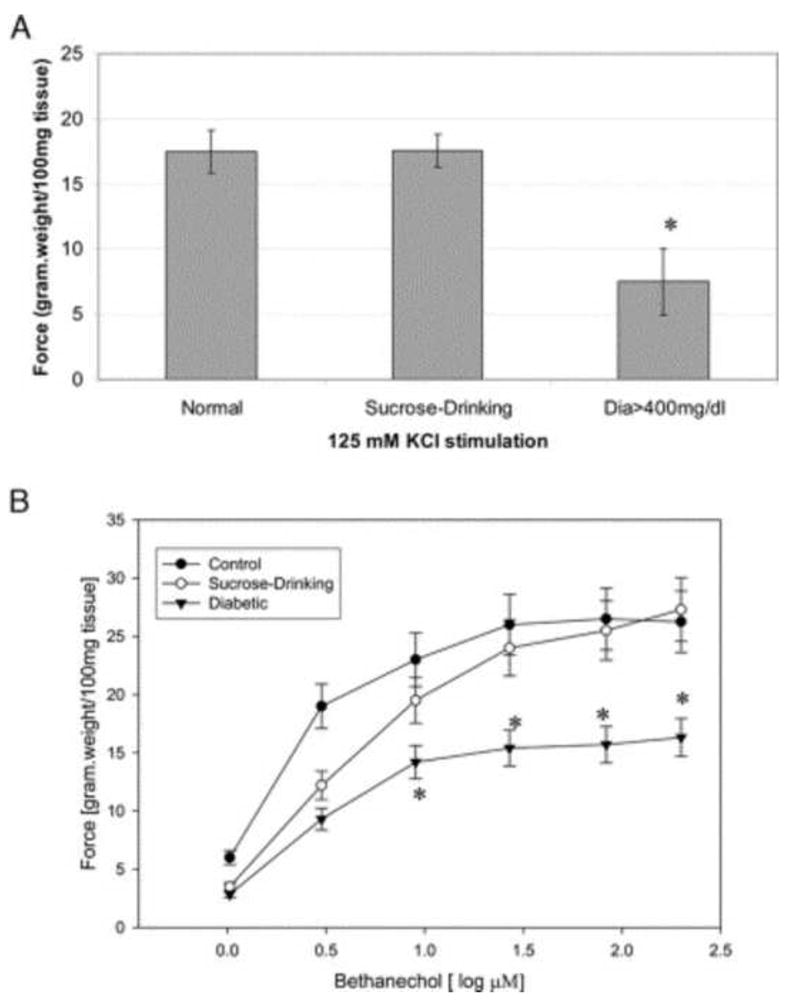
A, effects of 125 mM KCl on force generation by detrusor muscle from normal, sucrose drinking and diabetic (Dia>400mg/dl) rabbits. Force was significantly decreased for diabetes. Data are shown as mean ± SEM. Asterisk indicates p <0.05. B, bethanechol dose response curve of force generation by detrusor muscle from normal, sucrose drinking and diabetic rabbits. Force was significantly decreased for diabetes. Data are shown as mean ± SEM. Asterisk indicates p <0.05. (Reproduced from Changolkar et al., 2005 with permission.)
A major regulatory mechanism for smooth muscle contraction is the myosin-mediated regulation via phosphorylation-dephosphorylation of the regulatory myosin light chain (MLC20) by Ca2+-dependent myosin light chain kinase (MLCK) and the myosin light chain phosphatase (MLCP). The MLCP is inactivated by phosphorylation, catalyzed mainly by Rho-kinase, and also by binding to phosphorylated CPI-17. By lowering the activity of MLCP, these proteins retain the myosin in the phosphorylated state and maintain muscle tone in the absence of an elevation of cytosolic Ca2+. Studies performed on DSM from diabetic animals showed overexpression and overactivity of Rho-kinase and CPI-17 proteins involved in the Ca 2+-sensitization in smooth muscle (Figures 5 & 6). Interestingly, we also found high level of basal MLC20 phosphorylation in the diabetic detrusor (Figure 7). However, the molecular mechanisms for the diabetes-induced alteration in the expression of these proteins that regulate the myosin-mediated regulation of DSM contraction are not known.
Figure 5. Expression of Rho-kinase at both mRNA and protein level.
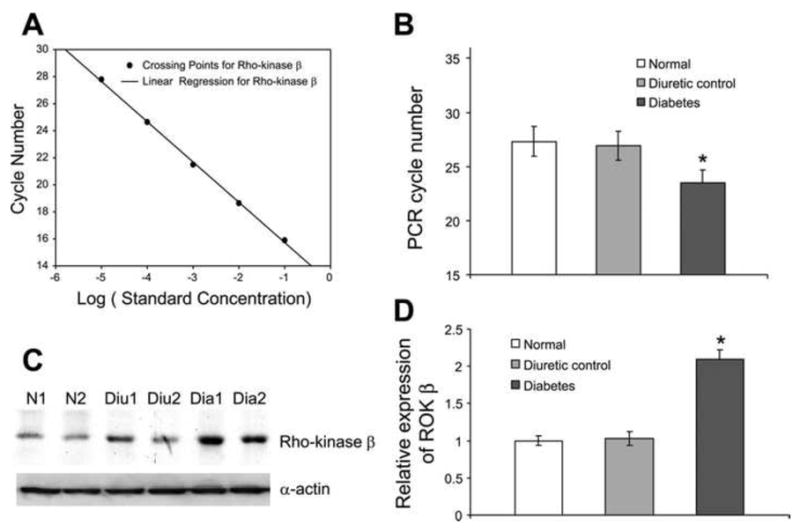
A: real-time PCR standard curve for Rho-kinase B: average of required PCR cycle numbers to reach crossing threshold. The required PCR cycle numbers was 27.3 for normal samples, 26.9 for diuretic controls, and 23.5 for diabetic samples. A significantly lower number of PCR cycles for the diabetic sample indicated that diabetic DSM sample had more copies of Rho-kinase transcript. C: Western blot for Rho-kinase and smooth muscle -actin. D: bar graph showing the average of relative protein expression level. There was almost a 2.1-fold higher protein expression of Rho-kinase in diabetic detrusor compared with normal and diuretic samples. *Significant difference between samples (n = 4, P < 0.01). (Reproduced with permission from Chang et al, 2006.).
Figure 6. Expression of CPI-17 at both mRNA and protein level.
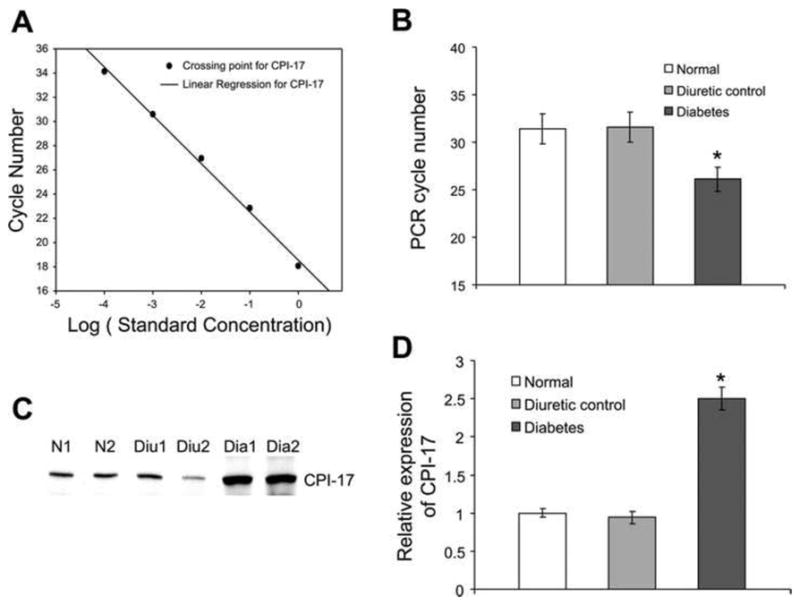
A: standard curve of real-time PCR for CPI-17. B: average of the PCR results. The required PCR cycle numbers was significantly decreased in diabetic DSM samples compared with normal or diuretic controls. It was 31.4 cycles for normal sample, 31.6 cycles for diuretic controls, and 26.1 cycles for diabetic samples. C: Western blot for CPI-17. D: bar graph showing the relative expression of CPI-17 at the protein level. There was almost a 2.5-fold higher expression of CPI-17 at the protein level in diabetic detrusor compared with normal and diuretic samples. *Significant difference between samples (n = 4, P < 0.01). (Reproduced with permission from Chang et al, 2006.).
Figure 7. Selected areas from 2-dimensional (2D) gel showing basal MLC20 phosphorylation in normal, diuretic, and diabetic detrusor smooth muscle (DSM).
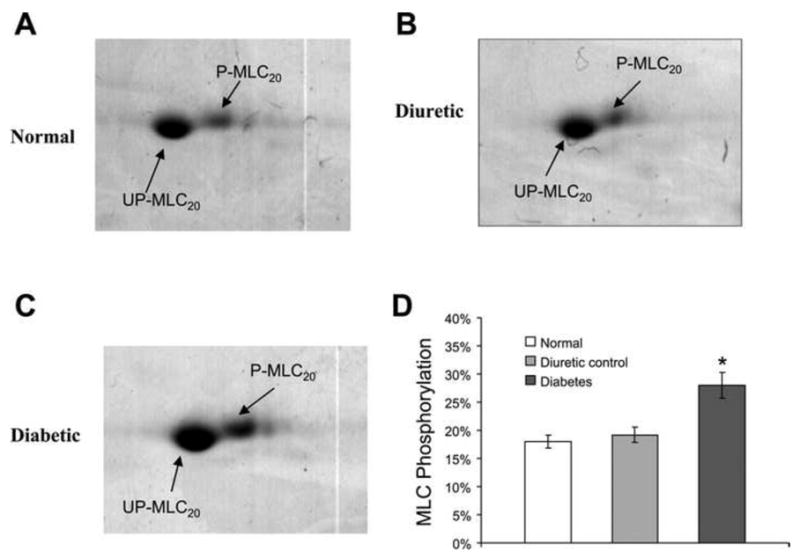
A: normal. B: diuretic. C: diabetic. D: bar graph showing the average values of myosin light chain (MLC) phosphorylation. Phosphorylated MLC20 (P-MLC20) runs slightly higher and more toward the acidic side than unphosphorylated MLC20 (UP-MLC20) in the gel. The basal phosphorylation of MLC20 was 18% in normal DSM. Diuretic control had a very similar level (19.2%) of MLC20 phosphorylation. However, the phosphorylation level was significantly increased to 28% in diabetic detrusor. *Significant difference between samples (n = 3, P < 0.05). (Reproduced with permission from Chang et al, 2006.).
Urothelial changes in diabetes
An important, but not well understood, function of epithelial cells is their ability to sense changes in their extracellular environment and then communicate these changes to the underlying nervous, connective and muscular tissues 39. This communication is likely to be important for tube- and sac-shaped organs such as blood vessels, the gut, and the bladder, whose normal function can be modulated by stimuli initiated within the epithelium. Though alterations in smooth muscle and nerve innervation have been shown in diabetic patients 4, there is little information regarding the involvement of the urothelium in the pathophysiology of DBD.
The small number of studies on the effects of DM on bladder urothelium, using the STZ-induced diabetic rat model, report an increase in urothelium proliferation 40,41 without an increase in the thickness of the urothelial lining itself 41. This increase in proliferation may divert the physiology of the urothelium cells from their normal inter-communication/two-way communication with the underlying bladder tissue, by modifying both urothelial cell receptor expression and the release of signaling molecules such as neurotransmitters. This in turn could impact/modify activity in underlying smooth muscle and nerve endings and contribute to the bladder function modification observed in DM. It has been reported that urothelial cell prostaglandin release is impaired, in STZ-DM rats 40. This might affect the barrier function of the urothelium. Prostaglandins are known to play an important role in the maintenance of musical integrity in the gut 42. In addition it has been proposed that the common occurrence of urinary tract infections seen in DM and attributed in part to the bladder stasis seen in the pathology, may be as a result of altered expression of adherence receptors for bacteria by urothelial cells 43.
Abnormalities in bladder urothelium could impact the LUT function by altering release of mediators as well as excitability of sensory fibers in the bladder. In addition, because many of these urothelial functions may be altered in diabetes, defects in urothelial cells may underlie, in part, changes such as detrusor instability and/or changes in bladder capacity. Thus, the urothelium is an active participant in the normal function of the bladder and exists as an integral part of a ‘sensory web’, in which it communicates the degree of bladder filling to the underlying nervous and muscular tissues and affects their functions. This communication is made possible by the input and output pathways of the urothelium, which allow it to respond to its chemical and physical environment and to engage in multi-directional communication with neighboring cells in subadjacent tissues. Defects in the urothelial expression of receptors or aberrant release of mediators may contribute to diabetes-associated bladder complications.
Neuronal changes in diabetes
The neuronal control of bladder function involves a sophisticated and complex interaction between the autonomic and somatic afferent and efferent pathways. One group reported an association of DBD with autonomic neuropathy detected by the sympathetic skin response in diabetic patients 44. Steers et al showed significant abnormalities in afferent pathways innervating the bladder in STZ induced diabetic rats 45. Adult rats treated with capsaicin, a C-fiber afferent neurotoxin, exhibit a number of similarities to diabetic rats 46. Since capsaicin is known to affect predominately small myelinated and unmyelinated afferents, it is temping to speculate that DM affects a similar afferent neuron population. On the other hand, it has also been suggested that DBD is initiated by neuropathy in the efferent limb of the micturition reflex 47.
Neurotrophic factors derived from target tissues can support the growth and survival of peripheral neurons. Rats with STZ-induced DM, 12 weeks after induction, showed significantly decreased levels of nerve growth factor (NGF), a member of the neurotrophin family, in the bladder and in L6 to S1 dorsal root ganglia, which contain bladder afferent neurons 48. Reports that diabetic rodents show loss of neurotrophic support to peripheral nerves have prompted studies to investigate the efficacy of neurotrophic factor supplementation on nerve disorders of diabetic rats. Unfortunately, NGF was not shown to be effective in a recently completed phase III trial 49. In addition, the use of exogenous neurotrophic factors as a therapy is limited by the need for non-oral delivery, the fiber selectivity of individual neurotrophins, limited delivery to the nervous system and concerns about harmful systemic actions of growth factors. The alternative approaches warrant further investigation.
CONCLUSIONS
The review of current literature indicates the following state of knowledge on DBD:
Temporal manifestation of DBD may explain the spectrum of bladder dysfunction seen in patients affected by type I or II DM.
Early phase of DBD is manifested by storage problems; such as urgency, urge incontinence manifested at the clinical level, whereas hypercontractility of the detrusor muscle, and neuronal changes are manifested at the experimental level. Early evidence indicates that polyuria plays a major role in pathogenesis of changes seen in the early phase of DBD.
The late phase of DBD is manifested by voiding problems leading to inability of the bladder to empty causing high post-void residual and overflow incontinence, at the clinical level and decompesated detrusor function at the experimental level. Accumulating evidence indicate a role for oxidative stress in pathogenesis of changes seen in the late phase of DBD.
Thus, it appears that the future investigative work should address the following issues:
Studies of time course of alterations in the function of urothelium, including impact of urine hyperosmolarity on urothelial remodeling.
Studies of afferent sensitivity during the early phase of DBD; and whether the observed experimental alterations translate to presence of overactive bladder manifested at the clinical level.
Mechanistic studies related to role of oxidative stress in DBD. Availability of genetically manipulated animals may facilitate such mechanistic studies.
Studies of time course of DBD in type II animal models, and whether type I and II mimic each other in DBD.
Key of Definitions for Abbreviations
- DBD
diabetic bladder dysfunction
- DM
diabetes mellitus
- DSM
detrusor smooth muscle
- LUT
lower urinary tract
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- NGF
nerve growth factor
- ROS
reactive oxygen species
- STZ
streptozotocin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Centers for Disease Control and Prevention. National diabetes fact sheet: general information and national estimates on diabetes in the United States. 2005 [Google Scholar]
- 2.Bladder Research Progress Review Group. Urologic complications of diabetes mellitus in overcoming bladder disease: a strategic plan for research. A report of the NIH-NIDDK bladder research progress review group. 2004 [Google Scholar]
- 3.Bladder Research Progress Review Group. Urologic complications of diabetes mellitus. 2002;133 [Google Scholar]
- 4.Daneshgari F, Moore C. Diabetic uropathy. Semin Nephrol. 2006;26:182. doi: 10.1016/j.semnephrol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Zderic SA, Chacko S, Disanto ME, Wein AJ. Voiding function: relevant anatomy, physiology, pharmacology, and molecular aspects. 2002 [Google Scholar]
- 6.Ueda T, Tamaki M, Kageyama S, Yoshimura N, Yoshida O. Urinary incontinence among community-dwelling people aged 40 years or older in Japan: prevalence, risk factors, knowledge and self-perception. Int J Urol. 2000;7:95. doi: 10.1046/j.1442-2042.2000.00147.x. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan SA, Te AE, Blaivas JG. Urodynamic findings in patients with diabetic cystopathy. J Urol. 1995;153:342. doi: 10.1097/00005392-199502000-00013. [DOI] [PubMed] [Google Scholar]
- 8.Brown JS, Nyberg LM, Kusek JW, Burgio KL, Diokno AC, Foldspang A, et al. Am J Obstet Gynecol; Proceedings of the National Institute of Diabetes and Digestive and Kidney Diseases International Symposium on Epidemiologic Issues in Urinary Incontinence in Women; 2003. p. S77. [DOI] [PubMed] [Google Scholar]
- 9.Liu G, Daneshgari F. Alterations in neurogenically mediated contractile responses of urinary bladder in rats with diabetes. Am J Physiol Renal Physiol. 2005;288:F1220. doi: 10.1152/ajprenal.00449.2004. [DOI] [PubMed] [Google Scholar]
- 10.Liu G, Daneshgari F. Temporal diabetes- and diuresis-induced remodeling of the urinary bladder in the rat. Am J Physiol Regul Integr Comp Physiol. 2006;291:R837. doi: 10.1152/ajpregu.00917.2005. [DOI] [PubMed] [Google Scholar]
- 11.Christ GJ, Hsieh Y, Zhao W, Schenk G, Venkateswarlu K, Wang HZ, et al. Effects of streptozotocin-induced diabetes on bladder and erectile (dys)function in the same rat in vivo. BJU Int. 2006;97:1076. doi: 10.1111/j.1464-410X.2006.06058.x. [DOI] [PubMed] [Google Scholar]
- 12.Daneshgari F, Huang X, Liu G, Bena J, Saffore L, Powell CT. Temporal differences in bladder dysfunction caused by diabetes, diuresis, and treated diabetes in mice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1728. doi: 10.1152/ajpregu.00654.2005. [DOI] [PubMed] [Google Scholar]
- 13.Daneshgari F, Liu G, Imrey PB. Time dependent changes in diabetic cystopathy in rats include compensated and decompensated bladder function. J Urol. 2006;176:380. doi: 10.1016/S0022-5347(06)00582-9. [DOI] [PubMed] [Google Scholar]
- 14.Longhurst PA. In vivo urinary bladder function in rats following prolonged diabetic and non-diabetic diuresis. Neurourol Urodyn. 1990;9:171. [Google Scholar]
- 15.Satriano J. Kidney growth, hypertrophy and the unifying mechanism of diabetic complications. Amino Acids. 2007;33:331. doi: 10.1007/s00726-007-0529-9. [DOI] [PubMed] [Google Scholar]
- 16.Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol. 2006;212:167. doi: 10.1016/j.taap.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 18.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 19.Bivalacqua TJ, Champion HC, Usta MF, Cellek S, Chitaley K, Webb RC, et al. RhoA/Rho-kinase suppresses endothelial nitric oxide synthase in the penis: a mechanism for diabetes-associated erectile dysfunction. Proc Natl Acad Sci U S A. 2004;101:9121. doi: 10.1073/pnas.0400520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beshay E, Carrier S. Oxidative stress plays a role in diabetes-induced bladder dysfunction in a rat model. Urology. 2004;64:1062. doi: 10.1016/j.urology.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 21.Changolkar AK, Hypolite JA, Disanto M, Oates PJ, Wein AJ, Chacko S. Diabetes induced decrease in detrusor smooth muscle force is associated with oxidative stress and overactivity of aldose reductase. J Urol. 2005;173:309. doi: 10.1097/01.ju.0000141583.31183.7a. [DOI] [PubMed] [Google Scholar]
- 22.Green DR, Bossy-Wetzel E, Finucane D, Marante-Mendes G. Understudy for the executioner: mitochondria versus caspases in the commitment to death. Biophys J. 1998;74:A2. [Google Scholar]
- 23.Fernyhough P, Huang TJ, Verkhratsky A. Mechanism of mitochondrial dysfunction in diabetic sensory neuropathy. J Peripher Nerv Syst. 2003;8:227. doi: 10.1111/j.1085-9489.2003.03028.x. [DOI] [PubMed] [Google Scholar]
- 24.Al-Shamsi M, Amin A, Adeghate E. Vitamin E ameliorates some biochemical parameters in normal and diabetic rats. Ann N Y Acad Sci. 2006;1084:411. doi: 10.1196/annals.1372.033. [DOI] [PubMed] [Google Scholar]
- 25.Ziegler D, Ametov A, Barinov A, Dyck PJ, Gurieva I, Low PA, et al. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care. 2006;29:2365. doi: 10.2337/dc06-1216. [DOI] [PubMed] [Google Scholar]
- 26.Frimodt-Moller C. Diabetic cystopathy: epidemiology and related disorders. Ann Intern Med. 1980;92:318. doi: 10.7326/0003-4819-92-2-318. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimura N, Chancellor MB, Andersson KE, Christ GJ. Recent advances in understanding the biology of diabetes-associated bladder complications and novel therapy. BJU Int. 2005;95:733. doi: 10.1111/j.1464-410X.2005.05392.x. [DOI] [PubMed] [Google Scholar]
- 28.Tammela TL, Leggett RE, Levin RM, Longhurst PA. Temporal changes in micturition and bladder contractility after sucrose diuresis and streptozotocin-induced diabetes mellitus in rats. J Urol. 1995;153:2014. [PubMed] [Google Scholar]
- 29.Poladia DP, Bauer JA. Functional, structural, and neuronal alterations in urinary bladder during diabetes: investigations of a mouse model. Pharmacology. 2005;74:84. doi: 10.1159/000083962. [DOI] [PubMed] [Google Scholar]
- 30.Liu G, Lin Y, Yamada Y, Daneshgari F. External urethral sphincter activity in diabetic rats. Neurourol Urodynam. 2008;27:429. doi: 10.1002/nau.20543. [DOI] [PubMed] [Google Scholar]
- 31.Torimoto K, Fraser MO, Hirao Y, De Groat WC, Chancellor MB, Yoshimura N. Urethral dysfunction in diabetic rats. J Urol. 2004;171:1959. doi: 10.1097/01.ju.0000121283.92963.05. [DOI] [PubMed] [Google Scholar]
- 32.Longhurst PA, Belis JA. Abnormalities of rat bladder contractility in streptozotocin-induced diabetes mellitus. J Pharmacol Exp Ther. 1986;238:773. [PubMed] [Google Scholar]
- 33.Waring JV, Wendt IR. Effects of streptozotocin-induced diabetes mellitus on intracellular calcium and contraction of longitudinal smooth muscle from rat urinary bladder. J Urol. 2000;163:323. [PubMed] [Google Scholar]
- 34.Mutoh S, Latifpour J, Saito M, Weiss RM. Evidence for the presence of regional differences in the subtype specificity of muscarinic receptors in rabbit lower urinary tract. J Urol. 1997;157:717. [PubMed] [Google Scholar]
- 35.DiSanto ME, Stein R, Chang S, Hypolite JA, Zheng Y, Zderic S, et al. Alteration in expression of myosin isoforms in detrusor smooth muscle following bladder outlet obstruction. Am J Physiol Cell Physiol. 2003;285:C1397. doi: 10.1152/ajpcell.00513.2002. [DOI] [PubMed] [Google Scholar]
- 36.Mannikarottu AS, Changolkar AK, Disanto ME, Wein AJ, Chacko S. Over expression of smooth muscle thin filament associated proteins in the bladder wall of diabetics. J Urol. 2005;174:360. doi: 10.1097/01.ju.0000161602.18671.c7. [DOI] [PubMed] [Google Scholar]
- 37.Su X, Changolkar A, Chacko S, Moreland RS. Diabetes decreases rabbit bladder smooth muscle contraction while increasing levels of myosin light chain phosphorylation. Am J Physiol Renal Physiol. 2004;287:F690. doi: 10.1152/ajprenal.00027.2004. [DOI] [PubMed] [Google Scholar]
- 38.Gupta S, Yang S, Cohen RA, Krane RJ, Saenz De Tejada I. Altered contractility of urinary bladder in diabetic rabbits: relationship to reduced Na+ pump activity. Am J Physiol. 1996;271:C2045. doi: 10.1152/ajpcell.1996.271.6.C2045. [DOI] [PubMed] [Google Scholar]
- 39.Birder LA, de Groat WC. Mechanisms of disease: involvement of the urothelium in bladder dysfunction. Nat Clin Pract Urol. 2007;4:46. doi: 10.1038/ncpuro0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinna C, Zanardo R, Puglisi L. Prostaglandin-release impairment in the bladder epithelium of streptozotocin-induced diabetic rats. Eur J Pharmacol. 2000;388:267. doi: 10.1016/s0014-2999(99)00833-x. [DOI] [PubMed] [Google Scholar]
- 41.Pitre DA, Ma T, Wallace LJ, Bauer JA. Time-dependent urinary bladder remodeling in the streptozotocin-induced diabetic rat model. Acta Diabetol. 2002;39:23. doi: 10.1007/s005920200008. [DOI] [PubMed] [Google Scholar]
- 42.Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology. 2008;135:41. doi: 10.1053/j.gastro.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 43.Geerlings SE, Meiland R, van Lith EC, Brouwer EC, Gaastra W, Hoepelman AI. Adherence of type 1-fimbriated Escherichia coli to uroepithelial cells: more in diabetic women than in control subjects. Diabetes Care. 2002;25:1405. doi: 10.2337/diacare.25.8.1405. [DOI] [PubMed] [Google Scholar]
- 44.Ueda T, Yoshimura N, Yoshida O. Diabetic cystopathy: relationship to autonomic neuropathy detected by sympathetic skin response. J Urol. 1997;157:580. doi: 10.1016/s0022-5347(01)65209-1. [DOI] [PubMed] [Google Scholar]
- 45.Steers WD, Mackway-Gerardi AM, Ciambotti J, de Groat WC. Alterations in neural pathways to the urinary bladder of the rat in response to streptozotocin-induced diabetes. J Auton Nerv Syst. 1994;47:83. doi: 10.1016/0165-1838(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 46.Cheng CL, Ma CP, de Groat WC. Effects of capsaicin on micturition and associated reflexes in rats. Am J Physiol. 1993;265:R132. doi: 10.1152/ajpregu.1993.265.1.R132. [DOI] [PubMed] [Google Scholar]
- 47.Paro M, Prashar A, Prosdocimi M, Cherian PV, Fiori MG, Sima AA. Urinary bladder dysfunction in the BB/W diabetic rat: effect of ganglioside treatment on functional and structural alterations. J Urol. 1994;151:781. doi: 10.1016/s0022-5347(17)35087-5. [DOI] [PubMed] [Google Scholar]
- 48.Sasaki K, Chancellor MB, Phelan MW, Yokoyama T, Fraser MO, Seki S, et al. Diabetic cystopathy correlates with a long-term decrease in nerve growth factor levels in the bladder and lumbosacral dorsal root Ganglia. J Urol. 2002;168:1259. doi: 10.1016/S0022-5347(05)64636-8. [DOI] [PubMed] [Google Scholar]
- 49.Apfel SC, Schwartz S, Adornato BT, Freeman R, Biton V, Rendell M, et al. Efficacy and safety of recombinant human nerve growth factor in patients with diabetic polyneuropathy: A randomized controlled trial. rhNGF Clinical Investigator Group. JAMA. 2000;284:2215. doi: 10.1001/jama.284.17.2215. [DOI] [PubMed] [Google Scholar]