

Abstract
Escherichia coli SecA uses ATP to drive the transport of proteins across cell membranes. Glutamate 210 in the “DEVD” Walker B motif of the SecA ATP-binding site has been proposed as the catalytic base for ATP hydrolysis (Hunt, J. F., Weinkauf, S., Henry, L., Fak, J. J., McNicholas, P., Oliver, D. B., and Deisenhofer, J. (2002) Science 297, 2018–2026). Consistent with this hypothesis, we find that mutation of glutamate 210 to aspartate results in a 90-fold reduction of the ATP hydrolysis rate compared with wild type SecA, 0.3 s−1 versus 27 s−1, respectively. SecA-E210D also releases ADP at a slower rate compared with wild type SecA, suggesting that in addition to serving as the catalytic base, glutamate 210 might aid turnover as well. Our results contradict an earlier report that proposed aspartate 133 as the catalytic base (Sato, K., Mori, H., Yoshida, M., and Mizushima, S. (1996) J. Biol. Chem. 271, 17439–17444). Re-evaluation of the SecA-D133N mutant used in that study confirms its loss of ATPase and membrane translocation activities, but surprisingly, the analogous SecA-D133A mutant retains full activity, revealing that this residue does not play a key role in catalysis.
SecA is an essential component of the protein translocation machinery in prokaryotes whose function is to transport pre-proteins across the cytoplasmic membrane. According to current model mechanisms for protein translocation, SecA takes up a precursor of secretory protein from the chaperone SecB, or from the cytosol, and inserts it through a membrane translocon formed by SecYEG and SecDFyajC proteins via repeated cycles of membrane insertion and retraction associated with pre-protein binding and release. SecA possesses an ATPase activity that is essential for protein transport (see reviews Refs. 1–3). The coupling between SecA-catalyzed ATP binding, hydrolysis, and product dissociation events, and the translocon and pre-protein binding and release events underlies the mechanics of the protein transport process.
SecA has a high-affinity ATP-binding site that comprises Walker A and B motifs responsible for coordinating the α- and β-phosphates of the nucleotide and the Mg2+ ion, respectively (Fig. 1) (4). There also appears to be a low-affinity nucleotide-binding site on SecA, although it is not known whether this site is catalytically active and how it might contribute to the protein translocation mechanism (5–10). The high-affinity nucleotide-binding site is better defined, but even its exact role in protein translocation is not completely clear (5, 11–15). SecA has a low endogenous ATPase activity that is stimulated 5–10-fold by the presence of SecYEG translocon/membrane and pre-protein, highlighting the link between the ATPase and protein translocation activities (16–18). Studies probing this link have shown that binding of AMP-PNP1 (a non-hydrolyzable ATP analog) to the high-affinity site stabilizes the interaction between SecA and SecYEG/membrane (11, 14, 19–22). One interpretation of this result is that ATP binding to SecA triggers its insertion into the translocon. However, because the interaction of AMP-PNP with SecA is ~1000-fold weaker than that of ATP, it is not entirely clear whether this nucleotide analog can accurately mimic the effects of ATP binding to SecA (5). Thus, the question of what role ATP binding to SecA plays in the protein translocation mechanism remains open. Another notable finding is that ADP binding to the high-affinity site stabilizes a compact conformation of SecA (ground state) that has low affinity for the SecYEG/membrane (5, 8, 12, 23). This result suggests that following ATP hydrolysis, the ADP-bound SecA undergoes retraction from the translocon to complete one reaction cycle. However, the apo (nucleotide free) form of SecA can also exist in a compact conformation with low affinity for the translocon (5, 24). Moreover, ADP release from SecA is stimulated by SecYEG/membrane, raising the question whether SecA retraction from the membrane occurs in the ADP-bound form, the apo form, or both (16).
FIG. 1. SecA high-affinity nucleotide binding site.
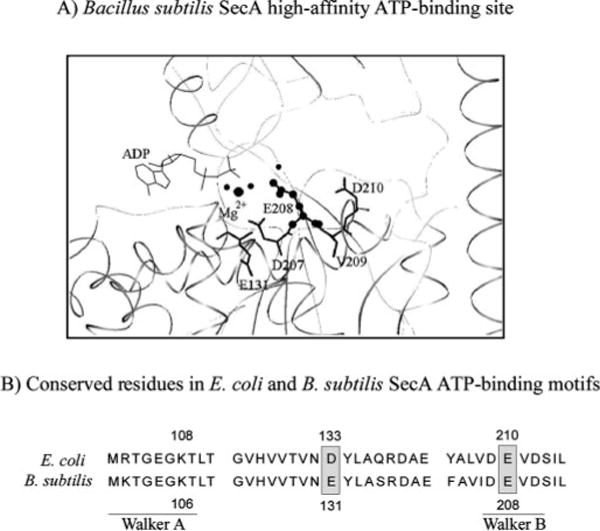
A, crystal structure of B. subtilis SecA showing ADP bound to the high-affinity site, as well as amino acids making up the Walker B motif. B, conserved amino acids in the Walker A and B ATP-binding motifs of E. coli and B. subtilis SecA, including glutamate 210 (E208 in B. subtilis SecA). Also noted is glutamate 131 (D133 in E. coli SecA), proposed previously to be the catalytic base for SecA ATPase activity.
One approach to resolving questions regarding the timing and stoichiometry of ATP binding and hydrolysis events as well as membrane insertion and retraction events, and how they relate to each other during protein translocation, is to directly measure the kinetic parameters that define the progress of the two reactions and the linkage between them (25). Here we report the kinetics of ATP binding, hydrolysis, and product release catalyzed by SecA, measured by pre-steady-state experiments. The results reveal that the SecA dimer rapidly binds and hydrolyzes two ATP molecules in one catalytic turnover (very likely at the high-affinity site), and that the slow step(s) limiting the steady-state turnover rate occur after phosphate release, either before or at ADP release from SecA.
Another objective of this study was to identify the catalytic base in the ATPase site that activates a water molecule for attack on the γ phosphate during ATP hydrolysis. Mutation of this residue might allow SecA to bind ATP with high affinity, but not catalyze hydrolysis. Such a mutant could be of value for characterizing the ATP-bound conformation of SecA, which is proposed to insert into the membrane. In a previous report, aspartate 133 (Asp-133) was proposed as the catalytic base, based on the finding that mutation of Asp-133 to asparagine drastically reduced SecA ATPase and protein translocation activity (26). However, in the crystal structure of Bacillus subtilis SecA, a homolog of Escherichia coli SecA, this residue does not appear in the correct position to serve a catalytic function (Fig. 1; Glu-131 in B. subtilis SecA); in fact, the structure indicates that glutamate 210 (Glu-210) is a strong candidate for this function (Glu-208 in B. subtilis SecA) (12). We tested the ATPase activities of several glutamate 210 mutants under pre-steady-state conditions and found that SecA-E210N and SecA-E210D are capable of binding ATP with similar affinity as wild type SecA, but their ability to catalyze ATP hydrolysis is severely disrupted, consistent with the hypothesis that Glu-210 is the catalytic base.
MATERIALS AND METHODS
Proteins and Other Reagents
Wild type SecA (pT7SecA2) was overproduced and purified from E. coli strain BL21.19 as described earlier (6, 27), with the following modifications: an SP-Sepharose column was used instead of Mono-S (protein eluted with 300 mM NaCl in 50 mM Tris-HCl, pH 7.5, 2 mM dithiothreitol) followed by a hexyl-agarose column (protein eluted with 400 mM (NH4)2SO4 in 50 mM Tris-HCl, pH 7.5, 2 mM dithiothreitol). Carboxyl terminus 6 histidine-tagged SecA (His-SecA) was prepared by inserting secA into pET29b in the polycloning site between NdeI/XhoI (pET29b-T7His-SecA), followed by overexpression in BL21.19 cells (growth to 0.6 A600 at 37 °C and a 3-h induction with 0.5 mM isopropyl 1-thio-β-D-galactopyranoside), and purification on a His-Bind resin column (Novagen) according to the manufacturer’s protocol (protein eluted with 1 M imidazole in 20 mM Tris-HCl, pH 7.9, 500 mM NaCl). His-tagged SecA-E210D, SecA-E210N, SecA-E210Q, and SecA-E210A were prepared by introducing mutations into pET29b-T7His-SecA using the QuikChange mutagenesis protocol (Stratagene). The mutant proteins were purified by nickel affinity chromatography similar to His-SecA. All SecA proteins were stored in 25 mM Tris-HCl, pH 7.5, 35 mM KCl, 0.5 mM EDTA, 10% glycerol at −80 °C. Protein concentrations were determined by the Bradford assay, absorbance at 280 nm in 6 M guanidinium-HCl, 20 mM potassium phosphate, pH 6.5 (SecA monomer molar extinction coefficient = 71,950 M−1 cm−1), and by amino acid analysis; all three methods yielded values within 20% of each other. Pro-OmpA (28) and urea-treated inverted membrane vesicles (29) were prepared as described. Phosphate-binding protein (PBP) was purified and labeled with 7-diethylamino-3-((((2-maleimidyl)ethyl)amino)carbonyl)coumarin (MDCC) as described (30, 31). Radioactive nucleotides, [α-32P]ATP and [35S]ATPγS, were purchased from PerkinElmer Life Sciences, [3H]ADP was purchased Amersham Biosciences, [α-32P]ADP was prepared as described previously (32), and non-radioactive nucleotides were purchased from Sigma. Polyethyleneimine cellulose-F TLC plates were purchased from EM Science and nitrocellulose membranes from Schleicher and Schuell.
In Vitro Protein Translocation Assays
For analysis of pro-OmpA translocation, 100-μl reactions were prepared with SecA (50 μg/ml), SecB (66 μg/ml), bovine serum albumin (200 μg/ml), ATP (2 mM), NADH (5 mM), and CK1801.4 urea-treated inverted membrane vesicles (100 μg/ml) in TL buffer (50 mM Tris-HCl, pH 8.0, 50 mM KCl, 5 mM MgCl2). Reactions were initiated with 125I-pro-OmpA (5 × 105 cpm/ml) and pro-OmpA (32 μg/ml) in 50 mM Tris-HCl, pH 8.0, 6 M urea, and incubated for 20 min at 37 °C. Samples were transferred to ice and digested with Proteinase K (1 mg/ml) in the presence or absence of Triton X-100 (0.1%) for 15 min. Proteins were precipitated by addition of 75 μl of 30% trichloroacetic acid, washed with acetone, re-suspended in 50 μl of SDS-PAGE buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 100 mM dithiothreitol, 10% glycerol, 0.1% bromphenol blue), heated for 5 min at 100 °C, followed by electrophoresis on 12% gels and autoradiography.
In Vivo Complementation Assays
Complementation was assessed by streaking Bl21.19 cells (containing a temperature-sensitive amber suppressor, an amber mutation in chromosomal secA and a plasmid-borne copy of wild type or mutant secA) on media containing 100 μg/ml ampicillin, followed by incubation at permissive (30 °C) or non-permissive temperatures (42 °C) for 16–24 h, and comparison of growth.
Nucleotide Binding Assays
ATP binding to SecA proteins was measured by nitrocellulose membrane filtration assays. The membranes were treated with 0.5 N NaOH for 2 min, washed with H2O, and equilibrated in nucleotide binding buffer (50 mM Hepes-NaOH, pH 7.5, 30 mM KCl, 10 mM MgOAc2). 15-μl reactions containing 2.5 μM SecA dimer and 0–250 μM ATP (+0.5 Ci of [α-32P]ATP per point) were incubated in binding buffer for 10 min at 4 °C. 10-μl aliquots of the reaction were filtered through the membrane. The membranes were washed before and after filtration with 150 μl of the binding buffer. One-μl aliquots were spotted onto a separate membrane to measure total nucleotide in the reaction. The molar amount of nucleotide bound to SecA dimer was determined by quantitation on a PhosphorImager (Amersham Biosciences) and plotted versus nucleotide concentration. The binding isotherms were fit to an equation describing 1:1 protein-ligand interaction, [N·M] = 0.5{(Kd + [Nt] + [Mt]) – [(Kd + [Nt] + [Mt])2 − 4[Nt][Mt]]1/2}, where N·M is the molar amount of ATP bound to SecA dimer, Nt and Mt are total ATP and SecA concentrations, respectively, and Kd is the apparent dissociation constant.
The rate of dissociation of ADP from SecA was measured by incubating 2.5 μM SecA dimer with 250 μM ADP (+0.5 μCi of [α-32P]ADP per point) in nucleotide binding buffer, at 25 °C for 10 min, followed by addition of 10 mM Mg2+-ADP chase and filtration through nitrocellulose membranes at times ranging from 20 s to 10 min. The molar amount of ADP bound to SecA was plotted versus time and fit to a single exponential function to determine the rate constant, koff.
Steady-state ATPase Assays
For colorimetric ATPase assays, 25-μl reactions were prepared with SecA (0.2 μM dimer) in buffer (50 mM Hepes-KOH, pH 7.5, 30 mM KCl, 30 mM NH4Cl, 0.5 mM MgOAc2, 1 mM dithiothreitol), in the absence of other ligands (endogenous ATPase) or in the presence of 50 μg of membrane protein/ml of CK1801 inverted membrane vesicle alone (membrane ATPase) or with 20 μM pre-protein PSN (chimera of alkaline phosphatase signal sequence and mature portion of staphylococcal nuclease; translocation ATPase). The reactions were initiated with 4 mM ATP and incubated for 15 min at 37 °C. ATP hydrolysis was measured using the malachite green assay (33) with the modifications described previously (6).
The steady-state ATPase rate constant was measured by radiometric assays with 1 μM SecA dimer and 1 mM ATP (+ 0.5 μCi of [α-32P]ATP per point) in ATPase buffer (50 mM Hepes, pH 7.5, 30 mM KCl, 10 mM MgOAc2) at 37 °C. At varying times, 5 μl of the reaction was quenched with 5 μl of 0.5 M EDTA and 1-μl aliquots were analyzed by polyethyleneimine cellulose thin-layer chromatography with 0.6 M potassium phosphate, pH 3.4. The molar amount of [μ-32P]ADP formed was plotted versus time, and the kcat value was obtained from velocity ÷ [SecA dimer].
Rapid-quench and Pulse-Chase ATPase Assays
Pre-steady-state ATPase assays were performed on a KinTek Corp. quench-flow instrument (Austin, TX) at 37 °C with 6 μM SecA dimer and 1 mM ATP (+ 2 μCi of [α-32P]ATP per point) in ATPase buffer. Sixteen μl of SecA was mixed rapidly with 16 μl of ATP and quenched after varying times (0.004–5 s) with 35 μl of 0.7 M formic acid (final concentrations were 3 μM SecA dimer and 0.5 mM ATP). One-μl aliquots of the reactions were spotted immediately on TLC plates and analyzed as above. Molar amounts of ADP formed were plotted versus time and fit to a single-exponential plus linear equation (burst equation), [ADP] = A(1-e−kt) + Vt, where A and k are burst amplitude and rate constant, respectively, V is steady-state velocity, and t is reaction time.
Pulse-chase experiments were performed similarly, except 35 μl of 10–20 mM unlabeled Mg2+-ATP chase was added to the reaction after various times (0.004–2 s). After chase time equivalents to 5–6 turnovers (25 s), the reactions were quenched with 70 μl of 0.7 M formic acid and analyzed as above.
Stopped-flow experiments were performed on a KinTek Corp. SF-2001 Stopped-Flow instrument (Austin, TX) to measure the rates of ATP hydrolysis and phosphate release catalyzed by SecA. Phosphate (Pi) release was assayed in real-time using fluorescent MDCC-labeled E. coli PBP as described previously (31, 34, 35). Changes in MDCC-PBP fluorescence upon Pi binding were monitored by excitation at 425 nm and monitoring emission above 450 nm (cut-off filter; Corion LL-450 F). A coupled enzyme reaction (Mop) containing 200 μM 7-methylguanosine and 0.03 units/ml purine nucleoside phosphorylase was used in all reactions to sequester contaminant Pi as ribose 1-phosphate, because MDCC-PBP is sensitive to micromolar concentrations of Pi. A Pi calibration curve relating the PBP-MDCC fluorescence signal to Pi concentration was generated prior to each experiment (31). All reactions and syringes were “mopped” for at least 45 min before each experiment.
Pi release experiments were carried out at 37 °C in ATPase buffer containing the Mop. A 60-μl solution of 2 μM SecA dimer, incubated with 16 μM MDCC-PBP, was mixed rapidly with an equal volume of 1 mM ATP, and the change in fluorescence monitored over time in the observation cell (final concentrations are 1 μM SecA, 500 μM ATP, 8 μM MDCC-PBP). Background fluorescence, measured for each reaction by omitting Mg2+ to prevent ATP hydrolysis, was subtracted from the traces. Raw data obtained by averaging at least 5 traces were divided by the slope from the Pi calibration curve to determine the molar amount of Pi released during the reaction. Data were fit to a burst equation or a linear equation.
RESULTS
The kinetics of ATP binding, hydrolysis, and product release catalyzed by wild type and mutant SecA proteins were measured to better define the mechanism of the ATPase reaction, which is necessary to understand how SecA uses ATP to drive transport of proteins across membranes.
Steady-state ATPase and Protein Translocation Activities of Wild Type and Mutant SecA Proteins
The proposed catalytic base, glutamate 210, in the Walker B “DEVD” motif of the high-affinity nucleotide-binding site was replaced by aspartate, asparagine, glutamine, or alanine residues, using site-directed mutagenesis (Fig. 1). A 6-histidine tag was introduced into the mutants to help completely purify these proteins from any contaminating ATPase activity. As a control, a 6-histidine-tagged version of wild type SecA was also prepared and its activity compared with that of untagged SecA; the two wild type SecA proteins functioned similarly in all assays. In a commonly used colorimetric assay used to measure ATP hydrolysis, none of the mutants had any detectable level of activity above background levels (Fig. 2A), alone (endogenous ATPase), or in the presence of inverted membrane vesicles that contain the SecYEG translocon and are stripped of endogenous SecA without (membrane ATPase) or with pre-protein (translocation ATPase) at 37 °C. As expected from previous studies, wild type SecA ATPase activity is stimulated about 10-fold in the presence of SecYEG/membrane and pre-protein (Fig. 2A) (22). A more sensitive and accurate radiometric assay used to measure the steady-state rate constants for wild type SecA (kcat = 0.2–0.3 s−1) and mutant SecA endogenous ATPase activity indicates that SecA-E210D retains some level of activity (kcat = 0.15 s−1), but SecA-E210A mutant activity is near background level (kcat = 0.04 s−1) (Fig. 2B). All the mutants, even SecA-E210D, fail to complement inactivated SecA in vivo (Fig. 2B), consistent with the results of an in vitro protein translocation assay in which the mutants cannot catalyze transport of pro-OmpA polypeptide across a membrane (Fig. 2C, lanes 3–6). These initial results suggest that glutamate 210 plays a key role in the ATPase reaction, and therefore the protein translocation activity of SecA. Before this residue can be implicated as the catalytic base, however, some significant caveats must be addressed. Because the steady-state assays described above do not measure individual ATP binding, hydrolysis, and product release steps in the reaction, they do not reveal which of these steps is affected by the mutations, and therefore cannot determine the role of Glu-210 in the catalytic mechanism. In addition, it is possible that the Glu-210 mutations affect SecA function for reasons other than direct disruption of catalytic chemistry (e.g. via some unrelated perturbation of SecA structure). The experiments described below address these concerns.
FIG. 2. SecA-E210D, SecA-E210Q, SecA-E210N, and SecA-E210A are deficient in ATPase and in vitro protein translocation activity.
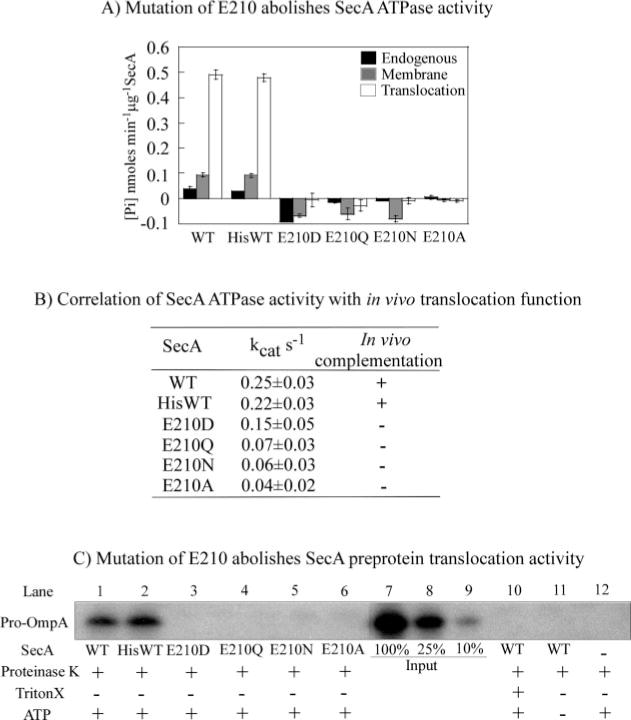
A, colorimetric assays measuring endogenous, membrane, and translocation ATPase activity of wild type (WT) SecA, His-SecA, and His-tagged SecA mutants at 37 °C indicate severe loss of activity upon mutation of glutamate 210. B, steady-state ATPase rates of SecA and Glu-210 mutants (measured by following hydrolysis of [α-32P]ATP) correlate with their in vivo function as assayed by complementation of inactivated endogenous secA. C, SecA-catalyzed protein translocation across membranes, assessed in vitro by comparing protection of pro-OmpA from proteinase K degradation. Lanes 3–6 show lack of mutant SecA activity, in contrast to wild type SecA (lanes 1 and 2). Lanes 7–9 show a calibration curve with 100, 25, and 10% of input 125I-pro-OmpA, respectively, and lanes 10–12 show control reactions for proteinase K activity under various reaction conditions.
Impact of Glu-210 Mutations on SecA Function
Wild Type SecA as Well as E210D and E210N Mutants Bind Two ATP Molecules Per Dimer
ATP binding to wild type and mutant SecA proteins was examined by nitrocellulose membrane filtration assays. 2.5 μM SecA (dimer concentration) bound 4.8 μM [α-32P]ATP at 4 °C, i.e. a ratio of nearly 2 ATP molecules per SecA dimer (there was no nonspecific ATP binding to the membrane up to 300 μM ATP) (Fig. 3A). The ATP molecules bind SecA with an apparent Kd of 1.4 μM, indicating that the interaction occurs at the high-affinity nucleotide-binding site. No additional ATP binding (i.e. to a low-affinity site on SecA) could be detected at 300 μM ATP, and data at higher ATP concentrations were not considered reliable because of increasing levels of nonspecific ATP binding to the membrane. The SecA-E210D mutant exhibits almost identical nucleotide binding characteristics, with 2 ATP molecules bound per dimer with an apparent Kd of 2.4 μM (Fig. 3B), and SecA-E210N also binds close to 2 ATP per dimer with an apparent Kd of 0.8 μM (Fig. 3C). These data suggest that substitution of glutamate 210 in SecA with aspartate or asparagine does not appreciably affect its nucleotide binding stoichiometry and affinity. In contrast, only a fraction of the SecA-E210A and SecA-E210Q mutant proteins appears capable of binding nucleotide, as their binding isotherms saturate at only ~1 ATP bound per protein dimer (Fig. 1, Supplemental Materials). Interestingly, a partial proteolysis assay of the proteins reveals that SecA-E210A and SecA-E210Q are also significantly more sensitive to trypsin digestion than wild type SecA and SecA-E210D and SecA-E210N (Fig. 1, Supplemental Materials). These data indicate variations in protein structure/stability between the mutants, and suggest that in the case of the SecA-E210A and SecA-E210Q mutants, the loss of ATPase and protein translocation activity may be attributable to reasons other than disruption of catalytic chemistry.
FIG. 3. High-affinity binding of two ATP molecules per SecA dimer.
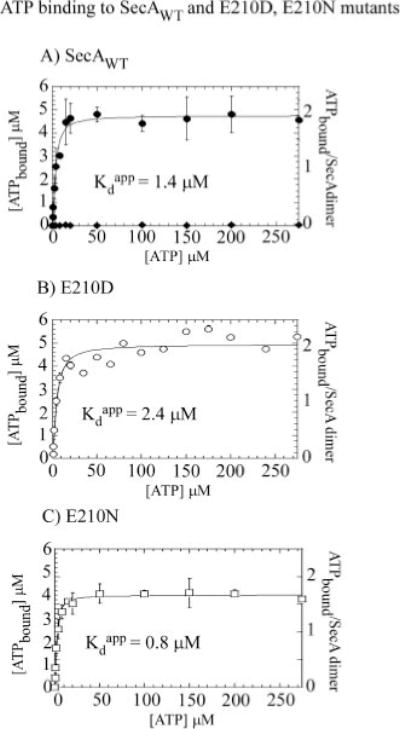
Membrane filtration experiments performed with 2.5 μM SecA dimer reveal that the wild type (WT) SecA (A) as well as SecA-E210D (B) and SecA-E210N (C) bind two ATP molecules per dimer with high affinity; apparent Kd 1.5 ± 0.3, 2.4 ± 0.8, 0.8 ± 0.1 M, respectively (nonspecific ATP binding to the membrane is negligible (◆)).
Wild Type SecA Rapidly Binds and Hydrolyzes Two ATP Molecules per Dimer, Unlike the E210D and E210N Mutants
To determine whether mutation of Glu-210 to aspartate or asparagine specifically impacts ATP hydrolysis rather than some other event(s) in the SecA-catalyzed ATPase reaction, it was necessary to directly measure the ATP binding, hydrolysis, and product release steps in the reaction. We first performed rapid quench experiments with 2.6–3 μM wild type SecA dimer and 500 μM [α-32P]ATP, and observed a burst of ATP hydrolysis in the first turnover at a rate constant of 27 ± 2.5 s−1, followed by a slow linear phase at a rate constant of 0.3 s−1 (similar to the steady-state kcat of 0.25 s−1) (Fig. 4A). The amplitude of the burst phase is 5.2 ± 0.1 μM, close to 2 ATP molecules per SecA dimer.
FIG. 4. Substitution of Glu-210 with Asp or Asn impairs rapid binding and hydrolysis of two ATP molecules by SecA dimer.
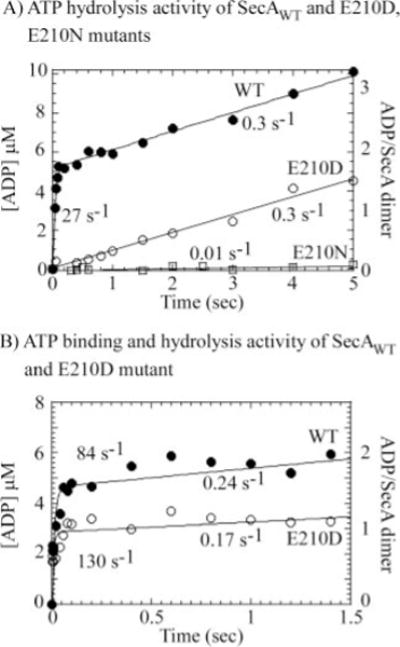
Pre-steady-state acid quench assays were performed with 2.6–3 μM SecA dimer and 500 μM ATP + [α-32P]ATP (final concentrations) at 37 °C. A, wild type (WT) SecA catalyzes rapid ATP hydrolysis (●), with a burst rate constant of 27 ± 2.5 s−1 and amplitude 5.2 ± 0.1 μM (1.8 ATP per dimer), followed by a slow steady-state phase at kcat = 0.3 s−1. In contrast, the SecA-E210N mutant displays no ATP hydrolysis activity in the first catalytic turnover (□). SecA-E210D appears to have residual ATPase activity manifest at a slow linear rate constant of 0.3 s−1 (○). B, pulse-chase analysis reveals rapid binding of 2 ATP molecules to the SecA dimer at an apparent binding rate constant of 0.2 × 106 M−1 s−1 (●; 80–90 s−1 500 M). SecA-E210D also binds ATP rapidly (0.25 × 106 M−1 s−1), but the reduced burst amplitude suggests a defect in ATP binding as well as ATP hydrolysis activity.
Next, a pulse-chase experiment was performed to measure the kinetics of ATP binding to SecA. The protein (2.6–3 μM dimer) was mixed with [μ-32P]ATP (500 μM) for varying times (mixing time), and chased with 20-fold excess unlabeled Mg2+-ATP for time equivalent to 5–6 turnovers (chase time). During the chase, bound [α-32P]ATP may be hydrolyzed to [α-32P]ADP + Pi or remain unhydrolyzed (either bound to SecA or released into solution). Any free [α-32P]ATP in solution is diluted upon addition of unlabeled ATP chase and is not available for further binding and hydrolysis. Thus, the pulse-chase experiment measures the rate of ATP binding to SecA and the fraction of the SecA·ATP complex that undergoes hydrolysis. The data reveal a burst of ATP binding at 80–90 s−1 with 4.8 μM amplitude, indicating that close to two ATP molecules per SecA dimer are bound at an apparent bimolecular rate constant of 2 × 105 M−1 s−1 and are hydrolyzed rapidly within the chase time; the steady-state rate constant is 0.24 s−1 (Fig. 4B).
In striking contrast to the wild type protein, the SecA-E210N mutant has barely detectable activity even in the first turnover, indicating that even though the protein appears capable of binding ATP with similar affinity as wild type SecA (Fig. 3C), it cannot catalyze ATP hydrolysis (Fig. 4A). SecA-E210D has a different phenotype, it displays a low level of ATPase activity, as observed also in the steady-state ATPase assay (Fig. 1B); however, even in the first turnover, this mutant appears to hydrolyze ATP at a very slow rate of 0.3 s−1 compared with the 27 s−1 ATP hydrolysis rate of wild type SecA (Fig. 4A). Thus, SecA short even a single carbon from the glutamate in the active site suffers a 90-fold loss in its ability to catalyze ATP hydrolysis, and removal of the carboxylate group completely abolishes catalytic activity.
Because SecA-E210D retains residual ATPase activity, we were able to perform a pulse-chase experiment to try and measure the kinetics of ATP binding to this mutant. The burst kinetics observed in Fig. 4B indicate rapid ATP binding (2.5 × 105 M−1 s−1) followed by slow hydrolysis; however, at 2.9 μM, the burst amplitude is only about 60% that of wild type SecA (4.8 μM). This result indicates heterogeneity in the SecA-E210D population in the reaction and can be interpreted as: (a) all SecA-E210D dimers in the reaction bind two ATP molecules in the mixing time, but only a fraction of the SecA-E210D ATP complexes undergo slow hydrolysis during the chase time; (b) only a fraction of SecA-E210D dimers in the reaction can bind ATP in the mixing time and these complexes undergo slow hydrolysis in the chase time. Both possibilities are consistent with the hypothesis that glutamate 210 in SecA plays a key role in ATP hydrolysis as a catalytic base, but the second possibility suggests a role for the residue in ATP binding as well as hydrolysis. At this time both possibilities are equally likely, although our finding that the SecA-E210D mutant binds two ATP molecules per dimer with similar affinity as wild type SecA is more consistent with the first one (Fig. 3B).
Dissociation of ADP, but Not Pi, Is Slower from the E210D Mutant Than from Wild Type SecA
Thus far, it appears that the glutamate 210 residue is important for SecA-catalyzed ATP hydrolysis, and it may also play some role in productive ATP binding to the protein (defined as ATP binding followed by hydrolysis). We tested the kinetics of product release from SecA, to determine whether the glutamate plays any role in this step of the ATPase reaction. We measured the rate of phosphate (Pi) release, using a real time Pi release assay developed previously by the Webb research group (30). In the assay, MDCC-labeled PBP (MDCC-PBP) is used as a fluorescent reporter of Pi in solution. MDCC-PBP binds Pi rapidly (1.4 × 108 M−1 s−1) and with high affinity (Kd = 0.1 μM), and its fluorescence increases ~7-fold upon interaction with Pi (36). The stopped-flow trace in Fig. 5A shows the increase in MDCC-PBP fluorescence as SecA (1 μM dimer) hydrolyzes ATP (500 μM ATP in the reaction) and releases Pi over time. The data reveal a burst of Pi formation at a rate constant of 26 s−1, followed by a slow linear phase at 0.3 s−1. Using a calibration curve to relate the fluorescence signal to known Pi concentrations (data not shown), we found that 2 Pi molecules are released per SecA dimer in the burst phase (burst amplitude = 2.1 μM). The rate and amplitude of Pi release are nearly identical to those measured for ATP hydrolysis in the rapid quench experiment in Fig. 4A, revealing that the Pi product dissociates rapidly from SecA following ATP hydrolysis. Apparently, a step after Pi dissociation, perhaps related to ADP dissociation from SecA, is responsible for limiting the steady-state ATPase rate.
FIG. 5. SecA turnover is not limited by phosphate release.
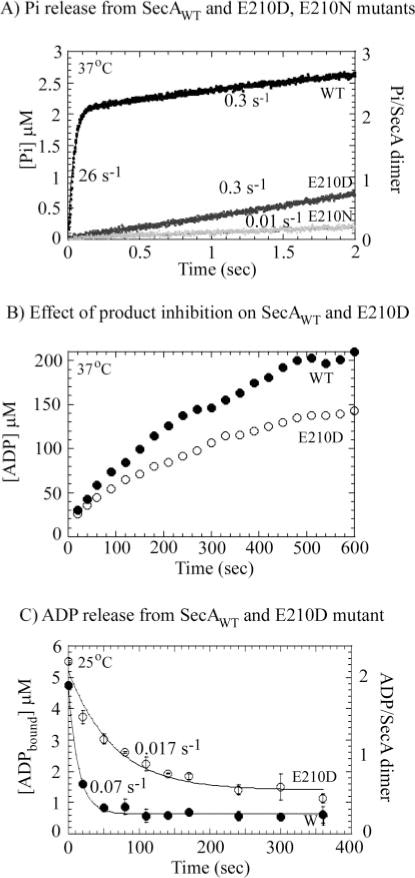
A, a fluorescent reporter assay for phosphate (Pi) performed at 37 °C shows a burst of Pi release from 1 μM wild type (WT) SecA dimer (black) at a rate of 26 ± 4 s−1 and amplitude of 2.1 ± 0.3 μM (2.1 ATP per dimer), followed by a steady-state phase at 0.3 s−1, indicating rapid Pi dissociation from SecA following ATP hydrolysis. SecA-E210D (gray) releases Pi at a 90-fold slower linear rate of 0.3 s−1 governed by the slow ATP hydrolysis rate (Fig. 5A), and Pi release from SecA-E210N (light gray) is negligible. B, measurement of steady-state ATPase activity over a prolonged period reveals greater product inhibition effects on SecA-E210D (○) than on wild type SecA (●). C, measurement of [α-32P]ADP dissociation from SecA (2.5 μM dimer) following 10 mM Mg2+-ADP chase shows that the E210D mutant releases ADP at a slower rate (0.017 s−1; ○) than wild type protein (0.07 s−1; ●) at 25 °C.
The SecA-E210D mutant releases Pi at 0.3 s−1 and SecA-E210N has negligible Pi release activity, similar to their ATP hydrolysis activity measured in Fig. 4A. Thus, in the case of SecA-E210D also, Pi appears to dissociate rapidly following ATP hydrolysis, suggesting that this step may not be unduly affected by mutation of glutamate 210 to aspartate.
At this point we also noted a small but consistent difference in the steady-state rate constants for SecA-E210D measured in the pre-steady-state experiments (shorter times) versus steady-state experiments (longer times); i.e. 0.3 s−1 and identical to wild type SecA (Figs. 4A and 5A) versus 0.15 s−1 and approximately half as active as SecA (Fig. 2B), respectively. To explore this issue further, the ATPase activity of both SecA and SecA-E210D was followed over an extended time period ranging from 20 to 600 s. As shown in Fig. 5B, initially the two proteins exhibit similar rates of turnover (0.17–0.2 s−1; 37 °C), but over time, SecA-E210D ATPase activity is reduced significantly more than that of SecA. Presumably the inhibition of activity occurs because of accumulation of ADP product in the reaction (total ATP in the reaction = 500 μM), and SecA-E210D has higher affinity for ADP than SecA, therefore its activity is inhibited at lower ADP concentrations (see below).
We measured the rate of ADP dissociation from the two proteins by chase experiments in which wild type SecA and SecA-E210D (2.5 μM dimer) were incubated with [α-32P]ADP (250 μM) and chased with excess unlabeled Mg2+-ADP (10 mM), and dissociation of [α-32P]ADP was measured by filtration through nitrocellulose membranes at varying times (Fig. 5C; these experiments were performed at 25 °C because of limitations with membrane filtration assays at higher temperatures). In the absence of unlabeled ADP chase (at time 0), the proteins bind two [α-32P]ADP molecules per dimer (consistent with the data in Fig. 3 for ATP binding to SecA). ADP dissociates from wild type SecA at a slow rate of 0.07 s−1, suggesting that ADP release could indeed limit the ATPase turnover rate. The difference between the 0.07 s−1 ADP dissociation rate constant, and 0.25 s−1 kcat may be because of the fact that the two reactions are carried out at different temperatures, 25 and 37 °C, respectively, or perhaps because of the difference in assay techniques (membrane filtration versus ATPase kinetics); temperature effects on SecA nucleotide binding activity have been noted previously (37). It is also possible that the SecA·ADP complex formed in membrane filtration experiments by directly incubating SecA with ADP is not exactly the same as that formed following ATP hydrolysis and Pi release. The rate of ADP release from the SecA-E210D mutant is 0.017 s−1, which is 4-fold slower than ADP release from the wild type protein (Fig. 5C), and, along with the ATPase inhibition data from Fig. 5B, indicates that substitution of glutamate 210 with aspartate stabilizes ADP in the active site. Thus, it is possible that in addition to catalysis, this glutamate residue plays a role in product release and turnover as well.
Aspartate 133 Does Not Function as the Catalytic Base in SecA
In a previous study, an aspartate residue (Asp-133) between the Walker A and B motifs was proposed as the catalytic base that activates water for a nucleophilic attack on the γ phosphate of ATP (Fig. 1A) (26). This hypothesis was supported by the loss of ATPase and protein translocation activity following mutation of aspartate 133 to asparagine. Given our data indicating that glutamate 210 likely serves as the catalytic base in the SecA ATPase site, we decided to re-evaluate the role of aspartate 133 in SecA function.
In addition to SecA-D133N, a second mutant, SecA-D133A, was prepared to test if any change in activity is dependent on the nature of the amino acid substitution (as observed in the case of the E210Q and E210A mutations; Fig. 2 and Supplemental Materials Fig. 1). The mutant proteins were assayed for both ATPase and protein translocation activities. Fig. 6A shows that SecA-D133N suffers a loss of ATPase activity (e.g. translocation of ATPase activity of SecA-D133N is ~5-fold lower than that of wild type SecA). This result mirrors that of the earlier study (26); however, we also found that mutation of the same aspartate 133 to alanine does not have any effect on SecA activity. Both wild type SecA and SecA-D133A have the same steady-state endogenous, membrane, and translocation ATPase rates (Fig. 6A), and in pre-steady-state assays, SecA-D133A shows a burst of ATP hydrolysis (and Pi release) at a rate of 20 s−1 and amplitude of 1.5 μM (1.5 ATP/SecA dimer), followed by a slow phase at 0.1 s−1 (Fig. 6B), similar to the wild type protein (Fig. 5A). In contrast, the ATPase activity of SecA-D133N appears severely compromised even in the first catalytic turnover (0.15 s−1 linear rate; Fig. 6B). These data reveal that the effects of aspartate 133 mutation on SecA are dependent on the substituent amino acid, and more importantly, they reveal that near complete removal of this residue (i.e. replacement with alanine) has a very small effect (possibly negligible within the margin of error) on SecA activity. Thus, it is highly unlikely that aspartate 133 functions as the catalytic base in the active site. As expected from the ATPase results, the D133N mutant is unable to catalyze protein translocation (Fig. 6C, lane 2), whereas D133A mutant activity is indistinguishable from that of wild type SecA (Fig. 6C, lane 3). It is possible that with SecA-D133N, as in the case of SecA-D210A and SecA-D210Q, some perturbation other than that of catalytic chemistry results in loss of activity. Consistent with this hypothesis, partial proteolysis assays indicate that SecA-D133N is slightly more sensitive to trypsin digestion than wild type SecA and SecA-D133A (data not shown).
FIG. 6. Aspartate 133 does not appear essential for SecA ATPase and protein translocation activity.
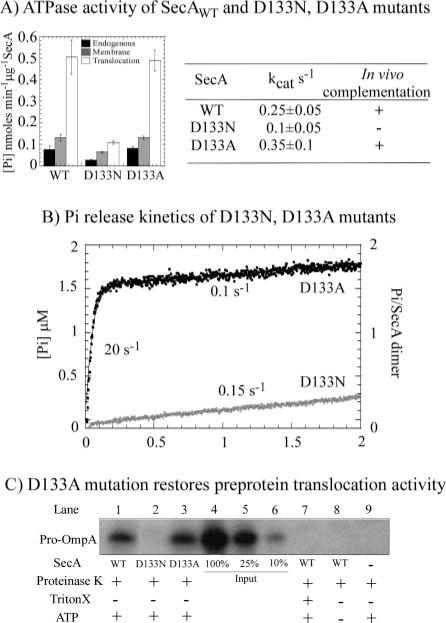
A, in endogenous, membrane, and translocation ATPase assays at 37 °C, the SecA-D133N mutant shows poor activity relative to wild type (WT) SecA, but SecA-D133A appears to retain full activity. B, Pi release kinetics in the first turnover reveal that SecA-D133N (black) indeed cannot catalyze ATP hydrolysis, whereas SecA-D133A (gray) retains near wild type ATP binding and hydrolysis activity at 37 °C (burst rate constant = 20 s−1; amplitude 1.6 μM; kcat 0.1 s−1). C, correspondingly, no pro-OmpA protein translocation activity can be detected with SecA-D133N in the reaction (lane 2), unlike that observed with SecA-D133A (lane 3) and wild type SecA (lane 1).
DISCUSSION
Here, we report a kinetic analysis of individual steps in the SecA-catalyzed ATPase reaction that fuels the protein translocation activity of this molecular machine. The ATPase reaction has been examined previously, with the focus primarily on the steady-state ATPase rate and how it is affected by the various ligands in the protein translocation reaction, i.e. precursor protein, signal peptide, SecB protein, membrane phospholipids, and SecYEG translocon (10, 16, 38–40). The resulting information has been used to generate model mechanisms of how SecA might use ATP to transport proteins across membranes. SecA is proposed to cycle between membrane-bound and free states, including a membrane/translocon-inserted state (in which it presumably carries a segment of precursor protein across the membrane channel), coupled to ATP binding and hydrolysis (19, 21, 41). The protein appears to adopt a compact “ground state” conformation when bound to ADP, and this state has low affinity for SecYEG/membrane, suggesting that at some point following ATP hydrolysis, SecA is prone to dissociating from the translocon (8, 23, 37). The nucleotide-free form of SecA appears to exist in multiple states in equilibrium, including the compact ground state and an extended “domain-dissociated” state that could have higher affinity for SecYEG/membrane (8, 12, 42). It is not clear yet which state of the protein, membrane-bound or membrane-free, is stabilized upon ATP binding to SecA. There is strong evidence that binding of the non-hydrolysable ATP analog, AMP-PNP, to SecA favors the membrane-bound state (11, 21), but this analog has 1000-fold lower affinity for SecA than ATP, which casts doubt on whether it can serve as a faithful mimic of ATP (5).
The steady-state ATPase rate of SecA is stimulated in the presence of SecYEG/membrane and precursor proteins in the reaction. Recently, ADP dissociation from SecA (and a related change in SecA conformation) has been identified as the rate-limiting step in the ATPase reaction (5). A key implication of this finding is that the steady-state ATPase rate is a measure of one or more post ATP hydrolysis events in the reaction, and does not yield any information on the ATP binding and hydrolysis steps. Another recent report in the literature indicates that SecYEG/membrane and precursor proteins influence the rate of ADP release from SecA (resulting in an increase in the ATPase rate), and this coupling may have an important role in the SecA-catalyzed protein translocation reaction (16). Further understanding of the mechanism of action of SecA requires knowledge of all events in the ATPase reaction and how they may be influenced by various components of the protein trans-location pathway.
Using pre-steady-state analysis, we have found that SecA rapidly binds two ATP molecules per dimer with an apparent binding rate constant of 2 × 105 M−1 s−1 (~100 s−1 at 500 μM ATP concentration). The SecA·ATP complex undergoes ATP hydrolysis at 27 s−1 at 37 °C, which is ~100-fold faster than the rate-limiting step at 0.2–0.3 s−1, associated with ADP release (Fig. 4). Dissociation of the phosphate product occurs almost instantaneously following ATP hydrolysis, resulting in an apparent release rate constant of 26 s−1. Thus, there are at least two points in the ATPase pathway when a fast step is followed by a pause and a relatively slow step, i.e. between ATP binding and hydrolysis, and between ATP hydrolysis and ADP release. It is possible that these transition points in the reaction are associated with changes in SecA conformation that constitute the mechanical motions required for protein translocation. For example, it is tempting to speculate that a slow step between ATP binding and hydrolysis may involve membrane insertion of the SecA·pre-protein complex, in keeping with previously proposed model mechanisms (19). Also, although the ADP-bound form of SecA has the most prolonged lifetime in the endogenous reaction cycle (in solution, in the absence of ligands), under protein translocation conditions the rate of ADP release from SecA is significantly faster (16). Rapid ADP release and turnover could be necessary for efficient retraction of SecA from the translocon (and correspondingly efficient re-insertion with the next segment of precursor protein); however, given that the ADP bound form of SecA has low affinity for the membrane, it is also possible that an increase in the ADP release rate forestalls complete dissociation of SecA from the translocon before the next ATP binding event, thus enabling processive protein translocation. These two very different yet at this time quite likely scenarios highlight the necessity of measuring the timing of individual events in both the protein translocation pathway and the associated ATPase pathway, to understand the SecA mechanism of action in detail. We anticipate that the kinetic parameters of endogenous ATP binding, hydrolysis, and product release reported here will facilitate future analyses of these events under protein translocation conditions.
Additionally, the pre-steady-state analysis revealed a complex role for glutamate 210 in the SecA ATPase mechanism. This residue is part of the conserved Walker B motif found in many different ATPases such as the ABC (ATP-binding cassette) proteins, including transporters and DNA repair proteins such as Rad50 and MutS, as well as helicases (43). The motif comprises a few hydrophobic residues followed by an aspartate that chelates the Mg2+ ion bound to the nucleotide, and then a glutamate that points directly toward the γ phosphate of the nucleotide bound in the active site (4). It has been proposed that in this position, glutamate serves to activate a catalytic water molecule for nucleophilic attack on the phosphate (12, 44–46). For example, mutation of the glutamate residue in MJ0796 and MJ1267 ABC transporters to alanine or glutamine results in loss of ATPase activity (as measured in steady-state ATPase assays), supporting the hypothesis that this residue is required for ATP hydrolysis (46). In the case of the B. subtilis transporter, BmrA, substitution of the glutamate with other amino acids resulted in entrapment of the nucleotide in the triphosphate form in the active site, also indicating disruption of the hydrolytic reaction (47). In an analysis of P-glycoprotein glutamate mutants, however, the data indicated entrapment of the nucleotide in the diphosphate form, and was interpreted to mean that the glutamate residue plays a role in ADP release and turnover, rather than catalysis (48, 49).
Our contribution to the debate regarding the role of the Walker B glutamate is evidence in that in E. coli SecA this amino acid is critical for catalyzing the ATP hydrolysis step in the reaction, but it also appears to play a role in ADP release following hydrolysis, and may play a role in productive ATP binding to the protein. These conclusions are based on direct measurements of individual ATP binding, ATP hydrolysis, and ADP and phosphate release events for both wild type and glutamate 210 mutants. Thus, we find that substitution of Glu-210 with aspartate or asparagine does not alter the stoichiometry or apparently the equilibrium of ATP binding to SecA (2 ATP molecules/SecA dimer; apparent Kd 1–2 μM). However, following ATP binding, both SecA-E210D and SecA-E210N mutants display defects in the ATP hydrolysis step. In the case of the E210N mutant, we cannot detect any significant ATPase activity even in the first catalytic turnover. In contrast, in steady-state assays, the E210D mutant exhibits only a 2-fold decrease in the ATPase rate compared with wild type SecA (measured at 37 °C). However, in pre-steady-state assays measuring ATP hydrolysis, rather than the rate-limiting step following hydrolysis, it is clear that the reaction rate drops substantially, from 27 s−1 for wild type SecA to 0.3 s−1 for SecA-E210D. Thus, even a conservative substitution of an aspartate residue for glutamate 210 results in nearly 100-fold reduction in the hydrolytic activity of SecA. Presumably, the carboxylate group present in SecA-E210D can still activate a water molecule for nucleophilic attack, and therefore the mutant has some residual ATPase activity; however, this activity is not sufficient to fuel protein translocation across membranes. This conclusion is consistent with the invariability of the glutamate residue in the Walker B DEVD motif of the nearly 50 SecA proteins whose sequences have been determined thus far.
Interestingly, SecA-E210D also exhibits a defect in ADP release; the ADP dissociation rate is ~4-fold slower for this mutant compared with wild type SecA (as measured at 25 °C). Perhaps the carboxylate group of glutamate 210 contributes to an unfavorable environment for the extra negative charge developed on the β phosphate following ATP hydrolysis, and thus plays a role in dissociation of ADP from SecA following ATP hydrolysis. Presumably, with the shorter aspartate residue the electrostatic repulsion effect is reduced, resulting in stabilization of ADP in the SecA-E210D active site relative to wild type SecA. Glutamate 210 also hydrogen bonds with another water molecule that participates in coordination of the Mg2+ cofactor, essential for both ATP binding and hydrolysis (12). If this coordination is disrupted because of the shifted position of the carboxylate group in SecA-E210D, the mutant could suffer a defect in ATP binding as well. We do not observe any significant difference in the stoichiometry or apparent affinity of interaction with ATP between the wild type and mutant proteins. However, in a pulse-chase experiment designed to measure the kinetics of productive ATP binding (defined as ATP binding followed by ATP hydrolysis), only a fraction of SecA-E210D protein in the reaction appears active (Fig. 4B). Although the underlying cause of this result in not completely clear yet, one possible explanation is that some SecA-E210D molecules in the reaction can bind ATP like the wild type protein and ATP hydrolysis is defective (slow or nonexistent), whereas others are defective in ATP binding and/or both ATP binding and hydrolysis.
This study also dispels an earlier proposal that aspartate 133 in SecA functions as the catalytic base in ATP hydrolysis. That conclusion was based on the loss of steady-state ATPase activity and protein translocation activity of SecA when the aspartate was mutated to an asparagine (26). However, as we show in this report, mutation of the same aspartate to alanine does not affect SecA-catalyzed ATP binding, hydrolysis, or product release, and does not affect its protein translocation activity either (Fig. 6). During our analysis of glutamate 210, we observed that substitution of this residue with aspartate, asparagine, glutamine, and alanine disrupted SecA activity. However, unlike the E210D and E210N mutants that suffered a specific defect in hydrolytic activity, SecA-E210Q and SecA-E210A appeared to have lost activity because of an alternate cause, perhaps because of perturbation of protein structure not directly related to alteration of the active site. Given that the SecA-D133A mutant appears fully functional, we propose that aspartate 133 does not play a key role in SecA ATPase and protein translocation activities, and some as yet unspecified reason underlies the loss of SecA-D133N function.
To summarize, kinetic measurements of SecA activity together with mutagenic analysis have revealed the timing of individual steps in the endogenous ATPase reaction and the importance of glutamate 210 in catalyzing ATP hydrolysis. Kinetic analyses of SecA activity in the presence of phospholipids, pre-protein, SecB, and SecYEG-reconstituted proteoliposomes should similarly define the ATPase reaction under protein translocation conditions, and help clarify how ATP binding, hydrolysis, and product release events are coupled to protein transport across membranes. Moreover, the discovery of mutant SecA proteins that apparently bind ATP with similar high affinity as wild type protein, but are severely compromised for ATP hydrolysis, can be expected to help define the specific role of ATP binding to SecA during the translocation process.
Supplementary Material
Acknowledgments
We thank Siying Chen for preparation of SecA mutants and Lucia Zito for protein preparations, and Emily Jacobs-Palmer for technical assistance and helpful discussions.
Footnotes
This work was supported by National Institutes of Health Grants GM64514 (to M. M. H.) and GM42033 (to D. B. O.).
The on-line version of this article (available at http://www.jbc.org) contains Figs. S1—S2.
The abbreviations used are: AMP-PNP, adenosine 5′ -(β,γ-imino)-triphosphate; ATPγS, adenosine 5′-3-O-(thio)triphosphate; PBP, phosphate-binding protein; MDCC, 7-diethylamino-3-((((2-maleimidyl)ethyl)-amino)carbonyl)coumarin.
References
- 1.de Keyzer J, van der Does C, Driessen AJ. Cell Mol Life Sci. 2003;60:2034–2052. doi: 10.1007/s00018-003-3006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mori H, Ito K. Trends Microbiol. 2001;9:494–500. doi: 10.1016/s0966-842x(01)02174-6. [DOI] [PubMed] [Google Scholar]
- 3.Economou A. Mol Membr Biol. 2002;19:159–169. doi: 10.1080/09687680210152609. [DOI] [PubMed] [Google Scholar]
- 4.Geourjon C, Orelle C, Steinfels E, Blanchet C, Deleage G, Di Pietro A, Jault JM. Trends Biochem Sci. 2001;26:539–544. doi: 10.1016/s0968-0004(01)01907-7. [DOI] [PubMed] [Google Scholar]
- 5.Fak JJ, Itkin A, Ciobanu DD, Lin EC, Song XJ, Chou YT, Gierasch LM, Hunt JF. Biochemistry. 2004;43:7307–7327. doi: 10.1021/bi0357208. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell C, Oliver D. Mol Microbiol. 1993;10:483–497. doi: 10.1111/j.1365-2958.1993.tb00921.x. [DOI] [PubMed] [Google Scholar]
- 7.van der Wolk JP, Boorsma A, Knoche M, Schafer HJ, Driessen AJ. Biochemistry. 1997;36:14924–14929. doi: 10.1021/bi971766n. [DOI] [PubMed] [Google Scholar]
- 8.den Blaauwen T, Fekkes P, de Wit JG, Kuiper W, Driessen AJ. Biochemistry. 1996;35:11994–12004. doi: 10.1021/bi9605088. [DOI] [PubMed] [Google Scholar]
- 9.van der Wolk JP, Klose M, de Wit JG, den Blaauwen T, Freudl R, Driessen AJ. J Biol Chem. 1995;270:18975–18982. doi: 10.1074/jbc.270.32.18975. [DOI] [PubMed] [Google Scholar]
- 10.Miller A, Wang L, Kendall DA. Biochemistry. 2002;41:5325–5332. doi: 10.1021/bi025639p. [DOI] [PubMed] [Google Scholar]
- 11.Economou A, Pogliano JA, Beckwith J, Oliver DB, Wickner W. Cell. 1995;83:1171–1181. doi: 10.1016/0092-8674(95)90143-4. [DOI] [PubMed] [Google Scholar]
- 12.Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, Deisenhofer J. Science. 2002;297:2018–2026. doi: 10.1126/science.1074424. [DOI] [PubMed] [Google Scholar]
- 13.van der Wolk J, Klose M, Breukink E, Demel RA, de Kruijff B, Freudl R, Driessen AJ. Mol Microbiol. 1993;8:31–42. doi: 10.1111/j.1365-2958.1993.tb01200.x. [DOI] [PubMed] [Google Scholar]
- 14.de Keyzer J, van der Does C, Kloosterman TG, Driessen AJ. J Biol Chem. 2003;278:29581–29586. doi: 10.1074/jbc.M303490200. [DOI] [PubMed] [Google Scholar]
- 15.Rajapandi T, Oliver D. Mol Microbiol. 1996;20:43–51. doi: 10.1111/j.1365-2958.1996.tb02487.x. [DOI] [PubMed] [Google Scholar]
- 16.Natale P, Swaving J, van der Does C, de Keyzer J, Driessen AJ. J Biol Chem. 2004;279:13769–13777. doi: 10.1074/jbc.M312892200. [DOI] [PubMed] [Google Scholar]
- 17.Sianidis G, Karamanou S, Vrontou E, Boulias K, Repanas K, Kyrpides N, Politou AS, Economou A. EMBO J. 2001;20:961–970. doi: 10.1093/emboj/20.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lill R, Cunningham K, Brundage LA, Ito K, Oliver D, Wickner W. EMBO J. 1989;8:961–966. doi: 10.1002/j.1460-2075.1989.tb03458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Economou A, Wickner W. Cell. 1994;78:835–843. doi: 10.1016/s0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 20.Eichler J, Wickner W. Proc Natl Acad Sci U S A. 1997;94:5574–5581. doi: 10.1073/pnas.94.11.5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Does C, Manting EH, Kaufmann A, Lutz M, Driessen AJ. Biochemistry. 1998;37:201–210. doi: 10.1021/bi972105t. [DOI] [PubMed] [Google Scholar]
- 22.Zito CR, Oliver D. J Biol Chem. 2003;278:40640–40646. doi: 10.1074/jbc.M308025200. [DOI] [PubMed] [Google Scholar]
- 23.Ulbrandt ND, London E, Oliver DB. J Biol Chem. 1992;267:15184–15192. [PubMed] [Google Scholar]
- 24.Shilton B, Svergun DI, Volkov VV, Koch MH, Cusack S, Economou A. FEBS Lett. 1998;436:277–282. doi: 10.1016/s0014-5793(98)01141-7. [DOI] [PubMed] [Google Scholar]
- 25.Gilbert SP, Mackey AT. Methods. 2000;22:337–354. doi: 10.1006/meth.2000.1086. [DOI] [PubMed] [Google Scholar]
- 26.Sato K, Mori H, Yoshida M, Mizushima S. J Biol Chem. 1996;271:17439–17444. doi: 10.1074/jbc.271.29.17439. [DOI] [PubMed] [Google Scholar]
- 27.Weinkauf S, Hunt JF, Scheuring J, Henry L, Fak J, Oliver DB, Deisenhofer J. Acta Crystallogr Sect D Biol Crystallogr. 2001;57:559–565. doi: 10.1107/s0907444901001202. [DOI] [PubMed] [Google Scholar]
- 28.Crooke E, Guthrie B, Lecker S, Lill R, Wickner W. Cell. 1988;54:1003–1011. doi: 10.1016/0092-8674(88)90115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhoads DB, Tai PC, Davis BD. J Bacteriol. 1984;159:63–70. doi: 10.1128/jb.159.1.63-70.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brune M, Hunter JL, Corrie JE, Webb MR. Biochemistry. 1994;33:8262–8271. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- 31.Jeong YJ, Kim DE, Patel SS. J Biol Chem. 2002;277:43778–43784. doi: 10.1074/jbc.M208634200. [DOI] [PubMed] [Google Scholar]
- 32.Antony E, Hingorani MM. Biochemistry. 2003;42:7682–7693. doi: 10.1021/bi034602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 34.Baird CL, Gordon MS, Andrenyak DM, Marecek JF, Lindsley JE. J Biol Chem. 2001;276:27893–27898. doi: 10.1074/jbc.M102544200. [DOI] [PubMed] [Google Scholar]
- 35.Bertram JG, Bloom LB, Hingorani MM, Beechem JM, O’Donnell M, Goodman MF. J Biol Chem. 2000;275:28413–28420. doi: 10.1074/jbc.M910441199. [DOI] [PubMed] [Google Scholar]
- 36.Brune M, Hunter JL, Howell SA, Martin SR, Hazlett TL, Corrie JE, Webb MR. Biochemistry. 1998;37:10370–10380. doi: 10.1021/bi9804277. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt M, Ding H, Ramamurthy V, Mukerji I, Oliver D. J Biol Chem. 2000;275:15440–15448. doi: 10.1074/jbc.M000605200. [DOI] [PubMed] [Google Scholar]
- 38.Triplett TL, Sgrignoli AR, Gao FB, Yang YB, Tai PC, Gierasch LM. J Biol Chem. 2001;276:19648–19655. doi: 10.1074/jbc.M100098200. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Miller A, Kendall DA. J Biol Chem. 2000;275:10154–10159. doi: 10.1074/jbc.275.14.10154. [DOI] [PubMed] [Google Scholar]
- 40.Ahn T, Kim H. J Biol Chem. 1998;273:21692–21698. doi: 10.1074/jbc.273.34.21692. [DOI] [PubMed] [Google Scholar]
- 41.van der Wolk JP, de Wit JG, Driessen AJ. EMBO J. 1997;16:7297–7304. doi: 10.1093/emboj/16.24.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding H, Hunt JF, Mukerji I, Oliver D. Biochemistry. 2003;42:8729–8738. doi: 10.1021/bi0342057. [DOI] [PubMed] [Google Scholar]
- 43.Hopfner KP, Tainer JA. Curr Opin Struct Biol. 2003;13:249–255. doi: 10.1016/s0959-440x(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 44.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. Mol Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hung LW, Wang IX, Nikaido K, Liu PQ, Ames GF, Kim SH. Nature. 1998;396:703–707. doi: 10.1038/25393. [DOI] [PubMed] [Google Scholar]
- 46.Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ. J Biol Chem. 2002;277:21111–21114. doi: 10.1074/jbc.C200228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orelle C, Dalmas O, Gros P, Di Pietro A, Jault JM. J Biol Chem. 2003;278:47002–47008. doi: 10.1074/jbc.M308268200. [DOI] [PubMed] [Google Scholar]
- 48.Sauna ZE, Muller M, Peng XH, Ambudkar SV. Biochemistry. 2002;41:13989–14000. doi: 10.1021/bi026626e. [DOI] [PubMed] [Google Scholar]
- 49.Urbatsch IL, Julien M, Carrier I, Rousseau ME, Cayrol R, Gros P. Biochemistry. 2000;39:14138–14149. doi: 10.1021/bi001128w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.