

Abstract
Epithelial renewal in the Drosophila intestine is orchestrated by Intestinal Stem Cells (ISCs). Following damage or stress the intestinal epithelium produces ligands that activate the epidermal growth factor receptor (EGFR) in ISCs. This promotes their growth and division and, thereby, epithelial regeneration. Here we demonstrate that the HMG-box transcriptional repressor, Capicua (Cic), mediates these functions of EGFR signaling. Depleting Cic in ISCs activated them for division, whereas overexpressed Cic inhibited ISC proliferation and midgut regeneration. Epistasis tests showed that Cic acted as an essential downstream effector of EGFR/Ras signaling, and immunofluorescence showed that Cic’s nuclear localization was regulated by EGFR signaling. ISC-specific mRNA expression profiling and DNA binding mapping using DamID indicated that Cic represses cell proliferation via direct targets including string (Cdc25), Cyclin E, and the ETS domain transcription factors Ets21C and Pointed (pnt). pnt was required for ISC over-proliferation following Cic depletion, and ectopic pnt restored ISC proliferation even in the presence of overexpressed dominant-active Cic. These studies identify Cic, Pnt, and Ets21C as critical downstream effectors of EGFR signaling in Drosophila ISCs.
Author Summary
Studies suggest that epidermal growth factor receptor (EGFR) signaling activation is a causal driver of many stem cell-derived epithelial cancers, including colorectal cancer. As in the human intestine, epithelial renewal in Drosophila intestine is orchestrated by intestinal stem cells (ISCs). EGFR signaling also plays an important role in regulating ISC proliferation in flies. However, the mechanism by which EGFR/Ras/MAPK signaling promotes ISC proliferation is poorly understood. Here we demonstrate that the transcriptional repressor, Capicua (Cic), mediates these functions of EGFR signaling. We found that the critical role of Cic as a negative regulator of cell proliferation in the fly midgut is consistent with its tumor suppressor function in mammalian cancer development. The direct target genes of Cic were identified by ISC-specific mRNA expression profiling and DNA binding mapping (DamID) method. Cic represses cell proliferation via regulating string (stg), Cyclin E (CycE), and the ETS domain transcription factors Ets21C and pointed (pnt). Using genetic tests we show that these interactions are meaningful for regulating stem cell proliferation. Combining our knowledge of Cic with what was previously known about CIC in tumor development, we propose that human CIC may regulate Ets transcription factors and cell cycle genes in Ras/MAKP-activated tumors.
Introduction
EGFR/Ras/MAPK signaling has diverse functions in regulating cell proliferation, growth, differentiation and survival in most animal cells [1]. Abundant studies also indicate that epidermal growth factor receptor (EGFR) activation is a causal driver of many cancers, including breast, lung, brain, and colorectal cancer [2]. Similarly, activating mutations in KRAS and BRAF, which are essential downstream effectors of the EGFR, are among the most common mutations found in a very wide range of human cancers [3,4]. However, despite much study, many questions remain to be answered to fully understand the impact of EGFR and its downstream effectors during normal cell function and in carcinogenesis. As many epithelial cancers arise through dysregulation of the stem cell self-renewal and homeostatic maintenance of the epithelium [5], understanding the precise functions of EGFR signaling in epithelial homeostasis is very important.
The Drosophila midgut is an outstanding model system to study the basis of epithelial homeostasis due to its simple structure, similarity to the mammalian intestine, and powerful genetics. As in the mammalian intestine, epithelial turnover in the fly midgut is carried out through a dynamic process mediated by intestinal stem cells (ISCs). ISCs undergo cell division to renew themselves and give rise to transient cells called enteroblasts (EBs), which can further differentiate into either absorptive enterocytes (ECs) or secretory enteroendocrine (EE) cells. When damaged or aged cells are lost from the fly’s gut epithelium, ISCs respond by dividing to replenish the epithelium [6,7,8]. During this response multiple Drosophila EGFR ligands, namely spitz (spi), vein (vn), and keren (krn) are induced in progenitor cells (EBs and ISCs), visceral muscle (VM) and ECs respectively. Thereby, the EGFR signaling pathway is activated in ISCs. This promotes ISC growth, division and midgut epithelial regeneration [9,10,11]. ISCs defective in EGFR signaling cannot grow or divide, are poorly maintained, and are unable to support midgut epithelial replenishment after enteric infection by the bacteria Pseudomonas entomophila (P.e.) [11] or Erwinia carotovora carotovora 15 (ECC15) [12]. Interestingly, the critical role of EGFR signaling in the Drosophila intestine is consistent with its role during mammalian gut homeostasis and colorectal cancer development [10,11,12,13]. EGFR signaling is required for murine ISC growth [14,15], and the deletion of Lrig1, a negative feedback regulator of EGFR signaling, causes excessive ISC proliferation [16]. Furthermore, adenoma formation in Apc min/+ mice was severely impaired in a genetic background with partial loss of function of EGFR (Egfr wa2) [17].
Despites its importance, the mechanism by which EGFR/Ras/MAPK signaling promotes ISC proliferation is poorly understood in this cell type. Indeed, despite decades of intensive study, the precise linkage between EGFR/Ras/MAPK signaling and cell growth and division is surprisingly obscure for animal cells in general [3]. Textbook models highlight a prevailing model in which EGFR/Ras signaling controls cell proliferation via a Ras-Myc-CyclinD-Rb pathway [18,19]. While this may have relevance in some human cancers it is clearly not the case in normal Drosophila cells, and so other mechanisms should be sought and characterized.
One potentially important downstream effector of EGFR signaling is the HMG-box transcriptional repressor Capicua (Cic). This highly conserved DNA binding factor has been shown to act downstream of receptor tyrosine kinase (RTK)/Ras/MAPK signaling in Drosophila eye and wing imaginal discs, embryos, and ovaries [20,21,22,23] where it regulates diverse RTK-dependent processes including cell proliferation, specification, and pattern formation. Cic orthologs from invertebrate and vertebrate species share two well-conserved regions: the HMG-box, presumed to mediate DNA binding at target promoters [21] and a C-terminal domain [24]. The C-terminal region of Drosophila Cic contains a “C1” motif important for repressor activity, and a “C2” motif that functions as a MAPK docking site responsible for downregulation of Cic following the activation of RTK signaling [25]. It has been proposed that MAPK phosphorylates Cic in its C2 motif, and that phosphorylated Cic is either degraded or re-localized to the cytoplasm [25]. Cic downregulation controlled by Torso and EGFR signaling varies in different Drosophila tissues [24]. For example, Torso RTK signaling, which also works via the Ras/Raf/MAPK pathway, apparently increases the rate of Capicua degradation by promoting its accumulation in the cytoplasm [26]. EGFR signaling has been reported to regulate Cic protein in distinct ways in different tissues. Wing and eye discs cell clones mutant for Egfr or Ras showed elevated levels of Cic protein [20,27]. In the ovary, in contrast, Cic protein localized to the cytoplasm in cells in which EGFR signaling was active, but in nuclei in cells in which EGFR signaling was inactive [25]. A recent study suggested that Cic actually undergoes a two-step process in releasing its target gene repression: slower changes in nuclear localization occur after a faster reduction of Cic repressor activity [28]. In cultured human cells, EGF stimulated dissociation of human CIC from importin-α4 (also known as KPNA3), an adaptor required for the nuclear import of many proteins. But full length GFP-CIC was nuclear even after EGF stimulation, and the N-terminal half of the CIC protein was found to be nuclear, even though it does not bind to importin-α4. Hence the biological significance of the CIC:importin association remains unclear [29].
CIC, the human homolog of Drosophila Cic, has been implicated in several human diseases including spinocerebellar ataxia type 1 (SCA1) neuropathology, oligodendroglioma (OD) [30] and Ewing-like sarcoma [31]. Human CIC is frequently mutated in samples from cancer genome studies such as The Cancer Genomic Atlas (TCGA) (S1 Fig) [32,33]. For instance CIC mutation was reported in 6 out of 7 brain tumors [30], 3 out of 11 breast cancers [34] and 6 out of 72 colorectal cancers [35]. The Drosophila work suggests that in these cases CIC loss might have the same downstream consequences (e.g. cell transformation) as oncogenic activation of the EGFR, RAS or BRAF, but this has not been rigorously evaluated.
During RNAi screening we discovered that depletion of Cic in Drosophila’s intestinal stem cells (ISCs) activates these cells for rampant proliferation [11]. Based on previous studies in other fly organs we hypothesized that Cic might act as an obligate repressor downstream of EGFR signaling, itself a central driver of normal ISC proliferation in both flies and mice, as well as in many human colorectal cancers, which are frequently mutant for RAS, BRAF, or CIC. However, until now this hypothesis had not been tested and the underlying mechanisms via which Cic might control ISC proliferation remained undefined. In this report we demonstrate that Cic acts as a critical negative downstream regulator of EGFR signaling to control ISC proliferation. We show that EGFR/Ras activity controls Cic nuclear localization, and we present RNA-Seq and DamID-Seq datasets that together constitute a genome-wide survey of potential Cic target genes in Drosophila ISCs. Our analysis indicated that Cic not only directly regulates cell cycle regulators such as string (cdc25) and Cyclin E, but also the ETS transcription factors pnt and Ets21C, all of which must be de-repressed to activate ISCs for growth and division.
Results
Cic inactivation promotes ISC proliferation
To investigate a potential role for Cic in regulating ISC proliferation, we used the esg-Gal4-UAS-2XEYFP; Su(H)GBE-Gal80, tub-Gal80 ts system (henceforth referred as esg ts ; Su(H)-Gal80) to express UAS-cic-RNAi specifically in ISCs. After 4 days of cic-RNAi induction, a dramatic increase in the number of YFP positive cells (Fig 1A and 1B) and a large increase in ISC mitoses were observed (Fig 1C). Most of the PH3+ cells were YFP+ [YFP+, PH3+ cells = 99.37% (nmidguts = 10 midguts, ncells = 994), YFP-, PH3+ cells = 0.63% (n = 10, ncells = 7)], indicating that Cic regulates ISC proliferation cell autonomously. When we used another ISC-specific driver Dl ts (tub-Gal80 ts UAS-GFP; Dl-Gal4) to knock down cic in ISCs specifically, we not only detected the same overporoliferation phenotype (S3A, S3B and S3E Fig) but also found that most of mitotic cells were GFP+ (S3F Fig).
Fig 1. cic inactivation promotes ISC proliferation and hyper-activation inhibits ISC proliferation.

(A,B) Knock down of Cic in ISCs using the esg ts ; Su(H)-Gal80 system. ISCs were marked by YFP (green). Samples were stained with anti-PH3 (red) for mitosis and DAPI (blue) for DNA. (A) Control adult midgut (B) Cic knock down midgut after 4 days induction 29°C. Increases in the number of YFP+ cells are observed in cic depleted midguts as was a large increase in mitotic cells. (C) Midguts were scored for PH3+ cells after 4 days of induction of cic-RNAi. A strong increase in numbers of ISC mitosis was observed in cic knockdown midguts. (D) Clone areas of cic mutant and control WT clones 10, 20, and 30 days after clone induction. Mutant ISCs divided faster and generated bigger clones. (E) Increased number of cells per clone was detected in cic mutant clones. Data was quantified 10 days after cic mutant clones were generated with the MARCM system. (F) Quantification of pH3-positive cells per adult midgut of the indicated genotype. cic transheterozygotes contained significantly more mitotic cells than controls. (G) Quantification of ISC proliferation after 12 hours P.e. infection. A decreased number of PH3+ cells, representing dividing ISCs, was observed in midguts overexpressing either cic or cic ΔC2 after P.e. infection. (H-J) Clones generated by the esg ts F/O system are marked with GFP (green), Cic over-expression was confirmed by anti-Cic (red) staining, and nuclei were visualized by DAPI (blue) staining. (E) Control adult midgut 12 days after clone induction (F) midgut overexpressing Cic (G) midgut overexpressing CicΔC2 12 days after clone induction. The size of clones marked by GFP was reduced after Cic or CicΔC2 overexpression. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Error bars represent standard deviations. Scale bars represent 20 μm in A-B and 50 μm in E-G.
Increased GFP+ cells and mitoses were also noticed when the esgGal4 UAS-GFP tub-Gal80 ts system (henceforth referred as esg ts) was used to express UAS-cic-RNAi in ISCs and their undifferentiated daughters, the EBs (S2A–S2B and S3 Figs). To further validate the specificity of this RNAi experiment, GFP-marked ISC clones homozygous for the loss-of-function allele cic fetU6 [22] were generated using the MARCM system [36] (S2C–S2H Fig). The size of marked ISC clones was quantified at intervals after clone induction by measuring GFP-labeled clone areas. cic mutant clones were larger than control clones at all time points assayed (Fig 1D). In addition, the numbers of cells per clone were increased in the cic mutant clones (Fig 1E). To further confirm Cic’s function in the midgut, we generated viable transheterozygotes using three different loss-of-function alleles of cic. cic fetE11 is a P-element insertion mutant, while both cic fetT6 and cic fetU6 are homozygous lethal EMS alleles [22]. In addition to the EGFR-related extra wing vein phenotype reported previously [27], these transheterozygote mutants showed increased mitoses in their midguts (Fig 1I). As the ISCs are the predominant dividing cell type in Drosophila midguts, these data further indicate a role for Cic as an obligate repressor of ISC proliferation.
To investigate the respective requirements of Cic in the ISC and EB cell types, the EB-specific driver Su(H) ts [Su(H)-Gal4,UAS-CD8-GFP; tub-Gal80 ts ] was used to knock down cic in EBs. Increased mitoses were observed after depleting cic in EBs (S3C–S3E Fig). However, in this case only a few GFP+ EBs were observed in mitosis, while most of the dividing cells marked by PH3 were GFP-negative (S3F Fig). These GFP-negative mitotic cells are likely ISCs. These data indicated that Cic has both cell autonomous and non-cell autonomous functions in regulating ISC proliferation. In this study we followed up on Cic’s cell autonomous effects on ISC proliferation, and the non-cell autonomous effect was not investigated further.
Increased Cic activity inhibits ISC proliferation and midgut epithelial regeneration
To determine whether increased Cic function yields a phenotype similar to that of EGFR loss-of-function, we generated transgenic flies harboring UAS-cic ΔC2 -HA or UAS-cic-HA. CicΔC2 is a Cic derivative carrying a deletion of the MAPK docking site-C2 motif, and has been shown to be a dominant repressor that escapes inactivation by MAPK [25]. Either cic or cic ΔC2 were over-expressed in progenitor cells using esg ts, and then the flies were fed Pseudomonas entomophila (P.e.) for 12 hours to generate an enteric infection. ISCs from control midguts, which expressed GFP only, showed regeneration-associated proliferation [8]. In contrast both cic and cic ΔC2 overexpressing midguts displayed an inhibition of regeneration after 12 hours P.e. infection (Fig 1J). To test if cic or cic ΔC2 overexpression could influence turnover of the midgut epithelium we used the esg ts F/O system (esg-Gal4; tubGal80 ts Act>Cd2>Gal4 UAS-flp UAS-GFP) [11] to mark all the ISC progeny produced during 12 days of cic overexpression. Normally, the posterior midgut epithelium renews it self within about 12 days [8]. Therefore, control midgut epithelia were almost completely replaced by large GFP+ clones that formed during 12 days. However, in the gain-of-function Cic conditions, growth of GFP-marked clones was significantly decreased, indicating that gut epithelial renewal was greatly suppressed (Fig 1H–1J).
Cic regulates ISC proliferation as a downstream effector of EGFR/Ras signaling
EGFR activates ISCs for growth and division via Ras/Raf/MAPK signaling. When an activated form of the EGFR (λTOP) [37] or activated Ras (Ras V12G) [38] is ectopically expressed in progenitor cells, ISC division is dramatically induced. Conversely, EGFR suppression by inducing Egfr-RNAi, Ras-RNAi, or MEK-RNAi in progenitor cells almost completely inhibits ISC division and growth [11,12]. Furthermore, inhibition of EGFR signaling suppresses the activation of ISC divisions after P.e. infection [10,11]. As demonstrated above, Cic knockdown and overexpression phenocopy these EGFR overexpression or knockdown phenotypes, respectively, suggesting that Cic may act as a downstream effector in the EGFR signaling in ISCs.
To test the function of Cic in EGFR signaling we performed epistasis tests. After 2 days of clone induction with the esg ts F/O system, control midguts generated only 2-cell clones, whereas clones overexpressing an activated variant of the EGFR, (λtop), grew very large and showed increased ISC division. However, when cic or cic ΔC2 was co-overexpressed along with λtop, clone sizes and ISC mitoses were significantly reduced (Fig 2A–2C and 2I). Overexpression of Cic or CicΔC2 could also partially inhibit the ISC growth effects of Ras V12S35, an activated allele that can activate RAF/MAPK signaling but not PI3K signaling [38] (Fig 2D, 2H, and 2J). Furthermore, we used esg ts to induce Egfr-RNAi, or Ras-RNAi in combination with cic-RNAi. The cic, Egfr or cic, Ras double RNAi animals exhibited increased ISC mitosis relative to controls expressing Ras-RNAi or Egfr-RNAi only (Fig 2E–2G and 2K), indicating that reduced ISC proliferation caused by the inactivation of EGFR signaling can be restored by Cic knock-down. These epistasis data further support the hypothesis that Cic acts as a negative downstream effector of EGFR to regulate ISC proliferation.
Fig 2. Cic regulates ISC proliferation as a downstream effector of EGFR signaling.

(A–C) Results of the λtop and cic epistasis tests, carried out using the esg ts F/O system, to co-express the indicated transgenes with GFP for 2 days at 29°C. (A) Control of adult midgut (B) λtop overexpresssing midgut (C) λtop and cic ΔC2 co-overexpresssing midgut. GFP+ clones (green) expressing λtop were much smaller when cic ΔC2 was co-overexpressed. Samples stained with anti-PH3 (red) and DAPI (blue). (D-F) Results of the epistasis test between cic and egfr, carried out using the esg ts system to express the indicated transgenes for 4 days at 29°C. (D) Control adult midgut, (E) Egfr-RNAi expressing midgut, (F) Egfr-RNAi and cic-RNAi co-expressing midgut. The number of GFP+ cells (green) still promoted by depleting cic in EGFR/Ras inactivated background. Samples were stained with anti-PH3 (red) and DAPI to visualize nuclei. (G-H) Results of epistasis tests between Ras V12S35 and cic, carried out using the esg ts F/O system. The transgenes were induced for 2 days at 29°C (G) Ras V12S35 over-expressing midgut (H) Ras V12S35 and cic ΔC2 co-over expressed midgut. Size of GFP+ clones (green) in Ras V12S35 and cic ΔC2 co-overexpressing midgut was significantly reduced. Samples were stained with anti-PH3 (red) and DAPI to visualize nuclei. (I-K) ISC mitoses as quantified by scoring PH3+ cells. (I) Quantification of ISCs mitoses for the λtop and cic epistasis test. The increase in mitoses induced by λtop was completely suppressed by cic or cic ΔC2 over expression. (J) Quantification of ISC mitoses from Ras V12S35/cic epistasis tests. The increase in mitosis induced by Ras V12S35 was partially suppressed by cic or cic ΔC2 over expression. (K) Quantification of ISCs mitosis in cic and either Egfr or Ras double knock down midguts. The increase in ISC mitoses induced by cic-RNAi is still observed when either Egfr or Ras RNAi is also expressed. Error bars represent standard deviations. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Scale bars represent 50μm (A-H).
EGFR signaling controls Cic subcellular localization
To understand how EGFR signaling controls Cic in ISCs, we expressed HA-tagged Cic or CicΔC2 protein in midgut progenitor cells (ISCs and EBs). As expected, HA-tagged Cic or CicΔC2 proteins were only detected in nuclei under normal conditions (Fig 3A–3A’ and 3B–3B’). However, HA-tagged Cic protein accumulated nearly exclusively in the cytoplasm when Ras V12S35 was co-expressed with it (Fig 3E–3E’). In contrast, CicΔC2 remained in the nucleus even following ectopic Ras V12S35 expression (Fig 3F–3F’). A similar but milder re-localization of Cic protein from the nucleus to the cytoplasm was observed following P.e. infection (Fig 3C and 3C’), a treatment known to increase MAPK signaling in the gut [11]. It is interesting to note that CicΔC2 did not completely suppress Ras V12S35 induced ISC proliferation, even though it remained localized to nuclei in Ras V12S35 expressing cells (Fig 2H and 2J). However, nuclear CicΔC2 lost its characteristic punctate localization in the presence of Ras V12S35 expression, and became more diffusely localized in the nucleoplasm (Fig 3F–3F’). These results suggest that, although EGFR signaling controls Cic nucleo-cytoplamic localization via the C2 motif, there may be a second MAPK-dependent mechanism to regulate Cic repressor activity, involving dissociation from chromatin, that is C2-independent.
Fig 3. EGFR signaling controls Cic subcellular localization in ISCs.

(A, B) Cic and CicΔC2 localized in the nuclei. Transgene expression was induced using the esg ts F/O system at 29°C for 2 days. HA-tagged Cic or CicΔC2 protein was detected by anti-HA antibody (red). Nuclear DNA is marked by DAPI staining (blue) (A) cic-HA overexpressing midgut. (B) cic ΔC2 -HA overexpressing midgut. (C, D) Cic but not CicΔC2 protein accumulated in the cytoplasm after P.e. infection. (C) cic-HA overexpressing midgut, exposed to P.e. bacteria for 16 hours. (D) cic ΔC2 -HA overexpressing midgut after 16 hours P.e. infection. (E, F) Cic protein accumulated in the cytoplasm when EGFR signaling was activated by Ras V12S35. (E) Ras V12S35 and cic-HA overexpressing midgut. (F) Ras V12S35 and cic ΔC2 -HA overexpressing midgut. Cic ΔC2 proteins stayed in the nucleus even after overexpressing Ras V12S35 to activate MAPK signaling. Scale bars represent 5μm.
Cic represses cell cycle genes in ISCs
Cic has been studied in several cell types from both Drosophila and humans. In human melanoma cells, CIC represses mRNA expression of the PEA3 subfamily of ETS transcription factors, namely ETV1, ETV4 and ETV5 [29]. In early Drosophila development post-transcriptional down-regulation of Cic by the Torso and EGFR pathways regulates terminal and dorsal-ventral patterning, respectively, by allowing expression of Cic target genes such as huckebein (hkb), intermediate neuroblasts defective (ind), and argos (aos) [39]. However, a genome-wide mapping of Cic target genes has not yet been reported.
To identify Cic target genes involved in ISC growth and proliferation we profiled Cic binding throughout the genome using the “TaDa” (Targeted DamID)” technique. The TaDa method involves low-level expression of a GAL4-inducible Dam methylase-fusion protein in a specific cell type, enabling cell-specific profiling without cell isolation [40,41]. Here, we induced a low level of Dam-only or Dam-Cic fusion protein in progenitor cells (ISC & EB) using the esg ts system and a 24-hour induction. Genomic DNA was extracted from isolated midguts, digested with Dpn I, which cuts only methylated GATCs, and amplified. The amplified gDNA fragments were subjected to high-throughput sequencing, rather than tiling microarrays as previously reported [40]. We identified 2279 binding sites that were highly enriched (log2 fold change > 3, false discovery rate<0.01%) when comparing Dam-Cic to Dam alone samples (S1 Table). These sites were non-randomly distributed in the genome, and were significantly over-represented ~500 bp 5’ to Transcription Start Sites (TSS; Fig 4A). Cic DamID was also performed on progenitor cells from P.e. infected midguts. After a 24 hours induction of Dam or Dam-Cic transgenes via the esg ts system, flies were fed P.e. bacteria for 16 hours. The number of highly enriched (log2 fold change > 3, FDR < 0.1%) peaks was reduced to 1903. In addition, the fold change of peaks (Dam-Cic vs Dam-alone) after P.e. infection was significantly decreased (Figs 4B–4C and S4A). The frequency of peaks adjacent to TSS was also significantly reduced in the P.e.-infected midgut sample (Fig 4A). We believe that this decrease was due to the change of Cic localization from the nucleus to cytoplasm, which was caused by the activation of EGFR/Ras/MAPK signaling after infection.
Fig 4. Cic targets genes in ISCs found by DamID-Seq.

(A) Graph showing the location of Cic binding relative to annotated transcript TSSs. The distance is from the summit of the Cic peaks to the nearest TSS. Dashed red line showed the summit of the graph is 500bp away from TSS. (B) Box plot showing fold change of peaks in CicDamID and P.e. infected CicDam. (C) Heatmap showing fold enrichment of Cic peaks from Cic DamID-Seq without or with P.e. infection. Y axis represents genes associated with the Cic binding peaks. (D) Expression heatmap of cell cycle regulators and DNA replication related genes from RNA-Seq data from cic-RNAi expressing FACS sorted progenitor cells. The names of the genes that had Cic binding sites by DamID are written in green. (E) Venn diagram showing the overlap between genes upregulated > 1.5 fold upon cic-RNAi (left) and genes associated with Cic binding peaks (right) in ISC/EBs. (F) Graph showing correlation between genes upregulated in Cic-depleted progenitor cells, and the Cic-DamID peaks that changed significantly upon P.e. infection (upper panel). Lower panel show genes ranked by absolute expression change, and then plotted for expression fold change (bottom). (G, H) Cic binding sites in the CycE and stg loci, as determined by Cic DamID-Seq in ISC/EBs from control (above) and P.e. infected (below) midguts. Vertical bars represent the log2 ratio of the Dam-fusion signal to the Dam-only signal. Red arrows indicate TGAATG(G/A)A motifs. (I-K) mRNA level fold changes of stg and CycE analyzed by qRT-PCR and normalized to β-Tub and Rp49. (I) stg and CycE fold enrichment from whole midguts after knocking down or over expressing cic in all cells using the tub ts (tubGal4; tubGal80ts) driver. Transcription of both stg and CycE was induced in cic knock-down midguts and inhibitied in cic over-expressing midgut. (J) stg and CycE expression is upregulated in cic-depleted, FACS-sorted progenitor cells (ISC &EB) and ISCs. (K) stg and CycE expression fold change in cic over expressing midguts after P.e. infection. The induction of stg and CycE by P.e. infection was suppressed by cic ΔC2 overexpression. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Error bars in each graph represent standard deviations.
To further understand how Cic regulates ISC proliferation we performed gene expression profiling using amplified mRNA from FACS-sorted esg+ progenitor cells that expressed cic-RNAi, and controls. As a way to identify potentially direct target genes of Cic, the RNA-Seq and DamID-Seq data sets were cross-compared. Amongst 439 transcriptionally up-regulated genes (>1.5 fold change, 90% CI) (S2 Table), a large fraction [134 genes, (S3 Table)] had Cic binding sites as defined by DamID (Fig 4E). We next examined the enrichment of the DamID peaks in the transcriptionally induced genes, ranked by absolute expression change in cic knockdown progenitor cells (see Materials and Methods). Cic binding peaks that were significantly reduced upon P.e. infection (< 2 fold change) were enriched in up-regulated genes from the RNA-Seq dataset (Fig 4F). Hence, the set of genes present in the overlapping set are likely to be direct target genes of Cic. Many cell cycle regulators and genes involved in DNA replication were upregulated in Cic-depleted progenitor cells (Fig 4D). In addition, a large portion of cell cycle control genes that were upregulated upon cic-RNAi, including string (stg, Cdc25) and Cyclin E (CycE), had Cic binding sites (Fig 4D, 4G, and 4H). To further assess the reliability of this approach we examined the occupancy of Cic on its previously characterized direct target gene-aos [39]. Our DamID-Seq data showed that aos contained two Cic binding sites within its enhancer, and that their occupancy was significantly reduced after P.e. infection (S4B Fig). The significant induction of aos transcription was verified both by RNA-Seq and qRT-PCR data from FACS-sorted progenitor cells expressing cic-RNAi (S4C Fig).
Having confirmed the reliability of our approach for identifying genes that are repressed by Cic in ISCs, we focused on genes likely to contribute to ISC proliferation. We were interested in stg and CycE because they are transcriptionally induced in proliferating ISCs [42], required for ISC divisions, and also sufficient to induce sustained ISC division when co-overexpresssed [42]. To further test whether Cic regulates the transcription of stg and CycE we measured their normalized expression ratios in gain- or loss-of-function Cic midguts via RT-qPCR (Fig 4I–4K). The stg and CycE mRNAs were significantly increased in Cic-depleted midguts, and decreased in midguts expressing the dominant active CicΔC2. Strong inductions of stg and CycE were also observed in Cic-depleted progenitor cells or ISCs purified using FACS (Fig 4J). Moreover, both the stg and CycE loci had multiple strong Cic-Dam-ID binding peaks containing TGAATG(G/A)A motifs, and binding these peaks were reduced by P.e. infection (Fig 4G and 4H). Consistently, the induction of stg and CycE transcription upon P.e. infection was significantly repressed by CicΔC2 overexpression (Fig 4K). These data support the notion that Cic controls ISC cell cycle progression by directly repressing transcription of stg and CycE via binding sites in their regulatory regions.
Cic represses pnt and Ets21C
It has been suggested that Cic might regulate the transcription of certain members in a subfamily of ETS transcription factors [29,31]. Consistent with this, we identified the Drosophila ETS transcription factors pnt and Ets21C as potential Cic direct target genes by both RNA-Seq and DamID-Seq (Figs 5 and S5). These genes contain Cic binding sites, were highly expressed in midgut progenitor cells, and were significantly induced upon infection or cic depletion or mutation. Notably, induction of pnt and Ets21C was detected in FACS-sorted ISCs depleted of Cic (Fig 5B). Moreover, the induction of pnt and Ets21C expression by P.e. infection was suppressed when the dominant active form, CicΔC2 was overexpressed (Fig 5D). Similar effects were observed when Cic was either depleted or overexpressed in whole midgut samples (Figs 5C and S5C). These data suggest that Cic also regulates pnt and Ets21C transcription in Drosophila midgut ISCs, by directly binding to these loci. As in the case of stg and CycE, this regulation appeared to be modulated by P.e. infection, most likely in a MAPK-dependent manner.
Fig 5. Cic regulates Pnt expression through binding to its genomic locus.

(A) Heatmap of mRNA levels indicating RPKM values from RNA-Seq data from control and cic-RNAi expressing, FACS-sorted progenitor cells. (B-D) Relative expression of pnt, pntP1, pntP2 and Ets21c as analyzed by qRT-PCR and normalized to β-Tub and Rp49. (B) Fold change of expression from the cic depleted FACS-sorted progenitor cells and ISCs. (C) pnt, pntP1, pntP2 and Ets21C were upregulated in cic knock down midguts and downregulated in cic overexpressing midguts. (D) Expression change in cic overexpressed midgut after P.e. infection. Error bars represent standard deviation. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). (E) Cic binding sites in the pnt locus, as determined by Cic DamID-Seq of esg+ progenitor cells from control and P.e. infected midguts. Vertical bars represent the log2 ratio of the Dam-fusion signal to the Dam-only signal. Peaks also found in Cic ChIP-Seq from embryos are marked by asterisks. Positions of EMSA probes from the pnt locus are indicated by blue bars. (F) Diagram of probes containing TGAATGAA sites. These sites were replaced with other sequences in probes 1 and 2 to generate probes 1mut and 2mut. (G) DNA binding of Cic and HMG-box mutated Cic to probe 1 or 1mut (left panel). DNA binding of Cic and HMG-box mutated Cic to probe 2 or probe 2mut (right panel). FP indicates “free probes.”
Cic represses pnt via a TGAATGAA motif
The HMG box of Human Cic binds to TGAATG(G/A)A octamers in vitro [31]. This motif was also verified as a Cic binding sequence in several Cic target genes in Drosophila embryos and wing discs [39]. Notably, the TGAATG(G/A)A motif was observed in 692/2279 Cic binding sites in our DamID-Seq dataset (p-value = 3.045475× 10−11). Each of the four Cic target genes discussed above contained more than one TGAATG(G/A)A motifs in its Cic binding sites. Moreover, TGAATGAA motifs found in the pnt locus also mapped to Cic binding sites that we determined from Drosophila embryo ChIP-Seq (Fig 5E). This suggests that Cic may bind to the pnt locus via TGAATGAA octamers, and that the occupancy of Cic at the pnt locus may also be conserved in different Drosophila cell types. To further evaluate this hypothesis we performed electrophoretic mobility shift assay (EMSA). Cic showed specific binding to two DNA fragments from the pnt locus that were identified as prominent in vivo Cic binding peaks by DamID-Seq and ChiP-Seq (Fig 5F and 5G). Importantly, the EMSA interaction was lost when the HMG-box in Cic was mutated, or when the TGAATGAA motifs were mutated. These data strongly support the idea that Cic directly regulates pnt transcription by directly binding to TGAATGAA motif in pnt locus.
Pnt regulates ISC proliferation as a direct target of Cic
Pnt is believed to be a downstream effector of EGFR signaling in developing Drosophila eyes [43,44,45]. The pnt locus produces two alternative transcripts that encode two different protein isoforms: PNTP1 and PNTP2 [44]. PNTP1 was proposed to be a constitutive activator of transcription, whereas PNTP2 has a PNT (pointed) domain that was reported to be phosphorylated by MAP kinase in vitro [45]. The mutant protein, PNTP2T151A, which cannot be phosphorylated in vitro, was unable to rescue pnt phenotype in eyes but instead enhanced the mutant phenotype, suggesting that the PNT domain is an auto-inhibitory domain that can be inactivated by MAPK-dependent phosphorylation [45]. Furthermore PNTP2 is thought to induce transcription of PNTP1, which might thereby encode the final nuclear effector of the EGFR pathway in eye discs [43]. In the midgut, we found an interesting interaction between Pnt and Cic: pntP1 and pntP2 were both induced when Cic was depleted, and both decreased when Cic was overexpressed (Figs 5B, 5C, and S5A). The expression of transcripts encoding both isoforms was also increased in P.e. infected guts (Figs 5D and S5B). This raises the possibility that pnt might be an important downstream effector of Cic in controlling ISC proliferation. To test this we over-expressed either pntP1 or pntP2 in progenitor cells using the esg ts or Dl ts driver systems. After 4 days of transgene induction a dramatic increase in ISC division was evident in response to either pntP1 or pntP2 (Figs 6A–6B, 6I, and S6A–S6B). Conversely, mutant clones that were generated using a pnt null allele (pnt Δ88) [46] did not grow past the 2-cell stage (S6D Fig). Moreover, when we depleted pnt in progenitor cells by expressing a pnt-RNAi that recognizes both isoforms, or generated homozygous pnt null mutant ISCs via MARCM, ISC proliferation after P.e. infection was suppressed (Figs 6C–6D, 6J, and S6E). Next, we investigated the functional significance of the inhibition of pnt expression by Cic. Whereas loss of Cic function induced massive ISC proliferation, inhibiting both isoforms of pnt in this context suppressed this over-proliferation (Figs 6G–6H, 6K and S6F–S6G). Conversely, when we over-expressed either pntP1 or pntP2 in ISCs that also overexpressed CicΔC2, the inhibitory effect of CicΔC2 on proliferation was bypassed and the cells divided (Figs 6F, 6L and S6C). Hence, a significant fraction of the ISC over proliferation caused by Cic knockdown can be attributed to Cic’s effects on pntP1 and pntP2
Fig 6. Cic controls ISC proliferation by regulating pnt transcription.

(A–E) Effects caused by pntP1 overexpression and RNAi’s. Transgenes were induced using the esg ts system at 29°C for 4 days, and samples were stained for GFP (green), DNA (blue) and mitoses (PH3, red). White arrows pointing out PH3 signals. (A) Control adult midgut. (B) pntP1 overexpressing midgut after 4 days induction at 29°C. (C) Control midgut after 12 hours P.e. infection. (D) pnt knockdown midgut after 12 hours P.e. infection. Fewer GFP+ and PH3+ cells are observed. (E) Ets21C overexpressing midgut, showing more PH3+ ISCs (arrows) and GFP+ ISCs and EBs (green). (F-H) Ectopic expression or loss of pnt bypasses ISC phenotypes caused by cic overexpression or depletion. (F) pnt and cic ΔC2 co-over-expressing midgut after 4 days induction at 29°C. GFP+ progenitor cells were still able to proliferate. (G) cic knockdown adult midgut and (H) pnt, cic double knockdown midgut. The increased number of progenitor cells marked by GFP upon cic knockdown was decreased by also knocking down pnt. (I-L) Quantification of PH3+ cells in adult midguts of the indicated genotypes. (I) pntP1, pntP2 or Ets21C overexpression driven by esg ts or Dl ts. All the pntP1, pntP2 and Ets21C overexpressing midguts contained more dividing ISCs. (J) pnt or Ets21C knockdown midguts after P.e. infection. ISC mitoses caused by P.e. infection were reduced in pnt or Ets21C knockdown midguts. (K) pnt and cic knock down using Dl ts system. Fewer mitotic ISCs were observed in the pnt and cic double knockdown midgut than the cic knockdown midgut. (L) pnt and cic ΔC2 co-overexpressing midguts. cic ΔC2 overexpression could not inhibit ISC mitoses caused by pnt overexpression. (M) Quantification of PH3+ cells from adult midguts following P.e. infection. MEK-RNAi completely blocked infection-driven ISC mitoses, but could not inhibit ISC proliferation driven by overexpressed Ets21c. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Error bars in each graph represent standard deviation. Scale bars represent 50μm.
Interestingly, mutant clones generated using a pntP1 specific mutant allele, pnt Δ33 [45,47], or a pntP2 specific mutant allele, pnt Δ78 [45,47], grew normally. However ISCs mutant for the pnt null allele pnt Δ88 did not expand (S6D Fig). In addition, pnt Δ33 and pnt Δ78 homozygous clones in which cic was depleted by RNAi had similar numbers of cells to cic-depleted control clones (i.e. they overgrew), whereas pnt Δ88 null mutant clones contained significantly fewer cells (S6G Fig). These data not only support our conclusion that pnt is required for ISC proliferation as a target of Cic, but show that PNTP1 and PNTP2 have redundant function in regulating ISC proliferation. Furthermore, pntP2 homozygous mutant ISCs did not appear to have any defect in proliferation upon P.e. infection (S6E Fig). Overall these results indicate that pntP2, the isoform proposed to be activated directly by MAKP phosphorylation [45], is not specifically required in ISC proliferation.
Pnt is the Drosophila ortholog of the human ETS2 transcription factor and has a conserved ETS-type DNA binding domain, while Ets21C is the Drosophila ortholog of the human proto-oncogene ERG. In addition to having Cic binding sites, RT-PCR and RNA-Seq data showed that Ets21C was highly induced upon P.e. infection (Figs 5A and S5C). Moreover RNAi mediated depletion experiments indicated that Ets21C was also required for ISC proliferation in response to P.e. infection (Fig 6J). Over-expression of Ets21C caused a strong increase of ISC division (Fig 6E and 6I) suggesting that transcriptional induction of Ets21C could promote ISC proliferation. Furthermore, ectopic expression of Ets21C in progenitor cells could bypass the strong growth-suppressive effect of depleting MEK (Fig 6M). These data indicated that Cic controls ISC proliferation in part by regulating Ets21C transcription.
Finally, we tested whether Yan, an inhibitory ETS type transcription factor, reported to be MAPK responsive and to compete with Pointed for binding to common sites on the DNA [45,48,49], had an opposite function in ISCs. Although yan mRNA is expressed in the midgut (Fig 5A), yan depletion from ISCs did not produce a detectable effect (S6F Fig). Two independent yan-RNAi lines were used, both of which were proven to be effective by qRT-PCR (S6H Fig). In summary these observations suggest that EGFR signaling controls ISC growth and division by regulating the activity of Cic, Pnt and Ets21C but not Yan, and that Cic directly represses pntP1, pntP2 and Ets21C in this context.
Discussion
It is well established that EGFR signaling is essential to drive ISC growth and division in the fly midgut [10,11,12]. However, the precise mechanism via which this signal transduction pathway activates ISCs has remained a matter of inference from experiments with other cell types. Moreover, despite a vast literature on the pathway and ubiquitous coverage in molecular biology textbooks, the mechanisms of action of the pathway downstream of the MAPK are not well understood for any cell type. From this study, we propose a model summarized in Fig 7. Multiple EGFR ligands and Rhomboid proteases are induced in the midgut upon epithelial damage, which results in the activation of the EGFR, RAS, RAF, MEK, and MAPK in ISCs. MAPK phosphorylates Cic in the nucleus, which causes it to dissociate from regulatory sites on its target genes and also translocate to the cytoplasm. This allows the de-repression of target genes, which may then be activated for transcription by factors already present in the ISCs. The critical Cic target genes we identified include the cell cycle regulators stg (Cdc25) and Cyclin E, which in combination are sufficient to drive dormant ISCs through S and M phases, and pnt and Ets21C, ETS-type transcriptional activators that are required and sufficient for ISC activation.
Fig 7. Model for Cic control of Drosophila ISC proliferation.
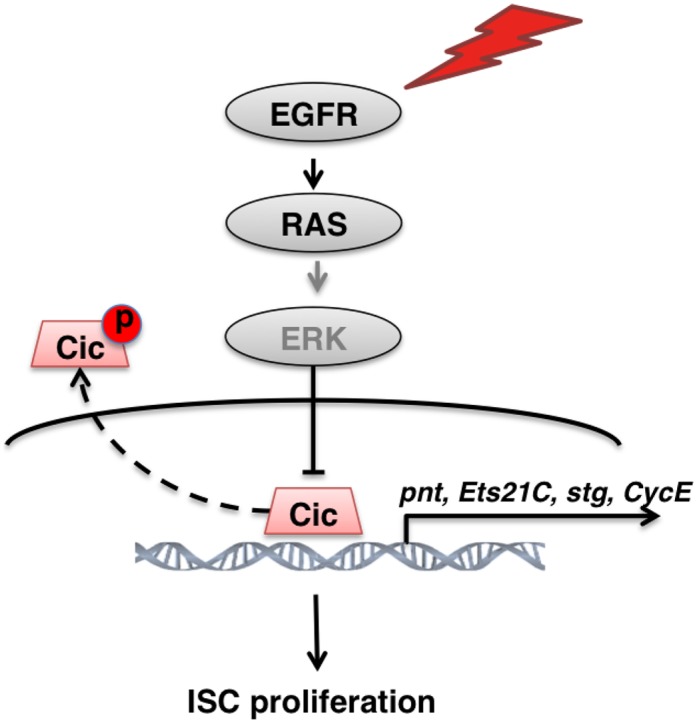
Upon damage, activated EGFR signaling mediates activation of ERK, which phosphorylates Cic, and relocates it to the cytoplasm. As a result, stg, CycE, Ets21C and pnt transcription are relieved from Cic repression, and induce ISC proliferation.
Although we found more than 2000 Cic binding sites in the ISC genome, not all of the genes associated with these sites were significantly upregulated, as assayed by RNA-Seq, upon Cic depletion. One possible explanation for this is that Cic binding sites from DamID-Seq were also associated with other types of transcription units (miRNAs, snRNAs, tRNAs, rRNAs, lncRNAs) that were not scored for activation by our RNA-Seq analysis. Indeed a survey of the Cic binding site distributions suggests this (S5 Table). This might classify some binding sites as non-mRNA-associated. However, it is also possible that many Cic target genes may require activating transcription factors that are not expressed in ISCs. Such genes might not be strongly de-repressed in the gut upon Cic depletion.
In other Drosophila cells MAPK phosphorylation is thought to directly inactivate the ETS domain repressor Yan, and to directly activate the ETS domain transcriptional activator Pointed P2 (PNTP2) [45,50]. In fact Pnt and Yan have been shown to compete for common DNA binding sites on their target genes [45,48,49]. Thus, previous studies proposed a model of transcriptional control by MAPK based solely on post-translational control of the activity of these ETS factors. However, we found that Pnt and Ets21C were transcriptionally induced by MAPK signaling, and could activate ISCs if overexpressed, and that depleting yan or pntP2 had no detectable proliferation phenotype. In addition, overexpression of PNTP2 was sufficient to trigger ISC proliferation, suggesting either that basal MAPK activity is sufficient for its post-translational activation, or that PNTP2 phosphorylation is not obligatory for activity. Moreover, pntP2 loss of function mutant ISC clones had no deficiency in growth (S6D Fig) even after inducing proliferation by P.e. infection, which increases MAPK signaling (S6E Fig). These observations indicate that the direct MAPK→PNTP2 phospho-activation pathway is not uniquely or specifically required for ISC proliferation. Or results suggest instead that transcriptional activation of pnt and Ets21c via MAPK-dependent loss of Cic-mediated repression is the predominant mode of downstream regulation by MAPK in midgut ISCs.
In addition to activating ISCs for division, EGFR signaling activates them for growth. Previous studies showed loss of EGFR signaling prevented ISC growth and division, and that ectopic Ras V12 expression could accelerate the growth not only of ISCs but also post-mitotic enteroblasts [11]. Similarly, our study shows that loss of cic caused ISC clones to grow faster than controls, by increasing cell number as well as cell size (Figs 1H and S2C–S2H). For instance, increased size of GFP+ ISCs and EBs was observed when cic-RNAi was induced by the esg ts or esg ts F/O systems (Figs 1B, 6G and S2B). Therefore, in our search for Cic target genes we specifically checked probable growth regulatory genes such as Myc, Cyclin D, the Insulin/TOR components InR, PI3K, S6K and Rheb, Hpo pathway components, and the loci encoding rRNA, tRNAs and snRNAs. We found that Cic bound to the InR, Akt1, Rheb, Src42A and Yki loci. However, of these only InR mRNA was significantly upregulated in Cic-depleted progenitor cells (S4 Table). In surveying the non-protein coding genome, we found that Cic had binding sites in many loci encoding tRNA, snRNA, snoRNA and other non-coding RNAs (S5 Table), though not in the 28S rRNA or 5S rRNA genes (S4 Table). Due to the method we used for RNA-Seq library production, our RNA expression profiling experiments could not detect expression of these loci, and so it remains to be tested whether Cic may regulate some of those non-coding RNA’s transcription to control cell growth. It is also possible that Cic controls cell growth regulatory target genes indirectly, for instance via its targets Ets21C and Pnt, which are also strong growth promoters in the midgut (Figs 6A–6B, 6E and S6A–S6B). But given that no conclusive model can be drawn from our data regarding how Cic restrains growth, one must consider the possibility that ERK signaling stimulates cell growth via non-transcriptional mechanisms, as proposed by several studies [51,52,53,54].
The critical role of Cic as a negative regulator of cell proliferation in the fly midgut is consistent with its tumor suppressor function in mammalian cancer development (S1 Fig). Also consistent with our findings are the observations that the ETS transcription factors ETV1 and ETV5 are upregulated in sarcomas that express CIC-DUX, an oncogenic fusion protein that functions as a transcriptional activator [31], and that knockdown of CIC induces the transcription of ETV1, ETV4 and ETV5 in melanoma cells [29]. Moreover the transcriptional regulation by ETS transcription factors is important in human cancer development (S7 Fig). Their expression is induced in many tumors and cancer cell lines. For example, ERG, ETV1, and ETV4 can be upregulated in prostrate cancers [55], and ETV1 is upregulated in post gastrointestinal stromal tumors [56] and in more than 40% of melanomas [57]. In addition, the mRNA expression of these ETS genes was correlated with ERK activity in melanoma and colon cancer cell lines with activating mutations in BRAF (V600E), such that their expression decreased upon MEK inhibitor treatment [58]. Furthermore, overexpression of the oncogenic ETS proteins ERG or ETV1 in normal prostate cells can activate a Ras/MAPK-dependent gene expression program in the absence of ERK activation [59]. These cancer studies imply that there is an unknown factor that links Ras/Mapk activity to the expression of ETS factors, and that some of the human ETS factors might act without MAPK phosphorylation, as does Drosophila PntP1. Combining our knowledge of Cic with what was previously known about CIC in tumor development, we propose that CIC is the unknown factor that regulates ETS transcription factors in Ras/MAKP-activated human tumors.
In summary, our study has elucidated a mechanism wherein Cic controls the expression of the cell cycle regulators stg (Cdc25) and Cyclin E, along with the Ets transcription factor Pnt, and perhaps also Ets21C, by directly binding to regulatory sites in their promoters and introns. Using genetic tests we show that these interactions are meaningful for regulating stem cell proliferation. Therefore, we suggest that human CIC may also lead to the transcriptional induction of cell cycle genes and ETS transcription factors in RAS/MAPK activated- or loss-of-function-CIC tumors such as brain or colorectal cancers.
Materials and Methods
Drosophila stocks and transgenes
esg ts: esg-Gal4/Cyo; tubGal80 ts UAS-GFP/TM6B [60]
esg ts F/O: esg-Gal4 tubGal80 ts UAS-GFP/Cyo;UASflp>CD2>Gal4/TM6B [8]
Tub ts: tub-Gal4; tubGal80 ts /TM3, ser [61](provided from Valeria Cavaliere lab)
esg ts ; Su(H)-Gal80: esg-Gal4-UAS-2XEYFP; Su(H)GBE- Gal80, tub-Gal80 ts (Gift from Steven Hou’s lab)
UAS-λTOP/FM7 [37]
UAS-RAS v12s35 [38]
UAS-Ras RNAi [11]
UAS-Egfr RNAi [11]
UAS-cic-RNAi/Cyo (VDRC KK103805)
UAS-cic-RNAi/Cyo (VDRC KK103012)
UAS-pnt.P1 (Bloomington Drosophila Stock Center 869)
UAS-pnt.P2 (Bloomington Drosophila Stock Center 399)
UAS-pnt-RNAi (Bloomington Drosophila Stock Center 31936)
UAS-pnt-RNAi (Bloomington Drosophila Stock Center 35808)
UAS-yan-RNAi (Bloomington Drosophila Stock Center 26759)
UAS-yan-RNAi (Bloomington Drosophila Stock Center 34909)
UAS-yan-RNAi (Bloomington Drosophila Stock Center 35404)
UAS-Ets21C-RNAi (VDRC KK103211)
FRT82B cic fetu6 / TM3, Sb, Se (gift from Jimenez lab, Barcelona)
w; cic fetT6 /TM3, Ser (gift from Nilson lab, Canada)
w; cic fetE11 / TM6b (gift from Nilson lab, Canada)
w; +; UAS-cic-HA
w; UAS-cic-HA; +
w; +;UAS-cic ΔC2-HA
w; UAS-cic ΔC2-HA; +
FRT82B pnt Δ33 [45,47] (gift from Joseph Bateman lab, Wolfson Centre for Age-Related Diseases)
FRT82B pnt Δ78 [45,47] (gift from Joseph Bateman lab, Wolfson Centre for Age-Related Diseases)
FRT82B pnt Δ88[45,47] (gift from Joseph Bateman lab, Wolfson Centre for Age-Related Diseases)
Generation of transgenic flies
The cic ΔC2 was amplified from the pCasper4—cic ΔC2 plasmid. The cic or cic ΔC2 cDNAs were inserted into pUASg-attB-HA [62] vector and used to generate transgenic flies. To generate UAS-cicDam transgenic flies, Cic was amplified from a cDNA library prepared from midgut. This cic cDNA was inserted into the pUASTattB-LT3-NDam plasmid (from Andrea brand lab), and transgenics were produced.
Ectopic expression
Ectopic expression of transgenes in the adult midgut was achieved using the temperature sensitive inducible UAS-Gal4 system [63], TARGET. Crosses were set up and maintained at 18°C, the permissive temperature. 3–7 day old flies were shifted to 29°C for different times as indicated.
Bacterial infection
Gut infections were performed by feeding flies live P.e. in 5% sucrose on Whatman filter paper and yeast paste at 29°C.
Clonal analysis
The MARCM system was used to generate ISC clones. In order to reduce heat shock dependent stress, the clones were induced by heat shocking 3–5 days old flies at 34°C for 20 minutes. The heat shocked flies were then kept at 25°C. Clone size was measured after 10, 20, 30 days of clone induction. The size of the clones was quantified by Fiji software measuring GFP+ area from z-projected confocal microscopy images.
Immunohistochemistry and microscopy
Female adult flies were dissected in 1×PBS. Midguts were fixed in 1×PBS with 4% paraformaldehyde for 30 minutes at room temperature. Samples were washed in PBS with 0.1% X-100 (PBST) for 3x10 minutes each. Then the tissues were blocked in PBS with 0.1% X-100, 2.5%BSA, 10% NGS for at least 30 min at room temperature. All samples were incubated with primary antibody overnight at the following dilutions: rat anti-HA (1:200; Roche), guinea pig anti-Cic (1:1000, generated by author), rabbit anti-PH3 (1:1000, Millipore). After washing 3 times 10 minutes each in PBST, samples were incubated with secondary antibodies for at least 2 hours at room temperature at a dilution of 1:1000. DNA was visualized with DAPI (0.1mg/ml, Sigma), diluted 1:200. Images of Figs 1A–1B and 2E–2H were acquired by Delta vision microscope and the rest of the fluorescence images were taken by Leica SP5 confocal microscope. Images were then processed using Fiiji and Adobe Photoshop software.
RT-qPCR
RNA was extracted from 10–12 female midguts using the RNAeasy kit (QIAGEN). RNA isolation from sorted cells was performed as previously described [64] and 100ng RNA (non-amplifed) used for reverse transcription. cDNA was synthesized by QuantiTect reverse transcription kit (QIAGEN). RT-qPCR was performed on a Light Cycler 480 II using SYBR Green I (Roche). Experiments were performed in biological triplicate. Relative fold differences in expression level of target genes were calculated as ratios to the mean of the reference genes rp49 [65] and tubulin [23]. Primer sequences are given in Supplementary Material and Methods.
RNA-Seq and data analysis
RNA isolation and amplification from sorted cells was performed as previously described [64]. Four independent biological replicates were used for sequencing. Raw reads were checked for quality using Fastqc and subsequently aligned using Tophat2, version 2.0.9, against the Flybase genome version 6. Mapped reads were counted using HTSeq-count version 0.5.4p5 [66] with mode „union“. Genes showing a cpm value below 1 in four samples per treatment were considered as poorly expressed and filtered out before conducting differential expression analysis using edgeR, version 3.2.4 [67]. Since our replicates were generated independently, we used a paired design and corrected the resulting p-values by the Benjamini-Hochberg method [68]. Subsequently, genes with a fold change of 1.5 and an adjusted p-value lower than 0.1 were considered as significantly deregulated.
DamID-Seq
UAS-Dam and UAS-cicDam transgenes were induced in esg+ cells for 24 hours at 29°C and 80 guts were dissected. Genomic DNA was extracted and methylated DNA was processed and amplified as previously described [69]. Sequencing libraries were prepared according to the protocol from Andrea Brand lab (personal communication), with the following modifications. Amplified DNA from experimental and Dam-only controls was fragmented in a Covaris-S2 then digested with Sau3AI to remove the adaptors. The Truseq DNA PCR-Free Sample Preparation kit (illumina) was used to prepare the sequencing library. The library was sent for Hiseq-2000 single-end 50bp sequencing.
DamID-Seq analysis
Raw reads were mapped to the Drosophlia genome (version 6.02, www.flybase.org) using Bowtie 2 [70] with default setting. After mapping, the uniquely mapped reads were extended 300 base pair (bp) toward 3' prime, and the genome were segmented with a unit of 75 bp window. Then the number of reads falling into each window was quantified. Normalization factors were computed based on the assumption that the mean log ratio between two experiments is equal to 1. The log ratio between treatment and background was observed to follow a normal distribution; the mean and standard deviation were estimated by using the fitdistr function in R (www.r-project.org). The statistical significance of the enrichment was then computed using the estimated mean and standard deviation. Using a sliding window approach, a binding site was called, where at least 4 continuous units (4x75bp windows) had a significant enrichment (p-value<0.01). The false discovery rate of the binding sites was calculated similarly to the previous publication [71].
Annotation of the peaks was carried out using two approaches. The first is based on the summit of the peak. The distance between the summit and the closest gene transcription start site (TSS) was computed with the gene orientation in consideration, and the closest gene was then assigned to the peak. The second approach is based on the entire peak. If the gene is found to be overlapping with the peak, then the gene is associated with the peak.
“TGAATG[AG]A” was searched in the peaks. The total number of occurrences was quantified, as well as the number of peaks that contained the searched pattern. In order to estimate the significance of the pattern, the background was generated by randomly moving the peaks in the genome for 1000 times. The occurrence of the pattern in the random sequences was then fitted to a Negative Binomial distribution by using the fitdistr function in R (www.r-project.org). The p-value was computed using the pnbinom function in R (www.r-project.org). To assess the association between the RNA-Seq and DamID-Seq hits, genome wide genes were ranked by fold change or absolute expression change, and corresponding number of genes which has binding sites were calculated by a moving sum (window size = 500). The absolute change is defined as the treatment value minus the background value.
in vitro DNA binding assays
EMSA experiments were conducted using derivatives of Cicmini, a minimal Cic protein that is functional in the embryo [25]. Wild-type and HMG-box mutant products were synthesized with the TNT T7 Quick Coupled Transcription/Translation System (Promega); the HMG-box mutant construct lacks the peptide sequence ILGEWW. DNA probes were amplified by PCR using primers carrying Not I restriction sites, digested with Not I and end-labeled with 32P-dCTP and Klenow Fragment, exo- (Thermo Scientific). Binding reactions were carried out in a total volume of 20 ml containing 60 mM Hepes pH 7.9, 20 mM Tris-HCl pH 7.9, 300 mM KCl, 5 mM EDTA, 5 mM DTT, 12% glycerol, ~1 ng probe, 1 mg poly (dI-dC), 1 mg BSA and 1 ml of programmed or non-programmed (control) TNT lysate. After incubation for 20 min on ice, complexes were resolved on 5% non-denaturing polyacrylamide gels run in 0.5X TBE at 4°C, and visualized by autoradiography.
RT-qPCR primers
Rp49 –Forward: TCGATATGCTAAGCTGTC
Rp49 –Reverse: GGCATCAGATACTGTCCCTTG
β-tubulin-Forward: ACATCCCGCCCCGTGGTC
β-tubulin-Reverse: AGAAAGCCTTGCGCCTGAACATAG
pnt-Forward: ACGCCCTATGATGCTCAATC
pnt-Reverse: TATCCAGACCCAAGGTGCTC
pntP1-Forward: CGACTGCGAACAATCTGGT
pntP1-Reverse: TTGCTGGTGTTGTAGCCTGT
pntP2-Forward: TTAGCCAATTGAACGGCATC
pntP2-Reverse: GCACAGATCCTTGCATCCAT
Ets21C-Forward: CCGGGCACTCAGGTACTACT
Ets21C-Reverse: CATACTGGAGGCCGGATCT
aos-Forward: AGAACCCATGGCTTACATGC
aos-Reverse: CGTCGCGGGTGTTAAGTTAC
yan-Forward: CTGCTGGTCATCGTGCTTAG
yan-Reverse: GACCTCAGTGTGAGCAGCAA
stg-Forward: CAGCATGGATTGCAATATCAGTA
stg-Reverse: CAACGTCGTCGTCGTAGAAC
CycE-Forward: ACAAATTTGGCCTGGGACTA
CycE-Reverse: GGCCATAAGCACTTCGTC
Supporting Information
The figure was reproduced from the cBioPortal for Cancer Genomics web page and modified to show only cancers with >3.3% alteration frequency. Asterisks mark colorectal cancer data from Genentech [35].
(TIF)
(A, B) RNAi-mediated depletion of Cic in ISCs and EBs using the esg ts system. esg+ progenitor cells (green), PH3+ (red) nuclear DNA (blue). (A) Control adult midgut (B) Cic knock down midgut after 4 days induction 29°C. Scale bars represent 50μm. (C-H) Cic mutant clones were analyzed using the MARCM system. ISC clones (green), DNA stained with DAPI (blue). Control (C, E, G) and mutant (D, F, H) ISC clones were induced with the MARCM system and examined 10 days, 20 days and 30 days later. Mutant ISCs divided faster and generated bigger clones. Scale bars represent 100μm.
(TIF)
(A, B) RNAi-mediated depletion of Cic in ISCs using the Dl ts system. ISCs are marked by GFP (green). Sample was also stained with anti-PH3 to detect mitoeses (red) and DAPI to detect nuclear DNA (blue). (A) Control adult midgut, (B) Cic depleted midgut after 4 days induction at 29°C. Dramatic increases in the number of GFP positive cells were observed in cic depleted midguts, as was large increase in ISC mitoses. (C, D) RNAi-mediated depletion of Cic in EBs using the Su(H) ts system. EB cells are marked by GFP (green). Samples were also stained with anti-PH3 (red) and DAPI (blue). (C) Control adult midgut (D) Cic depleted midgut after 4 days induction 29°C. Increases in the number of GFP positive cells and mitoses were observed in cic knockdown midguts. (E) Midguts as in A-C were scored for PH3+ cells after 4 days of induction of cic-RNAi in ISCs or EBs. (F) After 4 days induction of cic-RNAi in ISCs or EBs, midguts were scored for GFP+ or GFP- mitotic cells. Most mitotic cells were GFP+ when cic-RNAi was induced in ISCs using the Dl ts system, whereas in midguts in which cic was depleted in EBs, most of the mitotic cells were GFP- and likely ISCs. This indicates a non-cell autonomous effect. (G) Midguts were scored for PH3+ cells after 4 days of induction of cic-RNAi using the esg ts system, which targets gene expression to ISCs and EBs. Dramatic increases in the number of GFP positive cells were observed in cic knockdown midguts as was a large increase in ISC mitoses. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Error bars in each graph represent standard deviation. Scale bars represent 20μm.
(TIF)
(A) Graph showing fold change of peaks from Cic-DamID and P.e. infected Cic-DamID samples. (B) Cic binding sites in the aos locus from Cic-DamID-Seq using midgut ISCs. The black peaks are from control animals, and the grey peaks are from P.e. infected animals. Plot represents the log2 ratio between the Dam-fusion signal and the Dam-only signal. Red arrows point out TGAATG(G/A)A motifs. The aos transcription unit is shown below the graph. Yellow boxed regions indicate the ORF. (C) mRNA expression ratio change of aos was analyzed by qRT-PCR and normalized to β-Tub and Rp49 with non-amplified mRNA from FACS-sorted progenitor cells.
(TIF)
(A) mRNA expression heatmap of Ets transcription factors, showing fold change inductions from RNA-Seq data from whole midguts upon 6 hours P.e. infection. (B) Cic binding sites in the Ets21C locus from Cic-DamID-Seq from esg+ cells. Black peaks are from control samples and grey peaks are from P.e. infected midguts. The Y-axis represents the log2 ratio of the Cic-Dam fusion signal to the Dam-only signal. Red arrows point out TGAATG(G/A)A motifs. (C) Normalized mRNA expression fold change of pnt, pntP1, pntP2 and Ets21C in cic transheterozygous mutant midguts.
(TIF)
(A–C) Effect of pntP2 overexpression on ISC proliferation. Transgene expression was induced using the esg ts system at 29°C for 4 days. Samples were stained with anti-GFP (green), anti-PH3(red) and DAPI (blue) to mark DNA. (A) Control adult midgut. (B) pntP2 overexpressing midgut. The pntP2 over expressing midgut had more GFP+ ISCs and EBs (green). (C) pntP2 and cic ΔC2 over expressing midgut. GFP positive progenitor cells were still able to proliferate in the pnt, cic ΔC2 over-expressing midgut. (D) pnt mutant clones analyzed by the MARCM system. The size of the clones was quantified by counting cell numbers per clone. pnt Δ33 is a pntP1 specific mutant allele, pnt Δ78 is pntP2 specific mutant allele and pnt Δ88 is pnt null mutant allele that affect both isoforms. Only the pnt Δ88 detectably suppressed clone expansion. (E) Mitotic ratio of the pnt mutant clones was scored by calculating the average number of mitoses in each clone. (F) Quantification of ISC mitoses (PH3 positive cells) in pnt and cic depleted midguts or yan depleted midguts, using esg ts system. Fewer mitotic ISCs were observed in the pnt and cic double knock down midgut than in the cic knockdown midguts, showing that pnt is required downstream of cic. Yan depletion had no effect on ISC proliferation. (G) pnt mutant clones were generated in a cic depleted background using the MARCM system. The size of the clones was quantified by counting cell numbers per clone. Only the pnt Δ88 null allele suppressed the growth of cic-depleted ISC cell clones. (H) yan expression ratio as measured by qRT-PCR in yan-depleted midguts, using two different yan-RNAi lines. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Error bars in each graph represent standard deviation. Scale bars represent 50μm.
(TIF)
The figure was reproduced from the cBioPortal for Cancer Genomics web page and modified to show only cancers with >3.3% alteration frequency. (A) Cross-cancer alteration summary for EGR (the human orthologs of Drosophila Ets21C). (B) Cross-cancer alteration summary for EGR (the human orthologs of Drosophila Pnt).
(TIF)
Two lists of peaks from Cic DamID-seq and Cic DamID-Seq upon infection are included in this table. Each data sheet presents the specific genomic location of the peaks with detailed information for the Cic binding peaks such as, chromosome, Peak starting sites, Peak ending sites, Log2 fold change of CicDam/Dam-only and Summit of the peaks. (S1-1) Cic binding peaks from Cic DamID-Seq. (S1-2) Cic binding peaks from Cic DamID-Seq upon P.e. infection.
(XLSX)
The table included the full list of genes whose mRNA expression was significantly changed (>1.5-fold, at 90% confidence) in sorted esg ts UAS-GFP cells expressing cic-RNAi for 4 days. Listed data include Flybase gene ID, gene symbol, Log2-fold-change for each gene as well as significance (p-value with Benjamini-Hochberg correction).
(XLSX)
List of Flybase gene IDs for genes that were both upregulated upon cic-RNAi, and also associated with one or more Cic binding peak from the Cic DamID-Seq analysis. In addition, the normalized Log2 fold change of the peaks following P.e. infection, associated with the genes was also shown in the table. Some of the peaks disappeared after P.e. infection, so they were marked as #N/D (not detected). Derived from the data in S1 and S2 Tables.
(XLSX)
Information of well-known growth promoters was listed in the table with their log2 Fold change in RNA-Seq and significance (p-value with Benjamini-Hochberg correction) and numbers of Cic Dam-ID peaks, found in their introns or within 5kb range of the transcription start site. Genes that have Cic binding sites are shaded grey.
(PDF)
Three Lists of the non-protein coding RNAs such as tRNA, snRNA & snoRNA and non-protein coding genes that have Cic binding sites in their loci within the 5 kb range in the transcription start site were included in this table. Each data sheet shows the specific genomic location of each peak with detailed information for the Cic binding peaks such as, chromosome, Peak starting sites, Peak ending sites, Log2 fold change of CicDam/Dam-only, FlyBase ID and Gene symbol. (S5-1) Cic binding tRNAs. (S5-2) Cic binding snRNAs & snoRNAs. (S5-3) Cic binding non-protein coding genes.
(XLSX)
Acknowledgments
We would like to thank Andrea Brand for sharing the DamID-Seq experimental and analysis protocols. We specially acknowledge Jennifer Bandura for helping with the manuscript. We are also grateful to Gerado Jiménez, Laura Nilson, Iswar Hariharan, and the VDRC and Bloomington Drosophila Stock Centers for stocks and reagents. We also thank all the Edgar lab members, in particular Norman Zielke, Jerome Korzelius and Alexander Kohlmaier for stocks and discussion. We thank David Ibberson of the Heidelberg University Deep Sequencing Core Facility, Monika Langlotz from the ZMBH FACS Facility and Man Rao from Geno-Ming bioscience.
Data Availability
All raw and processed next generation sequencing data are available from the GEO database (accession number GSE74188).
Funding Statement
This work was supported by the DKFZ, DFG grant SFB 873, and ERC Advanced Grant 268515 (http://erc.europa.eu/starting-grants) to BAE. NH was supported by SFB638, SFB1036. MF and GJ were supported by research grants from the Spanish Government (BFU2011-23611) and Fundació La Marató de TV3 (20131730); GJ is an ICREA investigator.The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Herbst RS (2004) Review of epidermal growth factor receptor biology. International journal of radiation oncology, biology, physics 59: 21–26. [DOI] [PubMed] [Google Scholar]
- 2. Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, et al. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366: 2–16. [DOI] [PubMed] [Google Scholar]
- 3. Krasinskas AM (2011) EGFR Signaling in Colorectal Carcinoma. Pathology research international 2011: 932932 10.4061/2011/932932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nature reviews Cancer 3: 11–22. [DOI] [PubMed] [Google Scholar]
- 5. Radtke F, Clevers H (2005) Self-renewal and cancer of the gut: two sides of a coin. Science 307: 1904–1909. [DOI] [PubMed] [Google Scholar]
- 6. Amcheslavsky A, Jiang J, Ip YT (2009) Tissue damage-induced intestinal stem cell division in Drosophila. Cell stem cell 4: 49–61. 10.1016/j.stem.2008.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buchon N, Broderick NA, Poidevin M, Pradervand S, Lemaitre B (2009) Drosophila intestinal response to bacterial infection: activation of host defense and stem cell proliferation. Cell host & microbe 5: 200–211. [DOI] [PubMed] [Google Scholar]
- 8. Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, et al. (2009) Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137: 1343–1355. 10.1016/j.cell.2009.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang H, Edgar BA (2009) EGFR signaling regulates the proliferation of Drosophila adult midgut progenitors. Development 136: 483–493. 10.1242/dev.026955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buchon N, Broderick NA, Kuraishi T, Lemaitre B (2010) Drosophila EGFR pathway coordinates stem cell proliferation and gut remodeling following infection. BMC biology 8: 152 10.1186/1741-7007-8-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang H, Grenley MO, Bravo MJ, Blumhagen RZ, Edgar BA (2011) EGFR/Ras/MAPK signaling mediates adult midgut epithelial homeostasis and regeneration in Drosophila. Cell stem cell 8: 84–95. 10.1016/j.stem.2010.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu N, Wang SQ, Tan D, Gao Y, Lin G, et al. (2011) EGFR, Wingless and JAK/STAT signaling cooperatively maintain Drosophila intestinal stem cells. Developmental biology 354: 31–43. 10.1016/j.ydbio.2011.03.018 [DOI] [PubMed] [Google Scholar]
- 13. Jiang H, Edgar BA (2012) Intestinal stem cell function in Drosophila and mice. Current opinion in genetics & development 22: 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, et al. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265. 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- 15. Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, et al. (2011) Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418. 10.1038/nature09637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong VW, Stange DE, Page ME, Buczacki S, Wabik A, et al. (2012) Lrig1 controls intestinal stem-cell homeostasis by negative regulation of ErbB signalling. Nature cell biology 14: 401–408. 10.1038/ncb2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, et al. (2002) Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America 99: 1521–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weinberg R (2014) The Biology of Cancer: Garland Science. [Google Scholar]
- 19. Harvey Lodish A B, Kaiser Chris A., Monty Krieger, Anthony Bretscher, Hidde Ploegh, Angelika Amon, Scott Matthew P. (2012) Molecular Cell Biology: W. H. Freeman; [Google Scholar]
- 20. Tseng AS, Tapon N, Kanda H, Cigizoglu S, Edelmann L, et al. (2007) Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Current biology: CB 17: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jimenez G, Guichet A, Ephrussi A, Casanova J (2000) Relief of gene repression by torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes & development 14: 224–231. [PMC free article] [PubMed] [Google Scholar]
- 22. Goff DJ, Nilson LA, Morisato D (2001) Establishment of dorsal-ventral polarity of the Drosophila egg requires capicua action in ovarian follicle cells. Development 128: 4553–4562. [DOI] [PubMed] [Google Scholar]
- 23. Krivy K, Bradley-Gill MR, Moon NS (2013) Capicua regulates proliferation and survival of RB-deficient cells in Drosophila. Biology open 2: 183–190. 10.1242/bio.20123277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jimenez G, Shvartsman SY, Paroush Z (2012) The Capicua repressor—a general sensor of RTK signaling in development and disease. Journal of cell science 125: 1383–1391. 10.1242/jcs.092965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Astigarraga S, Grossman R, Diaz-Delfin J, Caelles C, Paroush Z, et al. (2007) A MAPK docking site is critical for downregulation of Capicua by Torso and EGFR RTK signaling. The EMBO journal 26: 668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grimm O, Sanchez Zini V, Kim Y, Casanova J, Shvartsman SY, et al. (2012) Torso RTK controls Capicua degradation by changing its subcellular localization. Development 139: 3962–3968. 10.1242/dev.084327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roch F, Jimenez G, Casanova J (2002) EGFR signalling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development 129: 993–1002. [DOI] [PubMed] [Google Scholar]
- 28. Lim B, Samper N, Lu H, Rushlow C, Jimenez G, et al. (2013) Kinetics of gene derepression by ERK signaling. Proceedings of the National Academy of Sciences of the United States of America 110: 10330–10335. 10.1073/pnas.1303635110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dissanayake K, Toth R, Blakey J, Olsson O, Campbell DG, et al. (2011) ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. The Biochemical journal 433: 515–525. 10.1042/BJ20101562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, et al. (2011) Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333: 1453–1455. 10.1126/science.1210557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, et al. (2006) Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Human molecular genetics 15: 2125–2137. [DOI] [PubMed] [Google Scholar]
- 32. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2: 401–404. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 6: pl1 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, et al. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314: 268–274. [DOI] [PubMed] [Google Scholar]
- 35. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, et al. (2012) Recurrent R-spondin fusions in colon cancer. Nature 488: 660–664. 10.1038/nature11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee T, Luo L (2001) Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends in neurosciences 24: 251–254. [DOI] [PubMed] [Google Scholar]
- 37. Queenan AM, Ghabrial A, Schupbach T (1997) Ectopic activation of torpedo/Egfr, a Drosophila receptor tyrosine kinase, dorsalizes both the eggshell and the embryo. Development 124: 3871–3880. [DOI] [PubMed] [Google Scholar]
- 38. Karim FD, Rubin GM (1998) Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 125: 1–9. [DOI] [PubMed] [Google Scholar]
- 39. Ajuria L, Nieva C, Winkler C, Kuo D, Samper N, et al. (2011) Capicua DNA-binding sites are general response elements for RTK signaling in Drosophila. Development 138: 915–924. 10.1242/dev.057729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Southall TD, Gold KS, Egger B, Davidson CM, Caygill EE, et al. (2013) Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Developmental cell 26: 101–112. 10.1016/j.devcel.2013.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vogel MJ, Peric-Hupkes D, van Steensel B (2007) Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nature protocols 2: 1467–1478. [DOI] [PubMed] [Google Scholar]
- 42. Kohlmaier A, Fassnacht C, Jin Y, Reuter H, Begum J, et al. (2014) Src kinase function controls progenitor cell pools during regeneration and tumor onset in the Drosophila intestine. Oncogene 0. [DOI] [PubMed] [Google Scholar]
- 43. Shwartz A, Yogev S, Schejter ED, Shilo BZ (2013) Sequential activation of ETS proteins provides a sustained transcriptional response to EGFR signaling. Development 140: 2746–2754. 10.1242/dev.093138 [DOI] [PubMed] [Google Scholar]
- 44. Klambt C (1993) The Drosophila gene pointed encodes two ETS-like proteins which are involved in the development of the midline glial cells. Development 117: 163–176. [DOI] [PubMed] [Google Scholar]
- 45. O'Neill EM, Rebay I, Tjian R, Rubin GM (1994) The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell 78: 137–147. [DOI] [PubMed] [Google Scholar]
- 46. Brunner D, Ducker K, Oellers N, Hafen E, Scholz H, et al. (1994) The ETS domain protein pointed-P2 is a target of MAP kinase in the sevenless signal transduction pathway. Nature 370: 386–389. [DOI] [PubMed] [Google Scholar]
- 47. Morimoto AM, Jordan KC, Tietze K, Britton JS, O'Neill EM, et al. (1996) Pointed, an ETS domain transcription factor, negatively regulates the EGF receptor pathway in Drosophila oogenesis. Development 122: 3745–3754. [DOI] [PubMed] [Google Scholar]
- 48. Xu C, Kauffmann RC, Zhang J, Kladny S, Carthew RW (2000) Overlapping activators and repressors delimit transcriptional response to receptor tyrosine kinase signals in the Drosophila eye. Cell 103: 87–97. [DOI] [PubMed] [Google Scholar]
- 49. Vivekanand P, Tootle TL, Rebay I (2004) MAE, a dual regulator of the EGFR signaling pathway, is a target of the Ets transcription factors PNT and YAN. Mechanisms of development 121: 1469–1479. [DOI] [PubMed] [Google Scholar]
- 50. Klaes A, Menne T, Stollewerk A, Scholz H, Klambt C (1994) The Ets transcription factors encoded by the Drosophila gene pointed direct glial cell differentiation in the embryonic CNS. Cell 78: 149–160. [DOI] [PubMed] [Google Scholar]
- 51. Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiology and molecular biology reviews: MMBR 68: 320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carriere A, Cargnello M, Julien LA, Gao H, Bonneil E, et al. (2008) Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Current biology: CB 18: 1269–1277. 10.1016/j.cub.2008.07.078 [DOI] [PubMed] [Google Scholar]
- 53. Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, et al. (2011) ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). The Journal of biological chemistry 286: 567–577. 10.1074/jbc.M110.159046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121: 179–193. [DOI] [PubMed] [Google Scholar]
- 55. Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, et al. (2007) Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448: 595–599. [DOI] [PubMed] [Google Scholar]
- 56. Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, et al. (2010) ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 467: 849–853. 10.1038/nature09409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jane-Valbuena J, Widlund HR, Perner S, Johnson LA, Dibner AC, et al. (2010) An oncogenic role for ETV1 in melanoma. Cancer research 70: 2075–2084. 10.1158/0008-5472.CAN-09-3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, et al. (2009) (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proceedings of the National Academy of Sciences of the United States of America 106: 4519–4524. 10.1073/pnas.0900780106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, et al. (2011) Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes & development 25: 2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Micchelli CA, Perrimon N (2006) Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature 439: 475–479. [DOI] [PubMed] [Google Scholar]
- 61. Romani P, Bernardi F, Hackney J, Dobens L, Gargiulo G, et al. (2009) Cell survival and polarity of Drosophila follicle cells require the activity of ecdysone receptor B1 isoform. Genetics 181: 165–175. 10.1534/genetics.108.096008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bischof J, Bjorklund M, Furger E, Schertel C, Taipale J, et al. (2013) A versatile platform for creating a comprehensive UAS-ORFeome library in Drosophila. Development 140: 2434–2442. 10.1242/dev.088757 [DOI] [PubMed] [Google Scholar]
- 63. Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415. [DOI] [PubMed] [Google Scholar]
- 64. Dutta D, Xiang J, Edgar BA (2013) RNA expression profiling from FACS-isolated cells of the Drosophila intestine. Current protocols in stem cell biology 27: Unit 2F 2 10.1002/9780470151808.sc02f02s27 [DOI] [PubMed] [Google Scholar]
- 65. Shaw RL, Kohlmaier A, Polesello C, Veelken C, Edgar BA, et al. (2010) The Hippo pathway regulates intestinal stem cell proliferation during Drosophila adult midgut regeneration. Development 137: 4147–4158. 10.1242/dev.052506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Anders S, Pyl PT, Huber W (2015) HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Benjamini Y, and Hochberg Y (1995) Controlling the False Discovery Rate—a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological 57: 289–300. [Google Scholar]
- 69. Papagiannouli F, Schardt L, Grajcarek J, Ha N, Lohmann I (2014) The Hox gene Abd-B controls stem cell niche function in the Drosophila testis. Developmental cell 28: 189–202. 10.1016/j.devcel.2013.12.016 [DOI] [PubMed] [Google Scholar]
- 70. Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nature methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Southall TD, Davidson CM, Miller C, Carr A, Brand AH (2014) Dedifferentiation of neurons precedes tumor formation in Lola mutants. Developmental cell 28: 685–696. 10.1016/j.devcel.2014.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The figure was reproduced from the cBioPortal for Cancer Genomics web page and modified to show only cancers with >3.3% alteration frequency. Asterisks mark colorectal cancer data from Genentech [35].
(TIF)
(A, B) RNAi-mediated depletion of Cic in ISCs and EBs using the esg ts system. esg+ progenitor cells (green), PH3+ (red) nuclear DNA (blue). (A) Control adult midgut (B) Cic knock down midgut after 4 days induction 29°C. Scale bars represent 50μm. (C-H) Cic mutant clones were analyzed using the MARCM system. ISC clones (green), DNA stained with DAPI (blue). Control (C, E, G) and mutant (D, F, H) ISC clones were induced with the MARCM system and examined 10 days, 20 days and 30 days later. Mutant ISCs divided faster and generated bigger clones. Scale bars represent 100μm.
(TIF)
(A, B) RNAi-mediated depletion of Cic in ISCs using the Dl ts system. ISCs are marked by GFP (green). Sample was also stained with anti-PH3 to detect mitoeses (red) and DAPI to detect nuclear DNA (blue). (A) Control adult midgut, (B) Cic depleted midgut after 4 days induction at 29°C. Dramatic increases in the number of GFP positive cells were observed in cic depleted midguts, as was large increase in ISC mitoses. (C, D) RNAi-mediated depletion of Cic in EBs using the Su(H) ts system. EB cells are marked by GFP (green). Samples were also stained with anti-PH3 (red) and DAPI (blue). (C) Control adult midgut (D) Cic depleted midgut after 4 days induction 29°C. Increases in the number of GFP positive cells and mitoses were observed in cic knockdown midguts. (E) Midguts as in A-C were scored for PH3+ cells after 4 days of induction of cic-RNAi in ISCs or EBs. (F) After 4 days induction of cic-RNAi in ISCs or EBs, midguts were scored for GFP+ or GFP- mitotic cells. Most mitotic cells were GFP+ when cic-RNAi was induced in ISCs using the Dl ts system, whereas in midguts in which cic was depleted in EBs, most of the mitotic cells were GFP- and likely ISCs. This indicates a non-cell autonomous effect. (G) Midguts were scored for PH3+ cells after 4 days of induction of cic-RNAi using the esg ts system, which targets gene expression to ISCs and EBs. Dramatic increases in the number of GFP positive cells were observed in cic knockdown midguts as was a large increase in ISC mitoses. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Error bars in each graph represent standard deviation. Scale bars represent 20μm.
(TIF)
(A) Graph showing fold change of peaks from Cic-DamID and P.e. infected Cic-DamID samples. (B) Cic binding sites in the aos locus from Cic-DamID-Seq using midgut ISCs. The black peaks are from control animals, and the grey peaks are from P.e. infected animals. Plot represents the log2 ratio between the Dam-fusion signal and the Dam-only signal. Red arrows point out TGAATG(G/A)A motifs. The aos transcription unit is shown below the graph. Yellow boxed regions indicate the ORF. (C) mRNA expression ratio change of aos was analyzed by qRT-PCR and normalized to β-Tub and Rp49 with non-amplified mRNA from FACS-sorted progenitor cells.
(TIF)
(A) mRNA expression heatmap of Ets transcription factors, showing fold change inductions from RNA-Seq data from whole midguts upon 6 hours P.e. infection. (B) Cic binding sites in the Ets21C locus from Cic-DamID-Seq from esg+ cells. Black peaks are from control samples and grey peaks are from P.e. infected midguts. The Y-axis represents the log2 ratio of the Cic-Dam fusion signal to the Dam-only signal. Red arrows point out TGAATG(G/A)A motifs. (C) Normalized mRNA expression fold change of pnt, pntP1, pntP2 and Ets21C in cic transheterozygous mutant midguts.
(TIF)
(A–C) Effect of pntP2 overexpression on ISC proliferation. Transgene expression was induced using the esg ts system at 29°C for 4 days. Samples were stained with anti-GFP (green), anti-PH3(red) and DAPI (blue) to mark DNA. (A) Control adult midgut. (B) pntP2 overexpressing midgut. The pntP2 over expressing midgut had more GFP+ ISCs and EBs (green). (C) pntP2 and cic ΔC2 over expressing midgut. GFP positive progenitor cells were still able to proliferate in the pnt, cic ΔC2 over-expressing midgut. (D) pnt mutant clones analyzed by the MARCM system. The size of the clones was quantified by counting cell numbers per clone. pnt Δ33 is a pntP1 specific mutant allele, pnt Δ78 is pntP2 specific mutant allele and pnt Δ88 is pnt null mutant allele that affect both isoforms. Only the pnt Δ88 detectably suppressed clone expansion. (E) Mitotic ratio of the pnt mutant clones was scored by calculating the average number of mitoses in each clone. (F) Quantification of ISC mitoses (PH3 positive cells) in pnt and cic depleted midguts or yan depleted midguts, using esg ts system. Fewer mitotic ISCs were observed in the pnt and cic double knock down midgut than in the cic knockdown midguts, showing that pnt is required downstream of cic. Yan depletion had no effect on ISC proliferation. (G) pnt mutant clones were generated in a cic depleted background using the MARCM system. The size of the clones was quantified by counting cell numbers per clone. Only the pnt Δ88 null allele suppressed the growth of cic-depleted ISC cell clones. (H) yan expression ratio as measured by qRT-PCR in yan-depleted midguts, using two different yan-RNAi lines. Statistical significance was determined by Student’s t test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Error bars in each graph represent standard deviation. Scale bars represent 50μm.
(TIF)
The figure was reproduced from the cBioPortal for Cancer Genomics web page and modified to show only cancers with >3.3% alteration frequency. (A) Cross-cancer alteration summary for EGR (the human orthologs of Drosophila Ets21C). (B) Cross-cancer alteration summary for EGR (the human orthologs of Drosophila Pnt).
(TIF)
Two lists of peaks from Cic DamID-seq and Cic DamID-Seq upon infection are included in this table. Each data sheet presents the specific genomic location of the peaks with detailed information for the Cic binding peaks such as, chromosome, Peak starting sites, Peak ending sites, Log2 fold change of CicDam/Dam-only and Summit of the peaks. (S1-1) Cic binding peaks from Cic DamID-Seq. (S1-2) Cic binding peaks from Cic DamID-Seq upon P.e. infection.
(XLSX)
The table included the full list of genes whose mRNA expression was significantly changed (>1.5-fold, at 90% confidence) in sorted esg ts UAS-GFP cells expressing cic-RNAi for 4 days. Listed data include Flybase gene ID, gene symbol, Log2-fold-change for each gene as well as significance (p-value with Benjamini-Hochberg correction).
(XLSX)
List of Flybase gene IDs for genes that were both upregulated upon cic-RNAi, and also associated with one or more Cic binding peak from the Cic DamID-Seq analysis. In addition, the normalized Log2 fold change of the peaks following P.e. infection, associated with the genes was also shown in the table. Some of the peaks disappeared after P.e. infection, so they were marked as #N/D (not detected). Derived from the data in S1 and S2 Tables.
(XLSX)
Information of well-known growth promoters was listed in the table with their log2 Fold change in RNA-Seq and significance (p-value with Benjamini-Hochberg correction) and numbers of Cic Dam-ID peaks, found in their introns or within 5kb range of the transcription start site. Genes that have Cic binding sites are shaded grey.
(PDF)
Three Lists of the non-protein coding RNAs such as tRNA, snRNA & snoRNA and non-protein coding genes that have Cic binding sites in their loci within the 5 kb range in the transcription start site were included in this table. Each data sheet shows the specific genomic location of each peak with detailed information for the Cic binding peaks such as, chromosome, Peak starting sites, Peak ending sites, Log2 fold change of CicDam/Dam-only, FlyBase ID and Gene symbol. (S5-1) Cic binding tRNAs. (S5-2) Cic binding snRNAs & snoRNAs. (S5-3) Cic binding non-protein coding genes.
(XLSX)
Data Availability Statement
All raw and processed next generation sequencing data are available from the GEO database (accession number GSE74188).