

Abstract
Heterotrimeric G proteins function at diverse subcellular locations, in addition to canonical signaling at the plasma membrane (PM). Gβγ signals at the Golgi, via protein kinase D (PKD), to regulate fission of PM-destined vesicles. However, the mechanism by which Gβγ is regulated at the Golgi in this process remains elusive. Recent studies have revealed that PAQR3 (Progestin and AdipoQ Receptor 3), also called RKTG (Raf Kinase Trapping to the Golgi), interacts with the Gβ subunit and localizes Gβ to the Golgi thereby inhibiting Gβγ signaling at the PM. Herein we show that, in contrast to this inhibition of canonical Gβγ signaling at the PM, PAQR3 promotes Gβγ signaling at the Golgi. Expression of PAQR3 causes fragmentation of the Golgi, while a Gβ binding-deficient mutant of PAQR3 does not cause Golgi fragmentation. Also, a C-terminal fragment of GRK2 (GRK2ct), which interacts with Gβγ and inhibits Gβγ signaling, and gallein, a small molecule inhibitor of Gβγ, are both able to inhibit PAQR3-mediated Golgi fragmentation. Furthermore, a dominant negative form of PKD (PKD-DN) and a pharmacological inhibitor of PKD, Gö6976, also inhibit PAQR3-mediated fragmentation of the Golgi. Importantly, expression of the Gβ binding-deficient mutant of PAQR3 inhibits the constitutive transport of the model cargo protein VSV-G from the Golgi to the PM, indicating the involvement of PAQR3 in Golgi-to PM vesicle transport and a dominant negative role for this mutant. Collectively, these results reveal a novel role for the newly characterized, Golgi-localized PAQR3 in regulating Gβγ at the non-canonical subcellular location of the Golgi and thus for controlling Golgi-to-PM protein transport via the Gβγ-PKD signaling pathway.
Keywords: heterotrimeric G protein, Golgi, membrane transport, vesicle trafficking, non-canonical signaling, subcellular localization
1. INTRODUCTION
The ubiquitously expressed heterotrimeric G proteins, composed of α, β and γ subunits, are central mediators of a vast number of signaling pathways and functions in the cell [1, 2]. Conventionally, G proteins transduce signals from G protein-coupled receptors (GPCRs) to downstream effector molecules, thereby initiating signaling pathways. Activation of GPCRs by their respective ligands catalyzes the release of GDP from Gα and the subsequent binding of GTP; the ligand-occupied GPCRs function as guanine-nucleotide exchange factors (GEFs) for Gα subunits. The binding of GTP to Gα results in the activation and dissociation of Gα from Gβγ; Gβ and Gγ subunits stay associated and function as a dimer while Gα functions alone. The intrinsic GTP hydrolysis activity of Gα returns Gα to the GDP bound state followed by re-association with the Gβγ subunits and the completion of the cycle.
In the classical view of G protein signaling, G proteins exert their functions from the cytoplasmic surface of the plasma membrane (PM); however, it is becoming increasingly evident that G proteins also function at non-canonical subcellular locations such as the ER, mitochondria, Golgi, endosomes and nucleus [3–7]. Studies have shown that G proteins traffic throughout the cell to reach various subcellular destinations via activation-dependent or constitutive trafficking pathways and have unique regulatory mechanisms by which they are regulated at their respective subcellular locations. A recent study, which focused on signaling at the Golgi, demonstrated that upon receptor activation Gβγ could translocate to the Golgi and enhance Golgi vesicle formation [8]. In another intriguing study it was shown that upon activation at the PM the β2-adrenergic receptor activates its cognate G protein Gs at the PM and then, upon internalization, generates a second wave of cAMP signaling via Gs localized at early endosomes [9]. With regards to the constitutive trafficking of G proteins, upon synthesis, G proteins traffic to subcellular locations such as the ER and the Golgi while en route to the PM, and assembly of the heterotrimeric G proteins occurs at these endomembranes, which is necessary for the delivery of the heterotrimer to the PM [10, 11]. It has also been shown that there is rapid and constitutive shuttling of G proteins between PM and endomembranes in basal state cells resulting in the constitutive localization of G proteins at endomembranes [12]. Recent reports have also provided unique insights regarding the mechanisms by which these G proteins, localized at diverse subcellular locations, are activated and regulated independently of cell surface receptor activation. For example, it was shown that a non-receptor GEF, a multimodular signal transducer known as GIV, activates heterotrimeric Gi at the Golgi and modulates structural organization of the Golgi and vesicle trafficking from the Golgi [13]. Another group of proteins known as AGS (Activators of G protein Signaling) proteins have been shown to activate G proteins independently of extracellular receptor input [14]. However, many of the mechanisms by which G proteins reach their subcellular locations and the details by which they are activated at these locales remain to be determined.
Recent studies have also unveiled a novel role for Gβγ at the Golgi where it regulates a signaling pathway resulting in the fission PM-destined transport vesicles from the trans Golgi network (TGN) [8, 15–17]. Many of the components of this pathway that function downstream of Gβγ at the Golgi have been described [18]. Phospholipase Cβ (PLC) is required for the generation of diacylglycerol (DAG) which in turn activates Golgi localized PKCη and also Golgi-recruitment of PKD, a central mediator in this pathway [16, 17, 19, 20]. Phosphorylation and activation of PKD by PKCη leads to the phosphorylation and activation of membrane fission machinery, including the activation of the lipid kinase, phosphatidyl inositol 4 kinase IIIβ (P4IKIIIβ) and the recruitment of proteins such as oxysterol binding protein 1 (OSBP1), and the ceramide transfer protein (CERT) [21–23]. Previous studies in our lab have focused on defining the importance and the exact localization of Gβγ in the activation of this pathway at the Golgi. To that extent we have shown that constitutively and inducibly Golgi-targeted Gβγ subunits promote fragmentation of the Golgi, indicating the over-activation of a normal signaling pathway at the Golgi by Gβγ and that while a Golgi-targeted Gβγ inhibitor, GRK2ct, inhibited Golgi-to-PM VSV-G transport, a PM-targeted GRK2ct did not inhibit transport [15]. Therefore, it became clear from our previous work that Gβγ is localized at the Golgi when activating this signaling pathway leading to fission of PM-destined Golgi vesicles. However, mechanisms by which Gβγ subunits are localized at the Golgi and regulated at the Golgi to activate this signaling pathway remain elusive.
Recent studies have shown that a newly characterized Golgi-localized membrane protein, PAQR3 (Progestin and AdipoQ Receptor 3), also known as RKTG (Raf Kinase Trapping to the Golgi), interacts with the Gβ subunit via the first 20 amino acids of its N-terminus and retains Gβγ at the Golgi [24]. PAQR3 is a 7-transmembrane protein belonging to the progestin and adipoQ receptor family; however, it should also be noted that PAQR3 does not possess the characteristics of GPCRs or GEFs [25, 26]. PAQR3 was first identified as a tumor suppressor protein due to the fact that it also has the ability to sequester Raf-1 to the Golgi and inhibit the Ras/Raf/MEK cell proliferative signaling pathway, and hence is also referred to as the aforementioned RKTG (Raf Kinase Trapping to the Golgi) [27, 28]. It was shown that by interacting with Gβγ via its N-terminus PAQR3 retains Gβγ at the cytoplasmic surface of the Golgi thereby interfering with and abrogating PM-localized Gβγ-mediated canonical signaling to downstream targets such as the activation of PI3K-AKT [24]. Therefore we raised the question as to whether PAQR3, in contrast to its ability to inhibit Gβγ signaling at the PM, could also promote Gβγ signaling at the Golgi to activate a Golgi localized signaling pathway. The N-terminus of PAQR3 (which has been shown to interact with Gβγ) is located at the cytoplasmic side of the Golgi, in a position where it is able to capture cycling Gβγ which are in constant flux in the cytoplasm. We therefore hypothesized that PAQR3 is involved in the regulation of the Gβγ-PKD mediated signaling pathway at the Golgi leading to fission of Golgi transport vesicles.
Our results show that the expression of PAQR3 enhances the vesicle formation process and fragments the Golgi, while molecular or pharmacological inhibition of Gβγ or PKD inhibits the fragmentation caused by PAQR3. Also, importantly, the Gβ binding-deficient mutant of PAQR3, PAQR3(NΔ20) did not cause the Golgi to vesiculate but did inhibit Golgi-to-PM transport of a model cargo protein, indicating a dominant negative function of this mutant. Here we show that PAQR3 is involved in the regulation of the Gβγ-PKD mediated signaling pathway at the Golgi and thus shed light onto a novel mechanism which would allow for regulation of Gβγ at the Golgi.
2. MATERIALS AND METHODS
2.1 Plasmids and Reagents
The GFP-tagged PAQR3 and PAQR3(NΔ20) constructs were kindly provided by Dr. Yan Chen (Chinese Academy of Sciences, Shanghai, China). The GRK2ct, mCherry-VSV-G, GFP-VSV-G and HA-KDELr-D193N expression plasmids were kindly provided by Drs. Jeffrey Benovic (Thomas Jefferson University, Philadelphia, PA), Elias T. Spiliotis (Drexel University, Philadelphia, PA), Jennifer Lippincott-Schwartz (NIH), and Piero Crespo (Universidad de Cantabria, Cantabria, Spain), respectively. The Gβγ inhibitor gallein was obtained from Tocris Bioscience and the PKD inhibitor (Gö6976) was gifted from Dr. Jeffrey Benovic (Thomas Jefferson University, Philadelphia, PA).
2.2 Cell Culture and Transfection
HeLa and HEK cells were grown in DMEM (Cellgro), supplemented with 10% Fetal Bovine Serum (FBS) and 1% Pen/Strep and maintained at 37 °C in a 95% air, 5% CO2-humidified atmosphere. Transfections were performed one day after cells were seeded in 6-well plates on coverslips using Lipofectamine 2000 (Invitrogen).
2.3 Immunofluorescence Microscopy
48 h after transfection cells were fixed with 3.7% formaldehyde, blocked with blocking buffer (2.5% nonfat milk in 1% Triton X-100/TBS) and incubated with primary antibody in blocking buffer for 1 h. Primary antibodies used were anti-TGN46 antibody (Bio-Rad AbD Serotec), anti-HA antibody (Santa Cruz Biotechnology), anti-VSV-G antibody (Sigma) and anti-GRK2 3A10 antibody (Sigma). Cells were then washed with blocking buffer and incubated for 30 min with the relevant Alexa fluor-conjugated secondary antibodies (Invitrogen), washed with 1% Triton X-100/TBS and mounted on glass slides with Prolong Antifade reagent (Invitrogen). Images were acquired using an Olympus BX-61 upright microscope and ORCA-ER cooled charge-coupled device camera (Hamamatsu, Bridgewater, NJ) controlled by Slidebook version 4.0 (Intelligent Imaging Innovations, Denver, CO).
2.4 Western Blotting
Cells were lysed with RIPA lysis buffer (150 mM NaCl, 1% Triton X-100, 0,5% Deoxycholic acid, 0.1% SDS, 50mM Tris pH 8) or immunoprecipitation (IP) lysis buffer (50mM Tris, 150mM NaCl, 0.5% Deoxycholic acid, 1% NP40 - for cell surface biotinylation experiments) and incubated on ice for 0.5 h and centrifuged for 10 min at 14,000rpm to clear nucleic acid and debris. Samples were run on SDS-PAGE gels and immunoblotting was performed using the indicated primary antibodies, anti-GFP antibody (Covance), anti-HSP90 (Santa Cruz Biotechnology) and anti-VSV-G antibody (Sigma) followed by HRP conjugated secondary anti-mouse or anti-rabbit antibodies (Promega) and detected using chemiluminescence reagent (Thermo Scientific).
2.5 VSV-G Transport Assay
HEK293 cells were transfected with an mCherry-tagged temperature sensitive mutant form of VSV-G alone or together with GFP-PAQR3 or GFP-PAQR3(NΔ20) and, GFP-tagged VSV-G (also the temperature sensitive mutant form) alone or together with HA-tagged Golgi-localized mutant version of KDLELr and incubated overnight at 37 °C. Cells were then shifted to the non-permissive temperature of 39 °C for 16–20 h at which temperature mis-folded VSV-G proteins accumulate in the ER. Cells were then moved to 20 °C for 2 h (after 1 h of which 10 μg/mL cyclohexamide was added), at which temperature VSV-G is folded properly in a chaperone-mediated manner, and transported from the ER to the Golgi. Cells were then shifted to the permissive temperature of 32 °C for 2 h to allow for Golgi to PM transport of VSV-G, upon which cells were fixed for immunofluorescence analysis.
2.6 Cell Surface Biotinylation
Surface biotinylation was performed to detect VSV-G at the PM upon completion of the VSV-G transport assay. Cells were grown in 6 cm plates and transfected with an mCherry-tagged temperature sensitive mutant form of VSV-G alone, or together with GFP-PAQR3, GFP-PAQR3(NΔ20) or a HA-tagged Golgi-localized mutant version of KDELr, incubated overnight at 37 C and a VSV-G transport assay was performed as described above. Upon incubation at the permissive temperature of 32 C for 2 h, cells were washed twice with ice cold PBS and incubated with 1mg/mL of cell impermeable EZ™-Link Sulfo-NHS-SS-Biotin [sulfosuccinimidyl-2-(biotinamido)ethyl1-1,3-dithiopropionate] (Pierce) for 0.5 h at 4 C with gentle shaking. Quenching solution (Pierce) was added to quench unreacted biotin and cells were collected, washed twice with TBS and lysed with immunoprecipitation (IP) lysis buffer (50mM Tris, 150mM NaCl, 0.5% Deoxycholic acid, 1% NP40). Biotinylated proteins were isolated by incubating with 50% slurry of Neutravidin agarose beads (Pierce) for 2 h at 4 C. The beads were then collected by centrifugation and washed three times with wash buffer (Pierce) and boiled for 5 min at 95 C in sample buffer containing 50mM DTT (dithiothreitol) to dissociate and elute bound protein complexes from the beads. Samples were run on SDS-PAGE gels and immunoblotted to detect PM localized VSV-G.
3. RESULTS
3.1 Expression of PAQR3 induces Golgi fragmentation while the Gβ binding-deficient mutant PAQR3(NΔ20) does not fragment the Golgi
Previous studies have shown that the overexpression of components of the Golgi-localized signaling pathway, such as Gβγ and PKD, which regulate the fission of PM-destined vesicles causes vesiculation/fragmentation of the Golgi [15, 16]. Such fragmentation is interpreted as an over-activation of the normal pathway and is used as an experimental readout of activation of this Golgi-localized pathway. In order to test our hypothesis that PAQR3 regulates the Gβγ-dependent pathway at the Golgi, HeLa cells were transfected with GFP-tagged PAQR3 or the Gβ binding-deficient mutant, PAQR3(NΔ20), and were analyzed for fragmented Golgi by fluorescence microscopy (Figure 1A and 1B). Expressed PAQR3 localized with the Golgi marker TGN46 and caused fragmentation of the Golgi (depicted by arrows, Figure 1A). In neighboring cells where PAQR3 is not expressed, the Golgi is not fragmented and remains intact. Strikingly, the Gβ binding-deficient mutant PAQR3(NΔ20) localized to the Golgi apparatus but did not cause fragmentation of the Golgi. Fragmentation of the Golgi was observed in >75% of PAQR3 expressing cells, while <5% of PAQR3(NΔ20) expressing cells showed fragmented Golgi (Figure 1C). Since the full length PAQR3 receptor containing the Gβ binding region caused the Golgi to fragment and the Gβγ binding-deficient mutant of PAQR3, PAQR3(NΔ20), left the Golgi intact, these data suggest that PAQR3 promotes Golgi fragmentation in a Gβγ-dependent manner.
Figure 1. Expression of PAQR3 induces Golgi fragmentation while the Gβ binding-deficient mutant PAQR3(NΔ20) does not fragment the Golgi.

A, HeLa cells were transfected with GFP-PAQR3 (top row) or GFP-PAQR3(NΔ20) (bottom row) for 48 h. Cells were fixed and processed for immunofluorescence microscopy using anti-TGN46 to detect Golgi morphology, and expressed PAQR3 and PAQR3(NΔ20) were detected by intrinsic GFP fluorescence. Arrows indicate Golgi fragmentation. Bar, 10 μm. B, To confirm equivalent expression of GFP-PAQR3 and GFP-PAQR3(NΔ20), immunoblotting was performed using HeLa cell lysates after transfection with pcDNA3, GFP-PAQR3 or GFP-PAQR3(NΔ20). An anti-GFP antibody was used to detect GFP-PAQR3 or GFP-PAQR3(NΔ20) (upper panel), and an anti-Hsp90 antibody was used as a gel loading control (lower panel). Consistent with the immunoblot shown, previous results have demonstrated that PAQR3 and PAQR3(NΔ20) display no detectable difference in mobility [24, 62]. C, Shown is a bar graph quantitating the effect of PAQR3, PAQR3(NΔ20), and the effect of Gβγ and PKD inhibitors in the presence of PAQR3 on Golgi fragmentation. Cells were transfected/treated as described in Figure Legends 1A, 2 and 3. Values are the means +/− S.D. for 3 separate experiments. 100 cells were counted in each experiment. Asterisks indicate statistical significance (p<0.001, t-test) compared to PAQR3 (first bar).
3.2 Molecular and pharmacological inhibition of Gβγ inhibits PAQR3-mediated Golgi fragmentation
To further demonstrate that the fragmentation of the Golgi upon the expression of PAQR3 requires Gβγ, we utilized the small molecule inhibitor of Gβγ, gallein, or expressed the C-terminal PH domain of GRK2 (GRK2ct), and examined whether inhibition of Gβγ in the presence of PAQR3 would prevent the fragmentation. GRK2ct binds to Gβγ and inhibits its ability to interact with downstream effectors; it is widely used to inhibit Gβγ-mediated signaling in cells [29–38]. Indeed, we have shown previously that GRK2ct is able to prevent the fragmentation caused by expression of Golgi targeted Gβγ [15]. Likewise, gallein has been widely used to inhibit Gβγ signaling, including its function at the Golgi [15, 39–42]. HeLa cells were transfected with GFP-PAQR3 and subsequently treated with gallein, or HeLa cells were transfected with GFP-PAQR3 together with GRK2ct (Figure 2). Both treatment with gallein and co-expression with GRK2ct prevented the Golgi-fragmented phenotype seen with GFP-PAQR3 expression alone (Figure 2). Only 21% and 16% of GFP-PAQR3 expressing cells contained fragmented Golgi when treated with gallein or when GRK2ct was expressed, respectively (Figure 1C). It should be noted that when inhibiting Gβγ with gallein in the presence of PAQR3, PAQR3 was expressed for ~36 h to allow for Golgi fragmentation, and then gallein was added for 8 more h before cells were fixed for staining. Addition of this pharmacological inhibitor for long periods of time such as the duration of the transfection period (36–48 h) proved to be toxic to cells. Therefore, the addition of gallein after expressed PAQR3 induces Golgi fragmentation shows that inhibition of Gβγ not only prevents PAQR3-mediated Golgi fragmentation, as observed with GRK2ct co-expression, but also reverses the fragmented Golgi phenotype. Thus, these data confirm that PAQR3-induced Golgi fragmentation requires Gβγ, supporting the hypothesis that PAQR3 is involved in the regulation of Gβγ at the Golgi and the subsequent signaling pathway at the Golgi that leads to fission of Golgi vesicles.
Figure 2. Gβγ inhibitors GRK2ct and gallein inhibit PAQR3-mediated Golgi fragmentation.
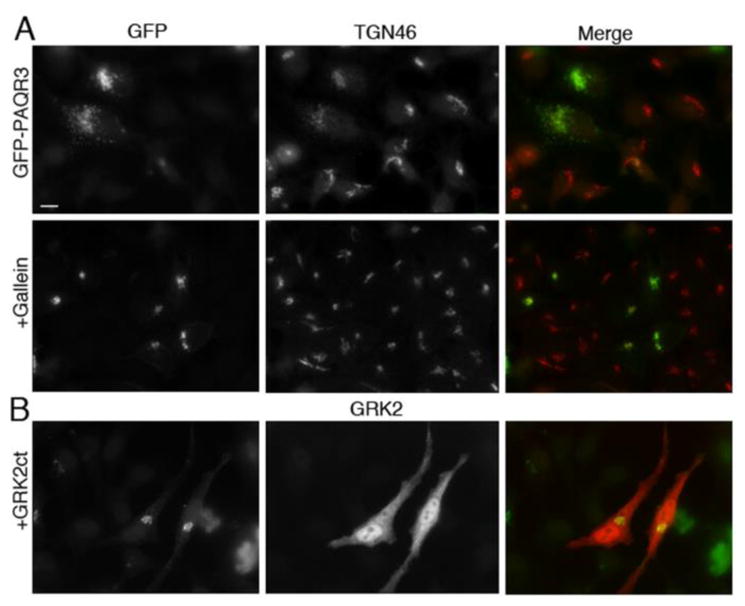
A, HeLa cells were transfected with GFP-PAQR3 for 36–48 h and then treated with 10 μM gallein (bottom row) or vehicle (top row) for 8 h prior to being fixed and processed for immunofluorescence microscopy using anti-TGN46 to detect Golgi morphology. Expressed GFP-PAQR3 was detected by intrinsic GFP fluorescence. B, HeLa cells were transfected with GFP-PAQR3 together with GRK2ct. Cells were fixed and stained with anti-GRK2, and the intrinsic GFP fluorescence allowed visualization of GFP-PAQR3, and, by extension, Golgi morphology. Bar, 10 μm.
3.3 Molecular and pharmacological inhibition of PKD inhibits PAQR3-mediated Golgi fragmentation
PKD is a key player in the signaling pathway at the Golgi that regulates vesicle formation; it phosphorylates and activates several proteins, including phosphotidylinositol 4-kinase IIIβ (PI4KIIIβ), at the Golgi, thus promoting membrane remodeling and the subsequent recruitment of membrane fission proteins [22, 43]. Previous studies in our lab have shown that the Golgi fragmentation observed by inducibly targeting Gβγ to the Golgi is prevented by inhibition of PKD. Since our hypothesis is that PAQR3 regulates Gβγ at the Golgi where it initiates a signaling pathway leading to the generation of Golgi to PM transport of vesicles via activation of PKD, we sought to investigate whether the generation of Golgi vesicles mediated by PAQR3 also requires PKD. In this experiment, we examined whether the fragmentation of the Golgi caused by expression of PAQR3 is prevented by the inhibition of PKD. HeLa cells were transfected with GFP-PAQR3 and subsequently treated with a small molecule inhibitor of PKD, Gö6976 (Figure 3A). This PKD inhibitor reversed the Golgi fragmentation observed in cells expressing GFP-PAQR3. As described above for gallein treatment (section 3.2), long term treatment (>24 h) with Gö6976 was toxic to cells, and therefore this inhibitor was added for the final 8 h of the experiment, after transfected GFP-PQAR3 had induced Golgi fragmentation. Importantly, this result (Figure 3A) shows that PKD inhibition, as with Gβγ inhibition (Figure 2A), reverses the observed PAQR3-induced Golgi fragmentation. In addition, HeLa cells were transfected with GFP-PAQR3 together with a dominant negative, kinase dead K618N mutant of PKD, PKD-DN (Figure 3B). GFP-PAQR3 was localized to compact Golgi when co-expressed with PKD-DN in contrast to the fragmented, dispersed Golgi localization when expressed alone. Only 13% and 19% of GFP-PAQR3 expressing cells displayed fragmented Golgi upon treatment with Gö6976 and co-expression of PKD-KD, respectively (Figure 1C). Therefore our results thus far suggest that PAQR3 is involved in the Golgi-localized Gβγ-PKD regulated pathway leading to the formation of Golgi vesicles.
Figure 3. PKD inhibitors PKD-DN and Gö6976 inhibit PAQR3-mediated Golgi fragmentation.
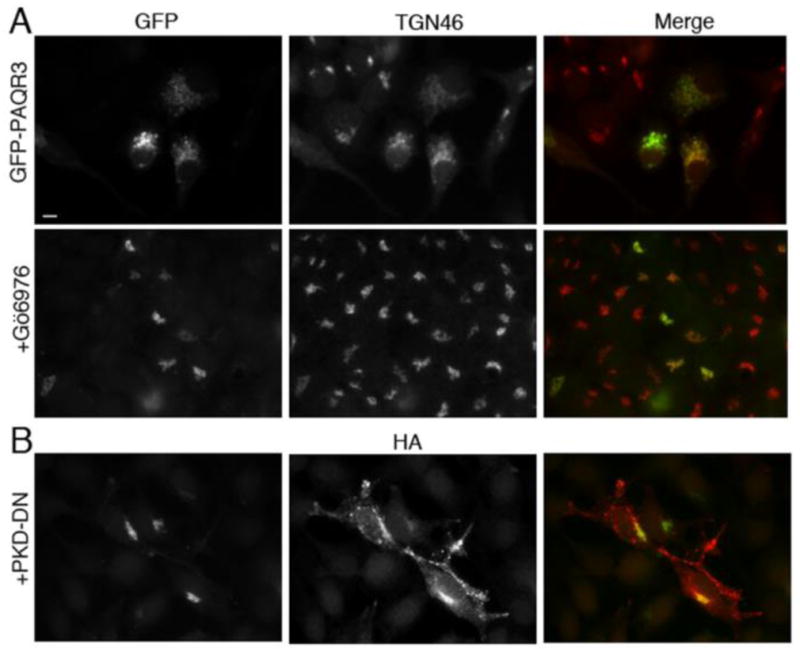
A, HeLa cells were transfected with GFP-PAQR3 for 36–48 h and then treated with 5 μM Gö6976 (bottom row) or vehicle (top row) for 8 h prior to being fixed and processed for immunofluorescence microscopy using anti-TGN46 to detect Golgi morphology. Expressed GFP-PAQR3 was detected by intrinsic GFP fluorescence. B, HeLa cells were transfected with GFP-PAQR3 together with HA-PKD-DN (PKD-K618N). Cells were fixed and stained with anti-HA, and the intrinsic GFP fluorescence allowed visualization of GFP-PAQR3, and, by extension, Golgi morphology. Bar, 10 μm.
3.4 Gβγ binding-deficient mutant PAQR3(NΔ20) inhibits VSV-G transport from the Golgi to the PM
So far we have shown that PAQR3 activates a signaling pathway at the Golgi via Gβγ and PKD leading to the formation of transport vesicles from the Golgi. Previous studies have shown that the Gβγ-PKD signaling pathway at the Golgi is involved in the fission of Golgi vesicles destined to the PM. In a recent study we demonstrated that inhibiting endogenous Gβγ with gallein or with the molecular inhibitor GRK2ct, inhibits Golgi to PM transport [15]. Since our results thus far indicate that PAQR3 is involved in the Gβγ-PKD signaling pathway leading to the formation of PM-destined transport vesicles from the Golgi, we sought to investigate the role of PAQR3 in Golgi to PM transport of cargo. In this experiment, an mCherry-tagged temperature-sensitive mutant form of the vesicular stomatitis virus glycoprotein (VSV-G) is used to follow cargo transport from the Golgi to PM within the cell. The VSV-G temperature-sensitive mutant is synthesized but does not fold properly at the non-permissive temperature of 39.5 °C and is retained in the ER. When transferred to 20 °C, VSV-G localizes in the Golgi, and, upon being transferred to the permissive temperature of 32 °C, VSV-G traffics from the Golgi to the PM. Therefore within 2 h incubation at 32 °C, VSV-G can be strongly detected at the PM. Initially, we used siRNA directed to PAQR3 to investigate its effect on cargo transport. These experiments did not show any change in VSV-G transport to the PM upon siRNA transfection; however, a lack of suitable antibodies to detect endogenous PAQR3 made such negative results difficult to interpret, even though PAQR3 mRNA levels were substantially decreased (data not shown). Next, we examined the effect of over-expressing PAQR3 or PAQR3(NΔ20) on VSV-G transport to the PM (Figure 4A and 4C). When transfected alone, VSV-G localizes at the PM in a majority of cells (> 80%) after 2 h at the permissive temperature of 32 °C. When transfected together with GFP-PAQR3, in a majority of cells, (>65%) VSV-G can be seen in puncta, likely Golgi-derived vesicles in close proximity to the Golgi, with some localization at the PM in the same cell; VSV-G is designated as being at Golgi+PM locales in these cells. Contrastingly, when the Gβγ binding-deficient mutant, PAQR3(NΔ20), was transfected along with VSV-G, PAQR3(NΔ20) did not fragment the Golgi, as also described above (Figure 1), and, importantly, VSV-G transport to the PM was inhibited as VSV-G was retained at the Golgi at the permissive temperature of 32 °C, in a majority of cells (75%) (Figure 4A and 4C). The fact that VSV-G transport is inhibited in the presence of the Gβγ binding-deficient mutant PAQR3(NΔ20) suggests that PAQR3(NΔ20) acts in a dominant negative manner, possibly negating a function of wild type, endogenous PAQR3 in regulating Golgi to PM cargo transport. As a control, we examined VSV-G transport upon expression of an unrelated Golgi-targeted transmembrane protein, the Golgi-localized mutant KDEL receptor [44, 45]. Expression of the mutant KDEL receptor (KDELr) did not cause any fragmentation of the Golgi and did not inhibit the Golgi to PM transport of VSV-G (Figure 4B and 4C), suggesting that the inhibition of VSV-G transport to the PM observed with PAQR3(NΔ20) is not simply an artefact of expression of any Golgi-targeted transmembrane protein, but instead is specific to PAQR3(NΔ20).
Figure 4. Gβ binding-deficient mutant PAQR3(NΔ20) inhibits VSV-G transport from the Golgi to the PM.

A, HEK293 cells were transfected with mCherry-VSV-G alone (top row) or together with GFP-PAQR3 (middle row) or GFP-PAQR3(NΔ20) (bottom row), incubated for 24 h and then transferred to the non-permissive temperature of 39 °C for 16 h. Cells were then shifted to 20 °C for 2 h (after 1 h of which cyclohexamide was added) at which temperature VSV-G localizes to and is retained at the Golgi. Cells were then transferred to the permissive temperature of 32 °C for 2 h for VSV-G transport to the PM. Cells were then fixed and stained with anti-VSV-G followed by Alexa 594-conjugated secondary antibody to better visualize mCherry-VSV-G. PAQR3 and PAQR3-(NΔ20) were detected by intrinsic GFP fluorescence. Bar, 10 μm. B, HEK293 cells were transfected with GFP-VSV-G alone (top row) or together with HA-KDELr (bottom row) and the VSV-G transport assay was performed as described in A. After 2 h incubation at the permissive temperature of 32 °C, cells were fixed and stained with anti-HA antibody to visualize KDELr. GFP-VSV-G was visualized by intrinsic GFP fluorescence. C, Shown is a bar graph quantitating the localization of VSV-G in the absence or presence of co-expressed PAQR3, PAQR3(NΔ20) or KDELr. VSV-G localization in individual cells was scored as predominantly PM (PM), both PM and Golgi (PM/Golgi), or predominantly Golgi (Golgi). Values depicted are the means (+/− S.D., vertical bars) for 3 separate experiments. 100 cells were counted in each experiment. Asterisks indicate statistical significance (p<0.001, t-test) of the indicated bar (*, PM; **, PM/Golgi; ***, Golgi) compared to the corresponding bar of the mCh-VSV-G only sample (first set of bars). D, HEK293 cells were transfected with mCherry-VSV-G together with pcDNA3, GFP-PAQR3, GFP-PAQR3(NΔ20) or HA-KDELr, as indicated, and cells were cultured as described above for A and B. After 2 h at 32 °C to allow transport of VSV-G to the PM, cell surface proteins were biotinylated and isolated as described under Materials and Methods. VSV-G in total cell lysates (upper panel) and cell surface biotinylated VSV-G (lower panel) were detected by immunoblotting with an anti-VSV-G antibody
To confirm the inhibition of VSV-G transport to the PM by PAQR3(NΔ20), we performed cell surface biotinylation assays (Figure 4D) [13]. After incubation of cells at the permissive temperature of 32 °C, VSV-G was detected at the cell surface, as determined by neutravidin bead isolation of cell surface biotinylated proteins. However, consistent with the immunofluorescence microscopy results (Figure 4A–C), expression of PAQR3 and PAQR3(NΔ20) inhibited the cell surface localization of VSV-G. A relatively small fraction of VSV-G was biotinylated when PAQR3 was co-expressed compared to VSV-G expression alone, likely due to the fact that some VSV-G reaches the PM (in cells with VSV-G designated as being at Golgi+PM locales in Figure 4C) when PAQR3 is overexpressed. Virtually no cell surface biotinylated VSV-G was isolated upon co-expression of PAQR3(NΔ20), reinforcing a dominant negative role for PAQR3(NΔ20) in the inhibition of VSV-G transport to the PM. Expression of the control KDELr did not effect the cell surface localization of VSV-G. Taken together with the results in Figures 1–3 indicating that PAQR3 promotes Golgi fragmentation in a Gβγ- and PKD-dependent manner, these results in Figure 4 suggest that PAQR3 regulates Golgi to PM transport via regulation of the Gβγ/PKD Golgi-localized pathway.
4. DISCUSSION
A previous study indicated that PAQR3, also referred to as RKTG, functions as a negative regulator of Gβγ-mediated signaling by sequestering Gβγ at the Golgi and thereby removing it from its active signaling location at the PM [24]. However, our studies here suggest that PAQR3 also has a positive function on Gβγ-mediated signaling; specifically, PAQR3 can promote Gβγ-mediated activation of the PKD-dependent vesicle fission signaling pathway at the Golgi. First, we show that while the expression of full length PAQR3 fragments the Golgi, the Gβ binding-deficient mutant, which lacks the first N-terminal 20 amino acids, does not fragment the Golgi (Figure 1), suggesting that Gβγ plays a role in PAQR3-stimulated Golgi vesicle formation. As described earlier (section 3.1), Golgi fragmentation here represents over-activation of a normal Gβγ- and PKD-mediated Golgi-localized signaling pathway. Secondly, inhibition of Gβγ by GRK2ct or gallein, in the presence of PAQR3, inhibited the Golgi fragmentation caused by PAQR3, confirming that the fragmentation caused by PAQR3 is mediated by Gβγ (Figure 2). Thirdly, PKD is a key regulator of the Gβγ-mediated Golgi vesicle fission signaling pathway, and inhibition of PKD, by a dominant negative, kinase dead mutant of PKD or by a pharmacological inhibitor of PKD, Gö6976, in the presence of PAQR3 inhibited Golgi fragmentation indicating further that PAQR3 plays a role in the Gβγ-PKD regulated pathway leading to Golgi vesicle formation (Figure 3). Lastly, we used the model cargo protein VSV-G to examine the role of PAQR3 in Golgi-to-PM transport. Importantly, the Gβ binding-deficient mutant of PAQR3, PAQR3(NΔ20), inhibited the transport of VSV-G to the PM (Figure 4). In this context, the Gβ binding-deficient mutant is likely exerting a dominant negative effect negating the function of endogenous, wild type PAQR3 in regulating Gβγ-PKD mediated Golgi vesicle fission.
The molecular details of how PAQR3 stimulates the Gβγ-dependent vesicle fission signaling pathway at the Golgi remain to be fully elucidated, but data here are consistent with a model that involves PAQR3 recruiting or retaining Gβγ at the Golgi. Previous work showed that Gβ co-immunoprecipitates with PAQR3 (there termed RKTG) but not with the deletion mutant version of this receptor which lacks the first 20 amino acids of the N-terminus (PAQR3(NΔ20)), and, importantly, Golgi-localized PAQR3, but not PAQR3(NΔ20), promoted Golgi localization of Gβγ [24]. Previous studies in our lab, which included constitutive and inducible targeting of Gβγ to the Golgi and specific inhibition of Golgi-localized endogenous Gβγ, established that Gβγ is localized at the Golgi when regulating the Golgi vesicle fission pathway [15]. Taken together, it is reasonable to postulate that PAQR3 regulates the level of Golgi-localized Gβγ for proper regulation of the generation of Golgi-to-PM transport vesicles.
G proteins are in constant flux in the cell via activation-dependent or constitutive trafficking pathways, cycling between the PM and endomembranes, and Golgi-localized PAQR3 is well positioned for the regulation of Gβγ at the Golgi. Studies have shown that Gβγ, along with at least some Gα, can reversibly translocate from the PM to intracellular membranes upon agonist stimulation of GPCRs [10, 46–50], while G protein subunits have also been shown to move constitutively between the PM and Golgi [12, 51]. Consequently, recent studies in our lab and other reports as well have revealed that a pool of Gβγ resides at the Golgi in basal state cells, likely being retained there while constitutively cycling between the PM and endomembranes, and available to regulate Golgi localized signaling pathways [12, 15]. PAQR3, being a Golgi-localized protein and able to interact with Gβ, is well poised to bind cycling Gβγ, retain the subunits at the Golgi and maintain a Golgi-localized Gβγ pool; thus, this also supports a mechanism for the constitutive regulation of Gβγ at the Golgi, independently of cell surface receptor activation. The biosynthetic pathway of G proteins represents another point at which PAQR3 could interact with Gβγ for Golgi retention. Upon synthesis, the nascent Gβγ dimer arrives at subcellular locations such as the Golgi for association with Gα and assembly of the heterotrimer [10, 11]. Therefore, the Gβγ dimer would be prevalent at the Golgi at this time, and it is possible that PAQR3 functions early in the lifetime of Gβγ to establish a Golgi pool. Regardless of the exact route that Gβγ takes to the Golgi, it is clear that the N-terminus of PAQR3 is strategically located at the cytoplasmic side of the Golgi membrane, positioned to capture Gβγ subunits traversing the Golgi. Our study provides evidence that PAQR3 does indeed promote the regulation of Gβγ-PKD mediated Golgi vesicle fission.
In our previous study it was shown that Golgi-localized Gβγ subunits regulate constitutive cargo transport from the Golgi to the cell surface [15]. This previous study also demonstrated that PM-targeted GRK2ct did not inhibit Golgi-to-PM transport of VSV-G, and thus it was unlikely that Gβγ required translocation from the PM. Also, the cargo transport assays were performed in serum-free medium which is mostly devoid of ligands for cell surface GPCRs, consistent with the idea that in our model of Gβγ regulated constitutive Golgi-to-PM cargo transport, Gβγ activation at the Golgi occurs independently of extracellular stimulation or cell surface receptor activation. As stated previously, the Golgi localized, Gβ-interacting, PAQR3 is well poised for the regulation of Gβγ at the Golgi and regulation of Gβγ by PAQR3 offers insight into a mechanism by which Gβγ can be regulated (at the Golgi) in a constitutive manner, independently of extracellular agonist stimulation, such as in our aforementioned model where Gβγ is regulated at the Golgi to control constitutive Golgi-to-PM cargo transport. It should also be noted however, that our current data do not rule out that PAQR3 might also play a key role in regulating the GPCR activation-dependent translocation of Gβγ from the PM to Golgi, such as in a recent study where it was demonstrated that, in a mouse pancreatic β cell line, regulated insulin secretion was mediated by M3 muscarinic receptor-stimulated translocation of Gβγ to the Golgi [8].
Previous biochemical studies have shown that PAQR3 specifically localized the Gβγ subunit to the Golgi but not the Gα subunit [24]. These studies also indicated that PAQR3 competes with GRK2ct to bind Gβ, and since GRK2ct binds to the interface of Gα/Gβγ, it is possible that PAQR3 directly binds the Gβγ heterodimer and localizes it to the Golgi where Gβγ would then be in its active form and ready to activate the Gβγ-PKD mediated signaling pathway. Although the simplest idea is that PAQR3 stimulates Gβγ-PKD signaling at the Golgi by recruiting or retaining Gβγ at the Golgi, it is also possible that PAQR3 takes a more active role in stimulating Gβγ function at the Golgi. Though two other members of the PAQR family, PAQR 7 and 8, have been shown to activate a Gi heterotrimeric G protein [52], PAQR3 does not possess the characteristics of GPCRs. In fact, although PAQR3 has seven transmembrane domains, its topology is reversed compared to GPCRs; the N-terminus of PAQR3 faces the cytoplasm, while the C-terminus is located in the lumen of the Golgi. Nonetheless, it remains to be tested if PAQR3 could directly activate a heterotrimeric G protein, possibly by functioning as a non-GPCR GEF, similar to what has been recently described for endomembrane G protein activation by GIV/Girdin [53].
It is also important to note that PAQR3 was first identified as a tumor suppressor protein as it had been shown to sequester Raf-1 to the Golgi and inhibit the Ras/MAPK signaling pathway suppressing cell proliferative activity [27, 28]. PAQR3 has been shown to function as a tumor suppressor mainly via its modulatory effects on the Ras/MAPK and PI3K/AKT signaling pathways. PAQR3 appears to play a tumor suppressor role in breast, gastric, bladder and colorectal cancer, as well as in osteosarcoma, melanoma and hepatocellular carcinoma [27, 54–59]. Moreover, PAQR3 inhibits angiogenesis by suppressing VEGF signaling and functionally cooperates with p53 to down regulate signaling leading to epithelial-mesenchymal transition (EMT) [60, 61]. In conformance with PAQR3’s role as a tumor suppressor and the evidence we show here of its role in the regulation of the Gβγ-PKD mediated secretory pathway at the Golgi, it would be interesting to determine whether PAQR3 also modulates this pathway in a tumor suppressive manner, such as by regulating the secretion of protective, anti-cancer proteins and/or excretion of toxic proteins.
PAQR3 is a ubiquitously expressed protein and future studies should be focused on determining its role in the positive regulation of Golgi-localized Gβγ signaling in a physiological context, such as the aforementioned regulation of insulin secretion by Golgi-localized Gβγ [8]. In another recent study using neonatal rat ventricular myocytes (NVRMs), it was shown that cardiac hypertrophy is regulated by Golgi-localized Gβγ. This study identified a signaling pathway in myocytes where Gβγ at the Golgi activates perinuclear PLCε, and PLCε subsequent hydrolyses PI4P, to activate nuclear PKD [30]. It will be of importance to determine whether PAQR3 plays a role in regulating the function of Golgi-localized Gβγ in biological contexts such as these to establish the role of PAQR3 as a positive regulator of Gβγ mediated signaling at the novel subcellular location of the Golgi.
5. CONCLUSION
In summary, we have shown that PAQR3 is involved in the regulation of a Gβγ-PKD signaling pathway at the Golgi that controls the fission of PM-destined transport vesicles. Expression of PAQR3, but not a Gβ binding-deficient mutant, induces fragmentation of the Golgi. Moreover, inhibition of Gβγ or PKD prevents PAQR3-mediated Golgi fragmentation. Lastly, PAQR3(NΔ20), the Gβ binding-deficient mutant, working in a dominant negative manner, inhibits cargo transport from the Golgi to PM. This work offers insight into a novel mechanism by which the function of Gβγ is regulated at a non-canonical, subcellular location.
Highlights.
Expression of PAQR3 enhances Golgi vesicle formation and fragments the Golgi.
A Gβ-binding deficient mutant of PAQR3 does not fragment the Golgi.
Inhibition of Gβγ and PKD inhibits PAQR3 mediated Golgi fragmentation.
Gβ binding-deficient mutant of PAQR3 inhibits Golgi-to-PM vesicle transport.
We identify a novel mechanism for regulation of Gβγ function at the non-canonical location of the Golgi.
Acknowledgments
We thank Drs. Yan Chen (Chinese Academy of Sciences, Shanghai, China), Jeffrey Benovic (Thomas Jefferson University, Philadelphia, PA) Elias T. Spiliotis (Drexel University, Philadelphia, PA), Jennifer Lippincott-Schwartz (NIH), and Piero Crespo (Universidad de Cantabria, Cantabria, Spain), for kindly providing plasmids and reagents. We thank Christopher Fischer (Thomas Jefferson University, Philadelphia, PA) for technical assistance. We would also like to extend our thanks to Dr. Pradipta Ghosh (University of California, San Diego) for advice on biotinylation assays and Lauren Klayman for critical and careful review of this manuscript
This work was supported by NIH grant GM56444 (P.W.)
Abbreviations
- PAQR
progestin and adipoQ receptor
- RKTG
Raf kinase trapping to the Golgi
- PKD
protein kinase D
- PM
plasma membrane
- VSV-G
vesicular stomatitis virus-glycoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Thamara Hewavitharana, Email: thamara.hewavitharana@jefferson.edu.
Philip B. Wedegaertner, Email: philip.wedegaertner@jefferson.edu.
References
- 1.Oldham WM, Hamm HE. Nat Rev Mol Cell Biol. 2008;9(1):60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 2.Milligan G, Kostenis E. Br J Pharmacol. 2006;147(Suppl 1):S46–55. doi: 10.1038/sj.bjp.0706405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hewavitharana T, Wedegaertner PB. Cell Signal. 2012;24(1):25–34. doi: 10.1016/j.cellsig.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boularan C, Kehrl JH. Cell Signal. 2014;26(6):1269–1282. doi: 10.1016/j.cellsig.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmed SM, Angers S. J Recept Signal Transduct Res. 2013;33(3):177–183. doi: 10.3109/10799893.2013.795972. [DOI] [PubMed] [Google Scholar]
- 6.Campden R, Audet N, Hebert TE. J Cardiovasc Pharmacol. 2015;65(2):110–122. doi: 10.1097/FJC.0000000000000198. [DOI] [PubMed] [Google Scholar]
- 7.Khan SM, Sleno R, Gora S, Zylbergold P, Laverdure JP, Labbe JC, Miller GJ, Hebert TE. Pharmacol Rev. 2013;65(2):545–577. doi: 10.1124/pr.111.005603. [DOI] [PubMed] [Google Scholar]
- 8.Saini DK, Karunarathne WK, Angaswamy N, Saini D, Cho JH, Kalyanaraman V, Gautam N. Proc Natl Acad Sci U S A. 2010;107(25):11417–11422. doi: 10.1073/pnas.1003042107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M. Nature. 2013;495(7442):534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marrari Y, Crouthamel M, Irannejad R, Wedegaertner PB. Biochemistry. 2007;46(26):7665–7677. doi: 10.1021/bi700338m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wedegaertner PB. Subcell Biochem. 2012;63:193–223. doi: 10.1007/978-94-007-4765-4_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chisari M, Saini DK, Kalyanaraman V, Gautam N. J Biol Chem. 2007;282(33):24092–24098. doi: 10.1074/jbc.M704246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo IC, Gupta V, Midde KK, Taupin V, Lopez-Sanchez I, Kufareva I, Abagyan R, Randazzo PA, Farquhar MG, Ghosh P. Dev Cell. 2015;33(2):189–203. doi: 10.1016/j.devcel.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blumer JB, Lanier SM. Mol Pharmacol. 2014;85(3):388–396. doi: 10.1124/mol.113.090068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irannejad R, Wedegaertner PB. J Biol Chem. 2010;285(42):32393–32404. doi: 10.1074/jbc.M110.154963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz Anel AM, Malhotra V. J Cell Biol. 2005;169(1):83–91. doi: 10.1083/jcb.200412089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diaz Anel AM. Biochem J. 2007 doi: 10.1042/BJ20070359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malhotra V, Campelo F. Cold Spring Harb Perspect Biol. 2011;3(2) doi: 10.1101/cshperspect.a005280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baron CL, Malhotra V. Science. 2002;295(5553):325–328. doi: 10.1126/science.1066759. [DOI] [PubMed] [Google Scholar]
- 20.Maeda Y, Beznoussenko GV, Van Lint J, Mironov AA, Malhotra V. Embo J. 2001;20(21):5982–5990. doi: 10.1093/emboj/20.21.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fugmann T, Hausser A, Schoffler P, Schmid S, Pfizenmaier K, Olayioye MA. J Cell Biol. 2007;178(1):15–22. doi: 10.1083/jcb.200612017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hausser A, Storz P, Martens S, Link G, Toker A, Pfizenmaier K. Nat Cell Biol. 2005;7(9):880–886. doi: 10.1038/ncb1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nhek S, Ngo M, Yang X, Ng MM, Field SJ, Asara JM, Ridgway ND, Toker A. Mol Biol Cell. 2010;21(13):2327–2337. doi: 10.1091/mbc.E10-02-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Y, Xie X, Zhang Y, Luo X, Wang X, Fan F, Zheng D, Wang Z, Chen Y. Mol Cell Biol. 2010;30(1):78–90. doi: 10.1128/MCB.01038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo X, Feng L, Jiang X, Xiao F, Wang Z, Feng GS, Chen Y. Biochem J. 2008;414(3):399–406. doi: 10.1042/BJ20080948. [DOI] [PubMed] [Google Scholar]
- 26.Tang YT, Hu T, Arterburn M, Boyle B, Bright JM, Emtage PC, Funk WD. J Mol Evol. 2005;61(3):372–380. doi: 10.1007/s00239-004-0375-2. [DOI] [PubMed] [Google Scholar]
- 27.Fan F, Feng L, He J, Wang X, Jiang X, Zhang Y, Wang Z, Chen Y. Carcinogenesis. 2008;29(6):1157–1163. doi: 10.1093/carcin/bgn119. [DOI] [PubMed] [Google Scholar]
- 28.Feng L, Xie X, Ding Q, Luo X, He J, Fan F, Liu W, Wang Z, Chen Y. Proc Natl Acad Sci U S A. 2007;104(36):14348–14353. doi: 10.1073/pnas.0701298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ. Science. 1992;257:1264–1267. doi: 10.1126/science.1325672. [DOI] [PubMed] [Google Scholar]
- 30.Malik S, deRubio RG, Trembley M, Irannejad R, Wedegaertner PB, Smrcka AV. Mol Biol Cell. 2015;26(6):1188–1198. doi: 10.1091/mbc.E14-10-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Y, Sondek J, Harden TK. Biochemistry. 2008;47(15):4410–4417. doi: 10.1021/bi800044n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirui JK, Xie Y, Wolff DW, Jiang H, Abel PW, Tu Y. J Pharmacol Exp Ther. 2010;333(2):393–403. doi: 10.1124/jpet.109.164814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong KC, Billington CK, Gandhi U, Panettieri RA, Jr, Penn RB. FASEB J. 2006;20(9):1558–1560. doi: 10.1096/fj.05-5622fje. [DOI] [PubMed] [Google Scholar]
- 34.Arai K, Maruyama Y, Nishida M, Tanabe S, Takagahara S, Kozasa T, Mori Y, Nagao T, Kurose H. Mol Pharmacol. 2003;63(3):478–488. doi: 10.1124/mol.63.3.478. [DOI] [PubMed] [Google Scholar]
- 35.Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ. Science. 1995;268(5215):1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 36.Nair LA, Inglese J, Stoffel R, Koch WJ, Lefkowitz RJ, Kwatra MM, Grant AO. Circ Res. 1995;76(5):832–838. doi: 10.1161/01.res.76.5.832. [DOI] [PubMed] [Google Scholar]
- 37.Yevenes GE, Peoples RW, Tapia JC, Parodi J, Soto X, Olate J, Aguayo LG. Nat Neurosci. 2003;6(8):819–824. doi: 10.1038/nn1095. [DOI] [PubMed] [Google Scholar]
- 38.Brinks H, Koch WJ. J Cardiovasc Transl Res. 2010;3(5):499–506. doi: 10.1007/s12265-010-9206-6. [DOI] [PubMed] [Google Scholar]
- 39.Casey LM, Pistner AR, Belmonte SL, Migdalovich D, Stolpnik O, Nwakanma FE, Vorobiof G, Dunaevsky O, Matavel A, Lopes CM, Smrcka AV, Blaxall BC. Circ Res. 2010;107(4):532–539. doi: 10.1161/CIRCRESAHA.110.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann DM, Seneviratne AM, Smrcka AV. Mol Pharmacol. 2008;73(2):410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathews JL, Smrcka AV, Bidlack JM. J Neurosci. 2008;28(47):12183–12189. doi: 10.1523/JNEUROSCI.2326-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Science. 2006;312(5772):443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- 43.Bard F, Malhotra V. Annu Rev Cell Dev Biol. 2006;22:439–455. doi: 10.1146/annurev.cellbio.21.012704.133126. [DOI] [PubMed] [Google Scholar]
- 44.Cole NB, Smith CL, Sciaky N, Terasaki M, Edidin M, Lippincott-Schwartz J. Science. 1996;273(5276):797–801. doi: 10.1126/science.273.5276.797. [DOI] [PubMed] [Google Scholar]
- 45.Arozarena I, Matallanas D, Berciano MT, Sanz-Moreno V, Calvo F, Munoz MT, Egea G, Lafarga M, Crespo P. Mol Cell Biol. 2004;24(4):1516–1530. doi: 10.1128/MCB.24.4.1516-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hynes TR, Mervine SM, Yost EA, Sabo JL, Berlot CH. J Biol Chem. 2004;279(42):44101–44112. doi: 10.1074/jbc.M405151200. [DOI] [PubMed] [Google Scholar]
- 47.Ajith Karunarathne WK, O’Neill PR, Martinez-Espinosa PL, Kalyanaraman V, Gautam N. Biochem Biophys Res Commun. 2012;421(3):605–611. doi: 10.1016/j.bbrc.2012.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akgoz M, Kalyanaraman V, Gautam N. J Biol Chem. 2004;279(49):51541–51544. doi: 10.1074/jbc.M410639200. [DOI] [PubMed] [Google Scholar]
- 49.Akgoz M, Kalyanaraman V, Gautam N. Cell Signal. 2006;18(10):1758–1768. doi: 10.1016/j.cellsig.2006.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saini DK, Kalyanaraman V, Chisari M, Gautam N. J Biol Chem. 2007;282(33):24099–24108. doi: 10.1074/jbc.M701191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsutsumi R, Fukata Y, Noritake J, Iwanaga T, Perez F, Fukata M. Mol Cell Biol. 2009;29(2):435–447. doi: 10.1128/MCB.01144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas P, Pang Y, Dong J, Groenen P, Kelder J, de Vlieg J, Zhu Y, Tubbs C. Endocrinology. 2007;148(2):705–718. doi: 10.1210/en.2006-0974. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Marcos M, Ghosh P, Farquhar MG. J Biol Chem. 2015;290(11):6697–6704. doi: 10.1074/jbc.R114.613414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Z, Ling ZQ, Guo W, Lu XX, Pan Y, Wang Z, Chen Y. Oncotarget. 2015;6(14):12357–12368. doi: 10.18632/oncotarget.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma Z, Wang Y, Piao T, Li Z, Zhang H, Liu Z, Liu J. Tumour Biol. 2015;36(5):3319–3324. doi: 10.1007/s13277-014-2964-z. [DOI] [PubMed] [Google Scholar]
- 56.Wang X, Li X, Fan F, Jiao S, Wang L, Zhu L, Pan Y, Wu G, Ling ZQ, Fang J, Chen Y. Carcinogenesis. 2012;33(11):2228–2235. doi: 10.1093/carcin/bgs245. [DOI] [PubMed] [Google Scholar]
- 57.Wu HG, Zhang WJ, Ding Q, Peng G, Zou ZW, Liu T, Cao RB, Fei SJ, Li PC, Yang KY, Hu JL, Dai XF, Wu G, Li PD. Oncol Rep. 2014;32(6):2687–2695. doi: 10.3892/or.2014.3532. [DOI] [PubMed] [Google Scholar]
- 58.Ling ZQ, Guo W, Lu XX, Zhu X, Hong LL, Wang Z, Chen Y. Ann Oncol. 2014;25(7):1363–1372. doi: 10.1093/annonc/mdu168. [DOI] [PubMed] [Google Scholar]
- 59.Xiu Y, Liu Z, Xia S, Jin C, Yin H, Zhao W, Wu Q. PLoS One. 2014;9(10):e109734. doi: 10.1371/journal.pone.0109734. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Jiang Y, Xie X, Li Z, Wang Z, Zhang Y, Ling ZQ, Pan Y, Chen Y. Cancer Res. 2011;71(8):2959–2968. doi: 10.1158/0008-5472.CAN-10-4077. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Jiang X, Qin X, Ye D, Yi Z, Liu M, Bai O, Liu W, Xie X, Wang Z, Fang J, Chen Y. Oncogene. 2010;29(39):5404–5415. doi: 10.1038/onc.2010.270. [DOI] [PubMed] [Google Scholar]
- 62.Qiao S, Guo W, Liao L, Wang L, Wang Z, Zhang R, Xu D, Zhang Y, Pan Y, Chen Y. Biochem J. 2015;469(3):469–480. doi: 10.1042/BJ20150253. [DOI] [PubMed] [Google Scholar]