

Abstract
A variety of water-soluble polymers, when attached to a liposome, substantially increase liposome circulation half-life in animals. However, in certain conditions, liposomes modified with the most widely used polymer, polyethylene glycol (PEG), induce an IgM response resulting in an accelerated blood clearance (ABC) of the liposome upon the second injection. Modification of liposomes with other water-soluble polymers: HPMA (poly[N-(2-hydroxypropyl) methacrylamide]), PVP (poly(vinylpyrrolidone)), PMOX (poly(2-methyl-2-oxazoline)), PDMA (poly(N,N-dimethyl acrylamide)), and PAcM (poly(N-acryloyl morpholine)), increase circulation times of liposomes; but a precise comparison of their ability to promote long circulation or induce the ABC effect has not been reported. To obtain a more nuanced understanding of the role of polymer structure/MW to promote long circulation, we synthesized a library of polymer diacyl chain lipids with low polydispersity (1.04–1.09), similar polymer molecular weights (2.1–2.5 kDa) and incorporated them into 100 nm liposomes of a narrow polydispersity (0.25–1.3) composed of polymer-lipid/hydrogenated soy phosphatidylcholine/cholesterol/diD: 5.0/54.5/40/0.5. We confirm that HPMA, PVP, PMOX, PDMA and PAcM modified liposome have increased circulation times in rodents and that PVP, PDMA, PAcM do not induce the ABC effect. We demonstrate for the first time, that HPMA does not cause an ABC effect whereas PMOX induces a pronounced ABC effect in rats. We find that a single dose of liposomes coated with PEG and PMOX generate an IgM response in rats towards the respective polymer. Finally, in this homologous polymer series, we observe a positive correlation (R = 0.84 in rats, R = 0.92 in mice) between the circulation time of polymer-modified liposomes and polymer viscosity; PEG and PMOX, the polymers that can initiate an ABC response were the two most viscous polymers. Our findings suggest that that polymers that do not cause an ABC effect such as, HPMA or PVP, deserve further consideration as polymer coatings to improve the circulation of liposomes and other nanoparticles.
Graphical abstract
INTRODUCTION
Poly(ethylene glycol) (PEG) modified materials are used with remarkable success to improve protein and liposome delivery in animals as well as humans.[1,2] The qualities that make PEG an appropriate candidate for drug/protein delivery applications include: high aqueous solubility, inexpensive to manufacture, pronounced biocompatibility, and it is a component of numerous FDA approved products.[3] The attribute that makes PEG the most commonly used polymer for use in parenteral drug delivery is its ability to extend the circulation lifetimes of therapeutics.[4,5] PEG accomplishes this by decreasing renal clearance through increased molecular weight and by acting as a steric shield to prevent the adsorption of serum opsonins. It can also reduce the interaction energy between the particle and cells, which greatly reduces uptake by the mononuclear phagocyte system (MPS).[6] PEG’s attachment to a therapeutic increase its circulation half-life, hence improving the pharmacokinetics (PK) and alters the materials biodistribution (BD). These attributes allow for prolonged therapeutic activity, increased efficacy by improving solid tumor uptake, and decreased off-target effects by reducing uptake into the MPS.[4,7] PEG, however, is not immunologically inert.[8] Between 0.6–25% of healthy blood donors exhibit preexisting anti-PEG IgM, presumably due to exposure to PEG at some point in the donor’s lifetime.[8,9,10] Moreover, a low dose of PEGylated material can initiate an immune response to generate anti-PEG IgM antibodies in various species.[9,11,12] It was subsequently observed that the first dose of a PEGylated material may initiate the production of anti-PEG IgM, and subsequent doses are rapidly cleared from circulation which is attributed to anti-PEG IgM binding to the PEG-shielded nanoparticle.[10] This results in the material rapidly accumulating in the liver and spleen.[11] The production of anti-PEG IgM in rats takes 3–5 days, and thus no accelerated blood clearance is observed on the initial dose of a PEGylated material.[10,13] However, many treatment strategies require multiple doses within the time window of the accelerated blood clearance effect (3–28 days post first dose).[13,14] In circumstances were this occurs, a therapeutic can completely lose its long circulating properties, and increased toxicity is possible due to altered biodistribution.[11,15]
A number of factors can alter or inhibit the ABC effect, including dose size,[15] therapeutic payload,[16] administration regimen,[13] and even PEG end-groups.[17] Still, many materials can initiate the ABC effect, and patients who have been previously exposed to PEG may exhibit anti-PEG IgM. Another possible method for avoiding the ABC effect is to use a polymer similar to PEG that does not result in an immune response but maintains the long circulation.
A number of other hydrophilic polymers have been reported to extend circulation times of drugs or liposomes. HPMA (poly[N-(2-hydroxypropyl) methacrylamide]),[18–20] PVP (poly(vinylpyrrolidone)),[21,22] PMOX (poly(2-methyl-2-oxazoline)),[23,24] PAcM (poly(N-acryloyl morpholine)),[25,26] PDMA (poly(N,N-dimethylacrylamide)),[27] polyglycerol,[28] and polyhydroxyethylaspartic acid[29] have all demonstrated the ability to increase the circulation half-life of various materials. PVP, PMOX, PAcM, and HPMA have been investigated as polymer coatings on liposomes, however, inconsistent experimental procedures among the various papers make it difficult to compare the extent to which they extend the circulation half-lives of liposomes (Table S1). Though materials conjugated to these polymers other than PMOX do not exhibit as long of circulation half-lives as PEG, other favorable properties could make them a better choice for certain drug delivery applications. Advantages of other polymers can include: improved physical properties, increased functional handles for drug loading, low viscosities and more versatile post-polymerization functionalization.[3] A handful of these polymers are currently undergoing clinical trials.[1,30,31]
Herein, we have synthesized a panel of polymers of similar molecular weights with low polydispersity and evaluated their in vivo behavior as polymer coatings for liposomes. The polymers evaluated are PEG, PMOX, HPMA, PVP, PDMA, and PAcM. We have compared the circulation times of liposomes modified with these polymers with similar molecular weights in mice and rats under consistent experimental conditions. We have also identified a correlation between polymer viscosity and circulation half-life. Furthermore, we investigated these polymers for their ability to avoid an accelerated blood clearance phenomenon and document that PMOX induces a pronounced ABC effect while HPMA and others do not cause the ABC effect.
MATERIALS AND METHODS
General
All reagents were purchased from commercial sources and used without further purification unless otherwise noted. 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (ammonium salt) (PEG-DSPE), hydrogenated L-α-phosphatidylcholine from soy (HSPC), cholesterol, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidomethyl)cyclohexane-carboxamide] (sodium salt) (MCC PE) were purchased from Avanti Polar Lipids (Alabaster, AL). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiD) was obtained from Life Technologies. HPMA was obtained from PolySciences Inc. and all other monomers were obtained from Sigma Aldrich. Cholesterol was recrystallized from acetone/hexanes before use. AIBN was recrystallized from methanol before use. N-(2-Hydroxypropyl)methacrylamide was recrystallized from acetone and all other monomers were distilled prior to polymerization. Water was purified to a resistance of 18 MΩ using a Barnstead NANOpure® Diamond™ purification system. N,N-dimethylformamide (DMF), triethylamine (TEA), chloroform (CHCl3) and methylene chloride (DCM) were purified by passing the solvents under nitrogen pressure through two packed columns of neutral alumina within a commercial solvent purification apparatus (Glass Contour, Laguna Beach, CA). All glassware was flame dried under vacuum or nitrogen purge prior to use and reactions were conducted under a nitrogen atmosphere. Unless otherwise noted, liquid reagents were introduced to the reaction flask via syringe or cannula. Volatile solvents were removed using a rotary evaporator under reduced pressure. All dialyses were performed in MeOH or H2O using Spectra/POR® regenerated cellulose dialysis bags.
Female CD-1 mice (6 – to 8-week old, 25 to 30 g in weight) were obtained from Harlan Laboratories, Inc. (Indianapolis, IN). Male Wistar rats (250–275 g) with jugular vein cannulas were obtained from Charles River Laboratories, Inc. (Wilmington, MA) and housed in the University of California San Francisco animal care facility with a 12-h light/dark cycle and allowed free access to water and food. All animals were handled in accordance with guidelines established by the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals, and with the approval of the Committee of Animal Research at the University of California, San Francisco. The studies described here were approved by the Committee on Animal Research, University of California, San Francisco.
Characterization
All NMR spectra were measured in CDCl3, CD2Cl2, MeOD (d-4), or D2O with TMS or solvent signals as the standards. 1H NMR spectra were recorded with a Bruker AVQ-300, an AVQ-400, a DRX-500, or an AV-600 and analyzed with Topspin software. MALDI-TOF data was collected on a Microflex Lt instrument (Bruker, Billerica, MA) in positive ion mode. MALDI samples were prepared using a matrix of a saturated solution of 2,5-hydroxybenzoic acid in MeOH unless otherwise specified. The spectra were analyzed with FlexAnalysis and PolyTools 1.0 software. Size exclusion was performed on two different systems, both calibrated with PEG standards. Polymer samples were dissolved in the solvent of the mobile phase at a concentration of 2 mg/ml and filtered through a 0.2 μm PVDF filter before injection. The first system consisted of two PSS columns (7.5 × 300 mm) with particle size of 5 μm with DMF as the mobile phase. The system consisted of a Waters 510 pump, a Waters U6K injector, a Waters 486 UV-Vis detector, and a Waters 410 differential refractive index detector. The columns were maintained at 70°C. The second system was run at 30°C in aqueous buffer (0.10 M NaNO3, 0.02 wt% NaN3) as eluent on three Waters Ultrahydrogel columns (exclusion limit = 3 × 106; pore size = 5000A; flow rate = 1 mL/min) equipped with a Viscotek 302 TDA. Fluorescence spectroscopy was measured on a FluoroLog-3 spectrofluorimeter equipped with a temperature-controlled stage (LFI-3751) with FluorEssence software (Horiba Scientific, Edison, NJ). Zeta potential and size measurements were carried out using a Nano-ZS Dynamic Light Scattering Instrument from Malvern (Westborough, MA). UV-VIS measurements were performed with a Lambda 35 UV-VIS spectrometer (PerkinElmer, Wellesley, MA). Measurements were performed in sealed, standard 1 cm quartz cells in the reaction solvent at room temperature.
General RAFT polymerization
All RAFT polymers were synthesized by adding monomer, RAFT chain transfer agent, solvent, and AIBN, in that order, to a flame dried flask. The solvent, RAFT agent, and molar equivalences varied by polymer, and are reported in the experimental details for each specific polymer. After 5 freeze-pump-thaw cycles the flask was backfilled with argon and stirred at 65°C. The polymerizations were monitored by MALDI by removing a small aliquot from the reaction flask. To quench RAFT polymerizations, the solution was exposed to air and cooled in an ice bath. All polymers were purified by dialysis. Yield is reported as % mass isolated for given conversion, based on molecular weight.
General post-polymerization modification of RAFT polymers
Aminolysis of all RAFT polymers yielded thiol terminated polymers, which were then conjugated to MCC PE in situ. The general procedure for post-polymerization functionalization was as follows. Polymer was added to a flame dried flask charged with a stir bar and dissolved in either THF or DMF at 100 mg/mL. The solution was subjected to 5 freeze-pump-thaw cycles and back filled with Ar. Propylamine was added to the solution dropwise (10 mol eq.). The reaction was stirred at room temperature for 3 hours at which point complete aminolysis was confirmed by UV-VIS spectrometry. Without exposure to oxygen, propylamine and solvent were removed under reduced pressure. After all liquids were removed, the flask was then backfilled with Ar. For polymers for viscosity measurements, an Ar-sparged solution of additional monomer (20 mol eq.) in THF (1:1 vol/vol) was added to the reaction flask. For polymers for liposome incorporation, an Ar-sparged solution of MCC PE (2.2 mol eq.) in THF or DMF was added to the remaining solids. The reaction was stirred overnight at room temperature. The final polymer-lipid conjugate was purified by dialysis in MeOH in 2 kDA MWCO regenerated cellulose bags, dialysis in water in 50 kDa MWCO regenerated cellulose bags, and reverse phase column chromatography with PrepSep™ SPE tubes (Fisher Scientific International Inc., Hampton, NH). Polymer lipid conjugates are labeled based on the molecular weight of the polymer.
PDMA (2.1 kDA)
Solvent: DMF. RAFT agent: 2-Cyano-2-propyl dodecyl trithiocarbonate Molar eq: N,N-dimethylacrylamide/RAFT agent/AIBN = 25/1/0.2. 1H NMR (300 MHz, CDCl3): δ 0.82–0.95 (t, 3H), 1.11–1.95 (br m, 70H), 2.40–2.83 (br m, 21H), 2.83–3.31 (br m, 132H). Mn,MALDI: 2.1 kDa, PDIMALDI: 1.04. Yield = 85 %.
PDMA-MCC PE (2.1 kDA)
1H NMR (300 MHz, CDCl3): δ 0.85–0.93 (t, 6H), 1.15–1.96 (br m, 96H), 2.41–2.81 (br m, 21H), 2.81–3.41 (br m, 128H). Mn,MALDI: 3.0 kDa, PDIMALDI: 1.06. Yield = 66 %.
PAcM (2.4 kDA)
Solvent: DMF. RAFT agent: 2-Cyano-2-propyl dodecyl trithiocarbonate Molar eq: acryloylmorpholine/RAFT agent/AIBN = 25/1/0.3. 1H NMR (300 MHz, CDCl3): δ 0.82–0.96 (t, 3H), 1.15–1.95 (br m, 58H), 2.34–2.60 (br m, 18H), 3.20–3.91 (br m, 146H). Mn,MALDI: 2.4 kDa, PDIMALDI: 1.04. Yield = 89 %.
PAcM-MCC PE (2.4 kDA)
1H NMR (300 MHz, CDCl3): δ 0.82–0.94 (t, 6H), 0.94–1.1 (br m, 4H), 1.15–2.06 (br m, 82H), 2.34–2.86 (br m, 18H), 3.16–4.19 (br m, 152H). Mn,MALDI: 3.3 kDa, PDIMALDI: 1.03. Yield = 54%
PVP (2.1 kDA)
Solvent: toluene. RAFT agent: Cyanomethyl N-methyl-N-phenyl dithiocarbamate Molar eq: vinyl-pyrrolidone/RAFT agent/AIBN = 25/1/0.3. δ 1.59–2.61 (br m, 120H), 2.98–3.48 (br m, 43H), 3.48–4.22 (br m, 21H), 7.21–7.70 (br m, 5H). Mn,MALDI: 2.1 kDa, PDIMALDI: 1.07. Yield = 76 %.
PVP-MCC PE (2.1 kDA)
1H NMR (300 MHz, CD2CL2): δ 0.79–0.96 (t, 6H), 1.03–2.58 (br m, 172H), 3.03–3.46 (br m, 44H), 3.46–4.32 (br m, 29H). Mn,MALDI: 3.1 kDa, PDIMALDI: 1.07. Yield = 41 %.
HPMA (2.5 kDA)
Solvent: t-Butanol. RAFT agent: 2-Cyano-2-propyl benzodithioate Molar eq: N-(2-hydroxypropyl)methacrylamide/RAFT agent/AIBN = 25/1/0.3. 1H NMR (300 MHz, MeOD): δ 0.89–1.41 (br m, 108H), 1.41–2.11 (br m, 34H), 2.91–2.31 (br m, 34H), 3.86–3.99 (br m, 17H), 7.33–7.90 (br m, 5H). Mn,MALDI: 2.5 kDa, PDIMALDI: 1.06. Yield = 81 %.
HPMA-MCC PE (2.5 kDA)
1H NMR (300 MHz, MeOD): δ 0.82–0.91 (t, 6H), 0.91–1.42 (br m, 148H), 1.51–2.11 (br m, 30H), 2.91–2.29 (br m, 30H), 3.80–4.06 (br m, 15H). Mn,MALDI: 3.4 kDa, PDIMALDI: 1.06. Yield = 69 %.
General PMOX Polymerization
A flame dried, high-pressure reaction vial was charged with a stir bar, methyl oxazoline, and acetonitrile to yield a 5 M methyl oxazoline solution. The solution was sparged with Ar for 15 min and cooled to 0°C. An Ar-sparged solution of methyltosylate in ACN was added to the reaction vial and the solution was heated at 90°C. The polymerization was monitored by MALDI by removing a small aliquot from the reaction flask and adding it to 0.01 M NaOH solution in water. At the target molecular weight, the polymerization was quenched with one of two alternative methods. Polymers for viscosity measurements and polymers for conjugation with 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-succinyl (succinyl PE) were quenched with 0.01 M NaOH solution in water. Polymers with a cationic linker to the lipid were quenched with a solution of dioctyldecylamine (5 mol eq to methyltosylate) in ACN (0.5 M) at 90°C. All polymers were purified by dialysis in MeOH.
PMOX-Dioctadecylamine (2.6 kDa)
1H NMR (300 MHz, CDCl3): δ 0.82–0.86 (t, 6H), 1.18–1.3 (br m, 62H), 2.01–2.16 (br m, 69H), 3.20–3.6 (br m, 95H). Mn,MALDI: 2.6 kDa, PDIMALDI: 1.09. Yield = 73 %.
PMOX-Succinyl PE (2.4 kDa)
PMOX-OH (300 mg, 1 eq) was added to a flame dried flask charged with a stir bar and DCM (2 mL). The solution was sparged with Ar for 10 min. Succinyl PE (112 mg, 1.1 eq.) was then added, followed by EDC (71.9 mg, 3 eq) and DMAP (3 mg, 0.2 eq.). The reaction was allowed to run for 14 h and was purified by dialysis in a 2 kDa MWCO bag (2× water, 2× MeOH, 2× 50 v/v % MeOH/DCM). (2.6 kDa): 1H NMR (300 MHz, CDCl3): δ 0.82–0.88 (t, 6H), 1.11–1.62 (br m, 56H), 2.01–2.76 (br m, 82H), 3.20–3.9 (br m, 110H). Mn,MALDI: 3.2 kDa, PDIMALDI: 1.04. Yield = 71 %.
Liposome formulation
Liposomes for in vivo experiments were prepared by an ethanol injection method followed by high pressure extrusion. A lipid solution in CHCl3 consisting of the polymer lipid conjugate, HSPC, cholesterol, and DiD at a molar ratio of 5.0: 54.5:40:0.5 (80 μmol total) was prepared. For conventional liposomes, polymer-lipid was replaced with POPG. The chloroform was removed via rotary evaporation to yield a thin film, which was subjected to high vacuum overnight. The lipid film was dissolved in ethanol (300 μL) at 65°C and added to 2.7 mL HEPES buffer (10 mM HEPES, 140 mM NaCl, pH 7.4) at 65°C in one portion. The resulting solution was extruded through 200 nm (×3), 100 nm (×3), and then 80 nm (×3) polycarbonate membranes. Liposomes were purified by dialysis in 100 × volume of 10 mM HEPES buffered saline pH 7.4 (HBS), diluted to desired concentrations in HBS for animal experiments, and then filtered through a 0.2 μm polyethersulfone sterile membrane. Endotoxin levels of selected liposome samples (PMOX initial ABC dose, PVP7 kDa and HPMA9 kDa) were measured with a Pierce ® LAL chromogenic endotoxin quantification kit and found to contain <0.15 EU/mg phospholipid.
PK and biodistribution in mice
Female CD-1 mice were administered DiD-labeled liposomes at 10 μmol phospholipid/kg via tail vein injection in 175–225 μL HEPES buffer. At selected time points (10 min, 30 min, 90 min, 4.5 h, 24 h, and 48 h), blood was collected into centrifuge tubes with 5 μL of heparin sulfate solution in PBS (5 mg/mL) by submandibular cheek bleeding. The blood samples were centrifuged for 5 min at 7,000 × g at 4°C. The supernatant (plasma) was collected and analyzed by spectrofluorimetry (excitation 644 nm, emission 664 nm) to detect the DiD label. Each group consisted of n = 3 mice.
At 24 h or 48 h after administration, animals were anesthetized with isoflurane. The liver, spleen, lung, and liver, of each mouse were excised and transferred into separate tubes with 1 mL of 0.075 M HCl in 90% isopropanol/10% water and 1 g zirconium beads. The tissues were then homogenized in a Mini-Beadbeater (Biospec Products, Inc., Bartlesville, OK) for 20 s. The homogenate was stored for 12 h at 4°C and then clarified by centrifugation in a microcentrifuge at 7,000 × g at 4°C for 5 min. The supernatant was collected and analyzed by spectrofluorimetry.
PK and biodistribubution in rats
Male Wistar rats were administered DiD-labeled liposomes at 10 μmol phospholipid/kg via jugular cannula in 200–300 μL HEPES buffer. After the liposome administration, the cannula was flushed with 500 μL saline including 10U/mL heparin followed by 100 μL of 50% dextrose including 500 U/mL heparin. This flushing process was performed after every blood withdrawal. At selected time points (10 min, 30 min, 90 min, 4.5 h, 24 h, and 48 h), blood was collected from the jugular vein cannula into BD Microtainer® tubes coated with lithium heparin. The blood samples were centrifuged for 5 min at 7,000 × g at 4°C. The supernatant was collected and analyzed by spectrofluorimetry. Each group consisted of n = 3 rats.
At 48 h after administration, animals were anesthetized with an overdose of pentabarbitol. The liver, spleen, lung, and liver, of each rat were excised and transferred into separate tubes with 0.075 M HCl in 90% isopropanol/10% water at a concentration of 1 g tissue/2.5 ml isopropanol solution. The tissues were then homogenized with an Omni THQ digital tissue homogenizer (Omni Int., Kennesaw, GA) for 120 s. The homogenate was stored for 12 h at 4 °C and then clarified by centrifugation at 2900 × g at 4°C for 5 min. The supernatant was collected and analyzed by spectrofluorimetry to detect the DiD.
ABC effect in rats
Male Wistar rats were administered non-labeled liposomes at 0.1 μmol phospholipid/kg via jugular cannula in 200–300 μL HEPES buffer. Seven days later, rats were administered DiD-labeled liposomes at 10 μmol phospholipid/kg via jugular cannula in 200–300 μL HEPES buffer. At selected time points (10 min, 30 min, 90 min, 4.5 h, 24 h, and 48 h), blood was collected from the jugular vein cannula into BD Microtainer® tubes coated with lithium heparin. The blood samples were centrifuged for 5 min at 7,000 × gat 4°C. The supernatant was collected and analyzed by spectrofluorimetry. Each group consisted of n = 4 rats. At 48 h, animals were anesthetized with an overdose of pentabarbitol and biodistribution was determined following the same methods reported for the rat PK studies.
Sample and data analysis
Supernatant of blood (plasma) and tissue homogenate after centrifugation (7,000 × g for blood, 2380 × g for tissue homogenate) included intact DiD-labeled liposome. Standard curves for each polymer-liposome were made by titrating DiD-labeled liposomes into the plasma and homogenized tissues from an untreated animal. DiD in each plasma and tissue sample was measured by spectrofluorimetry (excitation 644 nm, emission 664 nm) using DiD-labeled liposome standard curves prepared in the same matrix. Remaining percentage of liposome (% Inj Dose) at each time point was calculated by multiplying plasma DiD concentration, blood volume (6% for mouse or 7% for rat of total body weight) and plasma ratio (60% plasma in blood), and then divided by injected dose of liposome (10 μmol phospholipid/kg). The data for each liposome formulation group as a whole was analyzed using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). Area under the plasma percentage (% Inj Dose) vs. time curve (AUC) was calculated by a trapezoidal rule, and half-life was calculated by fitting each individual animal to the two-compartment model. Each parameter was represented as mean value +/− standard error. Once AUC, circulation half-life and accumulation in organs were found, p values were determined using the Student’s t-test. A p value ≤0.05 was considered significant (see supplemental information).
Anti-Polymer IgM Detection
The production of IgM against each polymer was evaluated by ELISA. Polymer-lipid conjugates were dissolved in ethanol and added to 96-well flat-bottom polypropylene plates (PlateOne) (50 μL, 20 nmol polymer-lipid/well). The wells were air-dried overnight. Serum samples were diluted with PBS-C (PBS, 0.5% casein) at a 1:25 dilution to identify samples with positive and negative serum IgM, or serially from 1:25 to 1:12800 to determine reciprocal endpoint titers. Diluted serum samples were added to the plate and the wells were incubated at 37°C for two hours and then washed six times with PBS-T (PBS, 0.1% Tween-20). Horseradish peroxidase-conjugated goat anti-rat IgM mu chain secondary antibody (Abcam) was then diluted 1:1000 in PBS-C and 100 μL was added to each well and incubated for 30 minutes at 37 °C. Following six washes with PBS-T, 100 μL of a tetramethylbenzidine substrate solution (Sigma Aldrich) was added to wells and incubated for 30 minutes at room temperature. The reaction was stopped with 100 μL of 0.5 M H2SO4 and the yellow product was monitored at 450 nm (Optimax, Molecular Devices, Sunnyvale, CA). Titer was defined as the reciprocal dilution of antisera yielding an optical density twice that of background. Samples were assayed in duplicate. Data analyses were performed using GraphPad Prism.
RESULTS
Synthesis
RAFT polymerizations were chosen for the vinyl polymers in order to achieve low polydispersity and a functional terminal handle for lipid conjugation (Scheme 1A). The selection of RAFT chain transfer agent and solvent for each polymerization were based upon previous literature reports, and proved critical for attaining low polydispersity polymers.[32] Following polymerization, RAFT endgroups were converted to thiols by aminolysis with propylamine under inert atmosphere. The free thiol end group was then either reacted with additional monomer for viscosity measurements, or MCC PE for liposome incorporation (Scheme 1B).
Scheme 1.
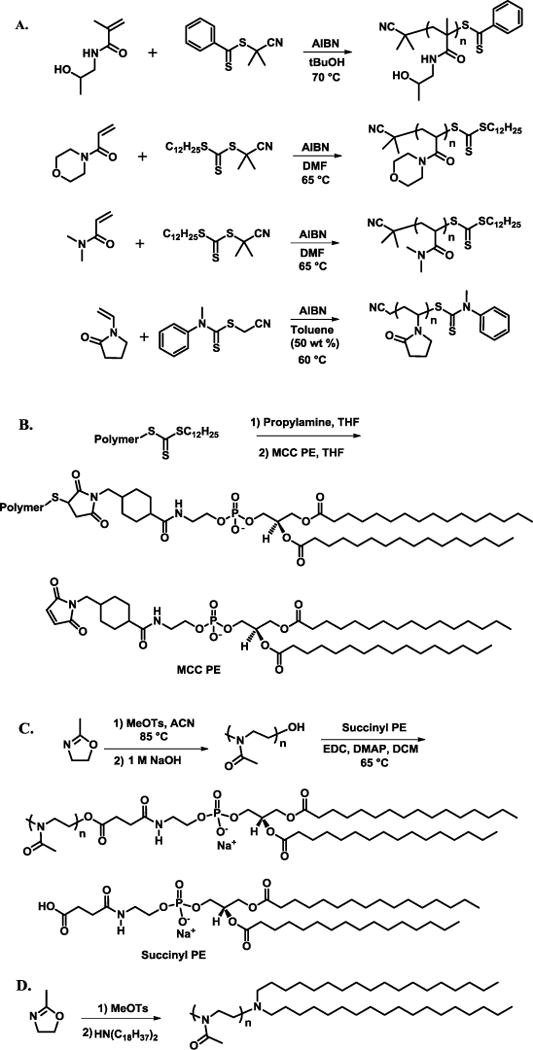
Synthetic schemes for HPMA polymerization, PAcM polymerization, PDMA polymerization, PVP polymerization (A, top to bottom), general polymer conjugation to MCC PE lipid (B), PMOX polymerization followed by lipid conjugation (C) (D).
Unlike the RAFT polymers, PMOX was synthesized through a cationic ring-opening polymerization according to previous literature procedures.[33] The polymerization of oxazolines is commonly initiated by a variety of alkylating agents and can be terminated with a wide variety of nucleophiles. In this case, PMOX polymerizations were terminated with basic water to yield a terminal hydroxyl on the polymer. For incorporation into liposomes, hydroxyl-terminated PMOX was functionalized with succinyl PE via carbodiimide coupling (Scheme 1C). In a second route, the PMOX-lipid conjugate was formed by terminating the polymerization with dioctadecylamine. This route yielded a polymer-lipid linkage with a tertiary amine, which is protonated and positively charged at physiological pH (Scheme 1D).
Liposome formulation
To investigate the ability of the selected polymers to extend circulation and induce an ABC effect, polymer-lipids were incorporated into a standard stable liposome composition that contained the DiD fluorophore. Liposomes are a versatile platform that can be tailored to yield desired in vivo properties, and have been extensively investigated for drug delivery applications.[34] When liposomes are formulated with a PEG-lipid, circulation half-life is greatly increased and uptake by the Mononuclear Phagocyte System (MPS) is greatly decreased.[35] Other polymers have also been shown to extend circulation when conjugated to the surface of liposomes (Table S1).[21]
Stable liposomes composed of HSPC, cholesterol, polymer-lipid conjugate, and DiD were formed through an ethanol injection method followed by high pressure extrusion. All liposome solutions were passed through a sterile filter and measured by DLS prior to in vivo studies. The diameter of the various modified liposomes used in these studies was ~100 nm +/− 10 nm with low polydispersity (Table 1).
Table 1.
Polymer-coated liposome diameters used in the in vivo studies. The variable in the diameter is +/− 10 nm.
| Polymer | Diameter (nm) | PDI |
|---|---|---|
| PEG | 107.7 | 0.13 |
| PMOX | 106.7 | 0.14 |
| PVP | 91.4 | 0.03 |
| HPMA | 110.0 | 0.07 |
| PAcM | 100.2 | 0.05 |
| PDMA | 99.8 | 0.13 |
| No Polymer | 98.7 | 0.06 |
Pharmacokinetics and Biodistribution of a single dose
The polymer modified liposomes prepared here increase liposome circulation in mice confirming previous reports (Supporting Information). However the increase in circulation half-life for certain polymers was longer than had previously been reported (Table S1).
The pharmacokinetics and biodistribution of each polymer-liposome at a 10 μmol phospholipid/kg dose were then evaluated in rats. All six polymers extended circulation in comparison to liposomes without polymer (Figure 1). Liposomes modified with PEG and PMOX provided the two longest circulating liposomes with circulation half-lives >30 h. Additionally, accumulation in the liver and spleen was reduced when all of the six polymers that were incorporated into the liposomes (Figure 2). Here again, PEG and PMOX reduced the amount of the injected dose in the liver and spleen better than the other polymer coatings, with less than 20% of the injected dose in the liver after 48 h. All liposomes incorporating polymers had <10% of the injected dose in the spleen. Moreover all polymer coatings had significantly longer half-lives in blood than did unmodified liposomes. Liposomes without polymer accumulated predominantly in the liver (31%) and spleen (11%). The fraction of the injected dose recovered in the plasma, liver, spleen, kidneys, and lungs was around 50% for each polymer liposome at 48 hours post dosing.
Figure 1.
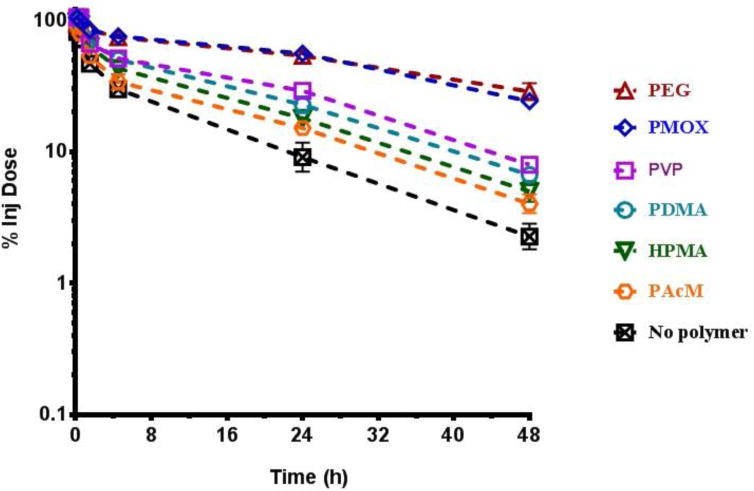
Elimination of a single dose of liposomes from blood in rats over 48 h. (Mean +/− SEM, n=3)
Figure 2.
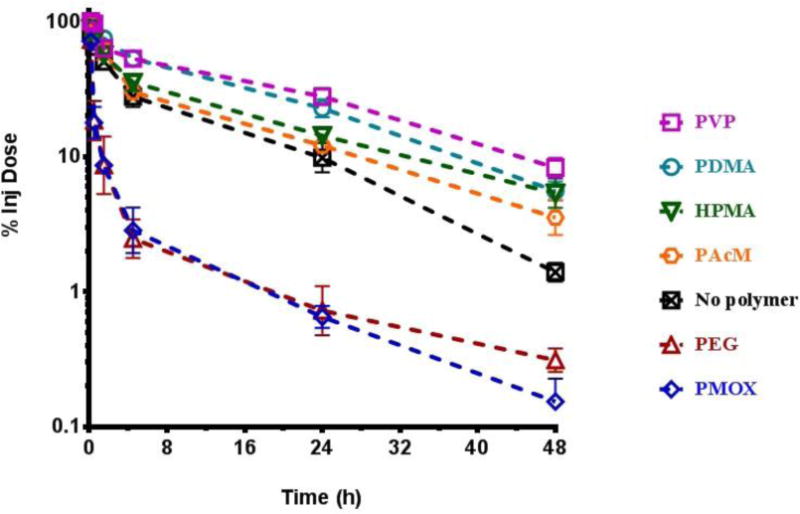
Elimination of the second dose of liposomes from blood in rats administered one week after the first dose measured over 48 h after administration of the second dose. (Mean +/− SEM, n=3–4)
Accelerated Blood Clearance in Rats
Each of the liposomes modified with the polymer lipids were then injected one week later into animals that had received the same polymer to determine if the polymer induced the ABC effect. The ABC effect is most prominent when a small (≤0.1 mol phospholipid/kg) first dose is followed 6–8 days later by a second dose.[13] When the ABC effect is elicited, the second dose is rapidly cleared from circulation and an increased accumulation in the liver and spleen is observed. If no ABC effect is observed, the second dose of a material will exhibit a similar pharmacokinetic profile and biodistribution as did the first dose.
To initiate the ABC effect, non-labeled polymer-coated liposomes were administered to rats at a concentration of 0.1 μmol phospholipid/kg. Seven days later, DiD-labeled polymer-coated liposomes were administered at 10 μmol phospholipid/kg. The concentration of liposomes in circulation was measured over a 48 hour period, at which point the animals were sacrificed and the concentration of liposomes in the liver, spleen, lung, and kidneys was determined. Liposomes without polymer and liposomes with HPMA, PVP, PDMA, and PAcM did not exhibit an ABC effect (Table 2). The pharmacokinetics and biodistribution of the second dose of each of these materials were very similar to those observed for a single dose (Figure 2).
Table 2.
Circulation half-lives of liposomes incorporating polymers for a single dose vs. the second dose (“PMOX+”=PMOX with cationic linker to lipid). (n = 3 per group for animals receiving a single dose, n = 4 per group for animals receiving 2 doses). Only the PEG and PMOX modified liposomes had significantly different circulation times compared at the first and second doses.
| Dose | Half-life (h) | AUC | |
|---|---|---|---|
|
|
|||
| PEG | 1 | 33.6 ± 1.3 | 2586 ± 12 |
| 2 | 1.66 ± 0.8** | 89 ± 18*** | |
|
|
|||
| PMOX | 1 | 30.5 ± 2.5 | 2605 ± 21 |
| 2 | 1.9 ± 0.5** | 89 ± 5*** | |
|
|
|||
| PVP | 1 | 20.7 ± 1.3 | 1524 ± 56 |
| 2 | 19.7 ± 2.6 | 1512 ± 56 | |
|
|
|||
| PDMA | 1 | 16.0 ± 1.2 | 1346 ± 54 |
| 2 | 15.0 ± 2.8 | 1387 ± 47 | |
|
|
|||
| HPMA | 1 | 16.0 ± 1.2 | 1138 ± 42 |
| 2 | 15.5 ± 2.8 | 953 ± 44* | |
|
|
|||
| PAcM | 1 | 16.4 ± 1.8 | 918 ± 36 |
| 2 | 16.3 ± 2.7 | 832 ± 16 | |
|
|
|||
| No Polymer | 1 | 12.0 ± 0.2 | 717 ± 40 |
| 2 | 12.3 ± 1.8 | 712 ± 37 | |
|
|
|||
| PMOX (+) | 1 | 30.5 ± 2.5 | 2570 ± 25 |
| 2 | 2.1 ± 0.5 | 80 ± 6*** | |
|
|
|||
, p < 0.05,
, p < 0.005,
, p < 0.001.
As expected, liposomes modified with PEG exhibited a pronounced ABC effect. Interestingly liposomes modified with PMOX also completely lost their long circulating character (half-life < 2 h) on the second dose and had increased accumulation in the liver and spleen (Figure 3).
Figure 3.
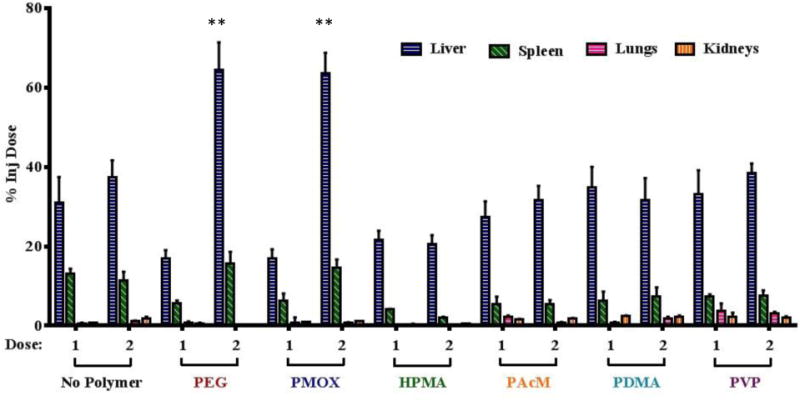
Biodistribution of the first and second dose of liposomes in rats 48 h after dosing. (n = 3 per group for animals receiving a single dose, n = 4 per group for animals receiving 2 doses). Only the PEG and PMOX modified liposomes had significantly different liposome accumulation in the liver and spleen over the two doses (**, p < 0.01).
Effect of Increased Molecular Weight
To obtain an accurate comparison between the chosen polymers, the molecular weight of each polymer evaluated was ~2000 Da. By holding the molecular weight constant, the hydrodynamic radius (which is a key factor for preventing opsonization of a liposome surface) of each polymer was similar. However, the actual chain lengths varied greatly. PEG and PMOX lack large pendant groups and have 3 polymer backbone atoms per monomer, where the other polymers in this study have large pendant groups and just two polymer backbone atoms per monomer. Thus, at Mw= 2000 Da, PEG has a chain length of 136 atoms ((2000 Da polymer/44 Da monomer)*3 backbone atoms per monomer, or 45 monomer segments) and PMOX has a chain length of 71 atoms. However, 2000 Da HPMA, for example, only has 28 atoms in its backbone and 14 monomer segments. To determine if chain segments impact the circulation time or ABC effect, two polymers, HPMA and PVP, were investigated at molecular weights corresponding to polymer backbone lengths of ~136 atoms (9,600 Da for HPMA and 7000 Da for PVP). The increased molecular weight polymers modestly increased circulation times over their 2000 Da counterparts, but still did not induce an observable ABC effect (Table 3, Figure S3).
Table 3.
Elimination half-lives of liposomes incorporating 7 kDa PVP and 9 kDa HPMA. (n = 3 per group, (Mean +/− SEM, no significant)
| Dose | Half-life (h) | AUC | |
|---|---|---|---|
|
|
|||
| PVP7 kDa | 1 | 23.1 ± 1.9 | 1533 ± 17 |
| 2 | 23.6 ± 2.3 | 1591 ± 101 | |
|
|
|||
| HPMA9 kDa | 1 | 18.7 ± 1.7 | 989 ± 57 |
| 2 | 17.4 ± 1.8 | 957 ± 42 | |
|
|
|||
Evaluation of cross-initiation of the ABC Effect
It has been reported that up to 25% of healthy blood donors exhibit anti-PEG IgM. Furthermore, we have shown that PMOX, a polymer similar to PEG in both chemical and physical properties induces from an ABC effect very similar to that of PEG. Therefore, determined if pre-existing anti-PEG IgM will result in the ABC of PMOX materials. We initiated anti-PEG IgM production in rats by pre-dosing with unlabeled PEGylated liposomes. Seven days later, the rats were treated with DiD-labeled PMOX liposomes and the pharmacokinetics and biodistribution were evaluated. Circulation times and biodistribution of the PMOX dose were consistent with a single dose of PMOX liposomes, indicating no cross reactivity between anti-PEG IgM and PMOX (Figure S4). These results were further confirmed through an ELISA assay.
Anti-Polymer IgM Detection
Anti-polymer IgM production was evaluated by ELISA. Serum from animals was added to the wells of a 96-well plate coated with the corresponding polymer-lipid conjugates. Pre-dose serum (before any injection) and post-dose serum (7 days after initiation dose) were compared. Results were consistent with those from the in vivo experiments. A pronounced IgM response was measured for animals treated with either PEG or PMOX liposomes, and no IgM response was observed in animals treated with the other polymer modified liposome (Table 4). Mean reciprocal endpoint titers were measured, and found to be 316 for PEG and 2094 for PMOX. Furthermore, cross-reactivity of the anti-polymer IgM was investigated by plating PEG and PMOX and testing the serum samples from rats treated with each of the other polymers. No cross-reactivity was observed (see supporting material). This was consistent with the absence of the in vivo cross-imitation study.
Table 4.
Determination of anti-polymer IgM by ELISA: sera from rats tested against same polymer rats were injected with. (Values are the mean +/− STD, n=4)
| A450 ELISA values of rat IgM | ||
|---|---|---|
| Rat Injections | Pre-dose | Post-dose |
| No polymer | 0.12 ± 0.01 | 0.12±0.01 |
| PEG | 0.19 ±0.04 | 2.94±0.65 |
| PMOX | 0.12±0.01 | 2.58±0.50 |
| PVP | 0.10±0.01 | 0.11±0.01 |
| HPMA | 0.10±0.01 | 0.10±0.01 |
| PAcM | 0.11±0.01 | 0.11±0.01 |
| PDMA | 0.12±0.00 | 0.11±0.01 |
DISCUSSION
We have evaluated a library of sterically stabilizing polymers to learn which may provide an extended circulation time yet avoid the ABC effect. All of the polymers studied extend circulation times. We show for the first time that pMOX induces an IgM response resulting in an ABC effect. We documented for the first time, that HPMA does not. Cause an ABC effect. Polymer modified liposomes were evaluated using consistent experimental conditions to allow a direct comparison of the effect on circulation half-life. The results are in accord with the previous literature that PEG and PMOX greatly extend the circulation half-life compared to the other polymers. However, when compared to unmodified, conventional liposomes, each polymer tested increased circulation half-life.
Although PEG is excellent at preventing interactions between the surface of a liposome and serum opsonizing factors, specific anti-PEG IgM can efficiently bind PEG. When anti-PEG IgM is present in circulation as either preexisting antibodies or as a response to a first dose of PEGylated material, PEG is rapidly removed from circulation and accumulates extensively in the liver and spleen. This can compromise treatments in which repeated administration is necessary and may increase toxicity towards organs where the PEGylated materials accumulate. We have identified four circulation-extending polymers (HPMA, PAcM, PDMA, and PVP) that do not induce the ABC effect upon repeated administration. However PMOX, a polymer with similar pharmacokinetics and biodistribution to PEG, also elicits an ABC effect.
It remains a mystery why some polymers induce an ABC effect and others do not. However, it is clear that the production of IgM plays an important role in the mechanism of the ABC effect.[36] In a critical step during the PEG ABC mechanism, PEGylated materials crosslink surface receptors on B-cells, which triggers the production of anti-PEG IgM. The quantity of anti-PEG IgM produced has been shown to correlate with the extent of the ABC phenomenon.[36] Furthermore, Ishihara et. al. found that PVP nanoparticles that did not exhibit an ABC effect also did not initiate the production of anti-PVP IgM.[37] We have corroborated those PVP results in our study, and shown that the other polymers in our study that do not display an ABC effect also do not initiate an IgM response. These results suggest that the polymers that do not initiate an ABC response may not be able to efficiently bind B-cell receptors or somehow interfere with IgM production through an alternative mechanism. The increased circulation times of polymer-coated liposomes in comparison to conventional liposomes is attributed to the ability of polymers to act as a steric shield. This ability to sterically shield the liposome surface can lead to decreased interaction with other membrane surfaces[38] and serum opsonizing factors.[39,40] Additionally, if opsonizing factors adsorb onto the surface of the liposome, sterically shielding polymers can still prevent those factors from interacting with the corresponding macrophage complement receptor.[41] When opsonins and other materials approach the surface of a polymer coated liposome, extended polymer chains are compressed into a higher energy conformation, which creates an opposing repulsive force.[6] Thus, differences in polymer molecular weight, flexibility, and conformation affect the ability of a polymer to repel approaching materials.[42] In examining the chemical nature of the hydrophilic polymers used in our work, one can identify polymers that only have hydrogen bond acceptors (PEG, PMOX, PVP, PACM) or have both hydrogen bond donors and acceptors (HPMA, PDMA). The polymers that only have hydrogen bond acceptors impart a longer half-life to the liposomes. However, hydrophobicity of the side chains, their interactions with water and chain-chain interactions when the polymer is anchored in a lipid membrane make it difficult to propose a structure-function relationship between chain chemistry and circulation half-live of the polymer modified liposomes.
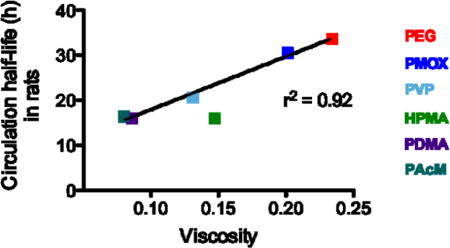
A physical parameter that is influenced by all of the interactions listed above is the intrinsic viscosity of the polymers. In fact, we observe a correlation when comparing polymer modified liposome circulation half-life and polymer viscosity (Figure 5). The two polymers with the highest circulation half-lives (PEG and PMOX) also have the highest viscosity as the free polymer. The polymer molecule weights used in this study are below the size where chain entanglement is predicted to arise[43] but the polymer surface density is in the brush regime[44] where polymer-polymer interactions result in an increase in the effective viscosity of the perimembranous region.[45] Increased polymer viscosity at the bilayer aqueous interface would in theory make it more difficult for macromolecules to interact with the liposome surface hence interfere with clearance mechanism mediated by opsonins.[41] Even if polymer viscosity is not the principal reason for the increase in circulation half-life, it could be a useful screen to identify other water-soluble polymers for potential half-life extenders. This trend is also seen in polymers of the same class as observed for HPMA and PVP (Table 3): as molecular weight increases, viscosity and circulation half-life modestly increase.
Figure 5.
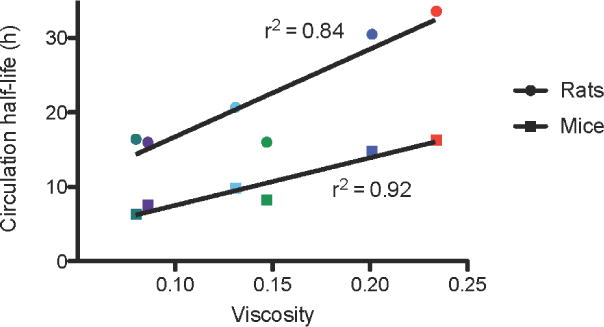
Viscosity of 10 kDa polymers in aqueous conditions vs the circulation half-life of liposomes incorporating 2 kDa polymers in rats (circles) and mice (squares). Red, PEG; dark blue,PMOX; irish green, HPMA; light blue, PVP; purple, PDMA; dark green, PAcM.
It is interesting, that the two most viscous polymers were also the two that initiated an ABC response. Thus, polymers with higher intrinsic viscosities may be more likely to induce an immune response, and our data suggests that there may be a minimum viscosity threshold for the IgM response to occur. Further investigation of the immune reactivity/B cell interaction of polymer modified liposome with viscosities greater than PEG or PMOX could shed light on this topic.
We compared pharmacokinetics and biodistribution of liposomes incorporating 5 mole percent of 2 kDa polymers. These values were chosen based on previous reports of the ideal mole percent and molecular weight of PEG for extending liposome circulation.[46,47] However, these may not be the optimal conditions for the other polymers investigated. In fact, we have shown that increasing the molecular weight of PVP and HPMA can modestly increase circulation half-lives (Table 3). The ideal graft density and molecular weight of polymers for the steric protection of liposomes in vivo is one that strikes a balance between providing complete surface coverage and allowing for sufficient chain mobility.[47] Because the polymers we have investigated have different chain flexibilities and degrees of polymerization, 5 mole percent and 2 kDa molecular weight may not be ideal parameters for each polymer. Polymers of optimal molecular weight and liposomal graft density may further improve the circulation half-lives, strengthening the case for their use rather than PEG in certain applications.
In this study we have evaluated the physical and in vivo properties of a panel of hydrophilic polymers for extended circulation. We have also confirmed that HPMA, PVP, PDMA and PAcM are able to extend the circulation half-lives of liposomes without eliciting the ABC effect upon repeated administration. We have found that PMOX, a polymer with very similar in vivo properties to PEG, initiates a pronounced ABC effect. Our results suggest that in specific cases where PEG’s ABC effect is detrimental to a delivery system or patient’s have pre-existing anti-PEG antibodies[8], other polymers can play the role of sterically stabilizing materials in vivo. These observations may assist the future design, selection and application of polymers for extended circulation and improved drug delivery.
Supplementary Material
Acknowledgments
We thank Dr. Charles Noble and Dr. Mark Hayes for their guidance with production of liposomes. We would also like to thank Dr. Aditya Kohli for his help on the measurement of liposomes in plasma and tissue samples. This work was supported by R01 GM061851 (FCS) and R01 EB002047 (FCS). VJ Venditto was funded by Ruth L. Kirschstein National Research Service Award from the National Institutes of Allergy and Infectious Diseases of the National Institutes of Health under Award Number F32AI095062.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Dr. Szoka declares a conflict of interest due to his involvement in a liposome company. The other authors declare no conflict of interest.
Contributions
P.H.K., J.M.JF. and F.C.S conceived and designed the experiments. P.H.K., T.C.C., S.K, V.J.B performed experiments. H.O. and S.K. conducted the animal experiments. P.H.K, V.J.V. and F.C.S. analyzed the data. P.H.K., V.J.V. and F.C.S. wrote the paper.
References
- 1.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 2.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed Engl. 2010;49:6288–6308. doi: 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- 4.Veronese FM. Peptide and protein PEGylation: a review of problems and solutions. Biomaterials. 2001;22:405–417. doi: 10.1016/s0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 5.Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of pegylation. Drug Deliv. 2006;13:399–409. doi: 10.1080/10717540600814402. [DOI] [PubMed] [Google Scholar]
- 6.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Veronese FM. PEGylated Protein drugs: basic science and clinical applications. Birkhäuser; Basel: 2009. [Google Scholar]
- 8.Richter AW, Åkerblom E. Polyethylene glycol reactive antibodies in man: titer distribution in allergic patients treated with monomethoxy polyethylene glycol modified allergens or placebo, and in healthy blood donors. Int Arch Allergy Immunol. 1984;74:36–39. doi: 10.1159/000233512. [DOI] [PubMed] [Google Scholar]
- 9.Ishida T, Harashima H, Kiwada H. Liposome clearance. Biosci Rep. 2002;22:197–224. doi: 10.1023/a:1020134521778. [DOI] [PubMed] [Google Scholar]
- 10.Ishida T, Ichihara M, Wang X, Kiwada H. Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J Control Release. 2006;115:243–250. doi: 10.1016/j.jconrel.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Dams ET, et al. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther. 2000;292:1071–1079. [PubMed] [Google Scholar]
- 12.Zhao Y, et al. Repeated injection of PEGylated solid lipid nanoparticles induces accelerated blood clearance in mice and beagles. Int J Nanomedicine. 2012;7:2891–2900. doi: 10.2147/IJN.S30943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laverman P, et al. Factors affecting the accelerated blood clearance of polyethylene glycol-liposomes upon repeated injection. J Pharmacol Exp Ther. 2001;298:607–612. [PubMed] [Google Scholar]
- 14.Suzuki T, et al. Accelerated blood clearance of PEGylated liposomes containing doxorubicin upon repeated administration to dogs. Int J Pharm. 2012;436:636–643. doi: 10.1016/j.ijpharm.2012.07.049. [DOI] [PubMed] [Google Scholar]
- 15.Ishida T, et al. Accelerated blood clearance of PEGylated liposomes following preceding liposome injection: effects of lipid dose and PEG surface-density and chain length of the first-dose liposomes. J Control Release. 2005;105:305–317. doi: 10.1016/j.jconrel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Ishida T, Atobe K, Wang X, Kiwada H. Accelerated blood clearance of PEGylated liposomes upon repeated injections: effect of doxorubicin-encapsulation and high-dose first injection. J Control Release. 2006;115:251–258. doi: 10.1016/j.jconrel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 17.Sherman MR, Williams LD, Sobczyk MA, Michaels SJ, Saifer MGP. Role of the methoxy group in immune responses to mPEG-protein conjugates. Bioconjug Chem. 2012;23:485–499. doi: 10.1021/bc200551b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lammers T, Ulbrich K, Duncan R, Vicent MJ. Do HPMA copolymer conjugates have a future as clinically useful nanomedicines? A critical overview of current status and future opportunities. Adv Drug Deliv Rev. 2010;62:272–282. doi: 10.1016/j.addr.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Duncan R. Development of HPMA copolymer-anticancer conjugates: clinical experience and lessons learnt. Adv Drug Deliv Rev. 2009;61:1131–1148. doi: 10.1016/j.addr.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Etrych T, et al. Biodegradable star HPMA polymer–drug conjugates: Biodegradability, distribution and anti-tumor efficacy. J Control Release. 2011;154:241–248. doi: 10.1016/j.jconrel.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 21.Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1:297–315. [PMC free article] [PubMed] [Google Scholar]
- 22.Zelikin AN, Such GK, Postma A, Caruso F. Poly(vinylpyrrolidone) for bioconjugation and surface ligand immobilization. Biomacromolecules. 2007;8:2950–2953. doi: 10.1021/bm700498j. [DOI] [PubMed] [Google Scholar]
- 23.Gaertner FC, Luxenhofer R, Blechert B, Jordan R, Essler M. Synthesis, biodistribution and excretion of radiolabeled poly (2-alkyl-2-oxazoline)s. J Control Release. 2007;119:291–300. doi: 10.1016/j.jconrel.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Zalipsky S, Hansen CB, Oaks JM, Allen TM. Evaluation of blood clearance rates and biodistribution of poly(2-oxazoline)-grafted liposomes. J Pharm Sci. 1996;85:133–7. doi: 10.1021/js9504043. [DOI] [PubMed] [Google Scholar]
- 25.Torchilin VP, et al. New synthetic amphiphilic polymers for steric protection of liposomes in vivo. J Pharm Sci. 1995;84:1049–1053. doi: 10.1002/jps.2600840904. [DOI] [PubMed] [Google Scholar]
- 26.Drotleff S, et al. Biomimetic polymers in pharmaceutical and biomedical sciences. Eur J Pharm Biopharm. 2004;58:385–407. doi: 10.1016/j.ejpb.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Kohori F, et al. Control of adriamycin cytotoxic activity using thermally responsive polymeric micelles composed of poly (N-isopropylacrylamide-co-N,N-dimethylacrylamide)-b-poly(d,l-lactide) Colloids Surfaces B Biointerfaces. 1999;16:195–205. [Google Scholar]
- 28.Abu Lila AS, Kiwada H, Ishida T. The accelerated blood clearance (ABC) phenomenon: Clinical challenge and approaches to manage. J Control Release. 2013;172:38–47. doi: 10.1016/j.jconrel.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 29.Romberg B, et al. Pharmacokinetics of poly (hydroxyethyl-l-asparagine)-coated liposomes is superior over that of PEG-coated liposomes at low lipid dose and upon repeated administration. Biochim Biophys Acta – Biomembr. 2007;1768:737–743. doi: 10.1016/j.bbamem.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 30.De Souza R, Zahedi P, Allen CJ, Piquette-Miller M. Polymeric drug delivery systems for localized cancer chemotherapy. Drug Deliv. 2010;17:365–75. doi: 10.3109/10717541003762854. [DOI] [PubMed] [Google Scholar]
- 31.Canal F, Sanchis J, Vicent MJ. Polymer–drug conjugates as nano-sized medicines. Curr Opin Biotechnol. 2011;22:894–900. doi: 10.1016/j.copbio.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Boyer C, et al. Bioapplications of RAFT polymerization. Chem Rev. 2009;109:5402–5436. doi: 10.1021/cr9001403. [DOI] [PubMed] [Google Scholar]
- 33.Woodle MC, Engbers CM, Zalipsky S. New amphipatic polymer-lipid conjugates forming long-circulating reticuloendothelial system-evading liposomes. Bioconjug Chem. 1994;5:493–496. doi: 10.1021/bc00030a001. [DOI] [PubMed] [Google Scholar]
- 34.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 35.Allen TM, Hansen C. Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim Biophys Acta – Biomembr. 1991;1068:133–141. doi: 10.1016/0005-2736(91)90201-i. [DOI] [PubMed] [Google Scholar]
- 36.Ishihara T, et al. Accelerated blood clearance phenomenon upon repeated injection of PEG-modified PLA-nanoparticles. Pharm Res. 2009;26:2270–2279. doi: 10.1007/s11095-009-9943-x. [DOI] [PubMed] [Google Scholar]
- 37.Ishihara T, et al. Evasion of the accelerated blood clearance phenomenon by coating of nanoparticles with various hydrophilic polymers. Biomacromolecules. 2010;11:2700–6. doi: 10.1021/bm100754e. [DOI] [PubMed] [Google Scholar]
- 38.Evans E, Klingenberg DJ, Rawicz W, Szoka F. Interactions between Polymer-Grafted Membranes in Concentrated Solutions of Free Polymer. Langmuir. 1996;12:3031–3037. [Google Scholar]
- 39.Jeon S, Lee J, Andrade J, De Gennes P. Protein—surface interactions in the presence of polyethylene oxide. J Colloid Interface Sci. 1991;142:149–158. [Google Scholar]
- 40.Müller BW, Müller HC. Particle size, surface hydrophobicity and interaction with serum parenteral fat emulsions and model drug as parameters related to RES uptake. Clin Nut. 1992;11:289–297. doi: 10.1016/0261-5614(92)90006-c. [DOI] [PubMed] [Google Scholar]
- 41.Moghimi SM, Hamad I, Andresen TL, Jørgensen K, Szebeni J. Methylation of the phosphate oxygen moiety of phospholipid-methoxy (polyethylene glycol) conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production. FASEB J. 2006;20:2591–2593. doi: 10.1096/fj.06-6186fje. [DOI] [PubMed] [Google Scholar]
- 42.Torchilin VP, Papisov MI. Why do polyethylene glycol-coated liposomes circulate so long? Molecular mechanism of liposome steric protection with polyethylene glycol: role of polymer chain flexibility. J Liposome Res. 1994;4:725–739. [Google Scholar]
- 43.Lee JCM, Santore M, Bates FS, Discher DE. From membranes to melts, rouse to reptation: diffusion in polymersome versus lipid bilayers. Macromolecules. 2002;35:323–326. [Google Scholar]
- 44.Marsh D, Bartucci R, Sportelli L. Lipid membranes with grafted polymers: physicochemical aspects, Biochim. Biophys Acta. 2003;1615:33–59. doi: 10.1016/s0005-2736(03)00197-4. (2003) [DOI] [PubMed] [Google Scholar]
- 45.Soong R, Macdonald PM. PEG molecular weight and lateral diffusion of PEG-ylated lipids in magnetically aligned bicelles. Biochim. et Biophys Acta. 2007;1768:1805–1814. doi: 10.1016/j.bbamem.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 46.Needham D, et al. Polymer-grafted liposomes: physical basis for the “stealth” property. J Liposome Res. 1992;2:411–430. [Google Scholar]
- 47.Storm G, Belliot SO, Daemen T, Lasic DD. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv Drug Deliv Rev. 1995;17:31–48. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.