

Summary
Heterozygous mutation of IDH1 in cancers modifies IDH1 enzymatic activity, reprogramming metabolite flux and markedly elevating 2-hydroxyglutarate (2-HG). Here, we found that 2-HG depletion did not inhibit growth of several IDH1 mutant solid cancer types. To identify other metabolic therapeutic targets, we systematically profiled metabolites in endogenous IDH1 mutant cancer cells after mutant IDH1 inhibition and discovered a profound vulnerability to depletion of the coenzyme NAD+. Mutant IDH1 lowered NAD+ levels by downregulating the NAD+ salvage pathway enzyme nicotinate phosphoribosyltransferase (Naprt1), sensitizing to NAD+ depletion via concomitant nicotinamide phosphoribosyltransferase (NAMPT) inhibition. NAD+ depletion activated the intracellular energy sensor AMPK, triggered autophagy and resulted in cytotoxicity. Thus, we identify NAD+ depletion as a metabolic susceptibility of IDH1 mutant cancers.
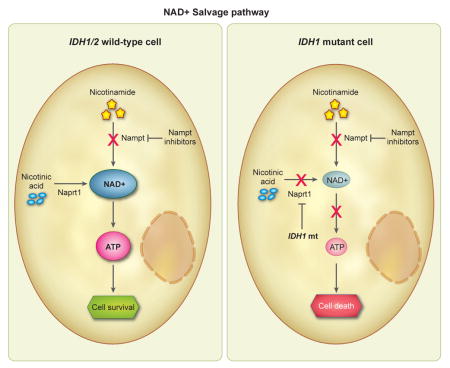
Graphical Abstract
INTRODUCTION
Accumulating evidence indicates that mutations in canonical metabolic enzymes can promote the development of cancer. Germline mutations in the tricarboxylic acid (TCA) cycle enzyme succinate dehydrogenase (SDH) give rise to familial paraganglioma and pheochromocytoma, and germline fumarate hydratase (FH) mutations result in familial forms of renal cell cancer (Kaelin, 2009). More recently, recurrent somatic mutations in the isocitrate dehydrogenase 1 (IDH1) and IDH2 genes have been identified in a large fraction of gliomas, acute myeloid leukemias (AML), angioimmunoblastic T-cell lymphomas (AITL), chondrosarcomas and cholangiocarcinomas.
In gliomas, approximately 90% of IDH1/2 mutations are arginine to histidine heterozygous substitutions in codon 132 of the IDH1 gene (R132H). A small fraction of the IDH1/2 mutations found in gliomas are different amino acid substitutions at the R132 position of IDH1 or substitutions of the structurally analogous R172 residue of the homolog IDH2 (Balss et al., 2008; Hartmann et al., 2009; Parsons et al., 2008; Yan et al., 2009). In contrast, the most common mutation in cartilaginous tumors is IDH1 R132C (40%) (Amary et al., 2011; Kipp et al., 2012; Wang et al., 2013a), whereas in AML and AITL, IDH2 mutations are most common (Marcucci et al., 2010; Mardis et al., 2009; Ward et al., 2012; Cairns et al., 2012). Essentially all of the mutations in IDH1/2 result in heterozygous amino acid substitutions within a few targeted hotspot loci, suggesting an oncogenic functional contribution of mutant IDH1/2 to tumorigenesis (Vogelstein et al., 2013). A shared consequence of these IDH1/2 mutations is the near-complete elimination of the normal oxidation and decarboxylation of isocitrate to alpha-ketoglutarate (a-KG) (Zhao et al., 2009; Yan et al., 2009) and the neomorphic overproduction of 2-hydroxyglutarate (2-HG) via NADPH-mediated reduction of a-KG (Dang et al., 2009). 2-HG is a byproduct metabolite that is otherwise produced at only modest levels in hypoxic states (Wise et al., 2011).
2-HG has therefore been proposed as an “oncometabolite” mediator of tumorigenesis in mutant IDH1/2 cancers. 2-HG has been shown to inhibit a range of a-KG-dependent dioxygenases, including hypoxia inducible factor (HIF) hydroxylases, histone demethylases and 5-methylcytosine hydroxylases, resulting in altered HIF activity, widespread chromatin alterations and a stem-like cell differentiation block (Figueroa et al., 2010; Koivunen et al., 2012; Losman et al., 2013; Lu et al., 2012; Saha et al., 2014; Chowdhury et al., 2011; Xu et al., 2011). A characteristic feature of IDH1 mutant cancers is the CpG island methylator phenotype (CIMP or G-CIMP) (Figueroa et al., 2010; Noushmehr et al., 2010; Sasaki et al., 2012), and in experimental models, introduction of mutant IDH1/2 and overproduction of 2-HG results in the subsequent development of global DNA hypermethylation (Figueroa et al., 2010; Sasaki et al., 2012; Turcan et al., 2012). Thus, inhibitors that decrease 2-HG levels are being investigated as potential therapeutics for IDH1 mutant cancers (Popovici-Muller et al., 2012; Rohle et al., 2013).
In addition to these 2-HG-mediated effects, mutant IDH1 alters canonical metabolic pathways (Reitman et al., 2014; Reitman et al., 2011; Zhao et al., 2009; Izquierdo-Garcia et al., 2014; Leonardi et al., 2012), including glutamine catabolism (Metallo et al., 2012; Seltzer et al., 2010) and the TCA cycle (Grassian et al., 2014). We therefore hypothesized that disruption of mutant IDH1 enzymatic activity would reveal metabolic dependencies that could be selectively targeted for therapeutic gain. Herein, we compared the metabolic profiles of IDH1 mutant glioma tumor initiating cell (TIC) tumorsphere lines with and without IDH1 inhibition to identify candidate metabolic vulnerabilities.
RESULTS
Inhibition of Mutant IDH1 in Endogenous IDH1 Mutant Cancer Cells Decreased 2-HG But Did Not Inhibit Cell Growth or Tumor Propagation
We recently developed in vitro and in vivo patient-derived tumor initiating cell (TIC) models of IDH1 mutant glioma that faithfully recapitulate the histological phenotypes and genetic complexity of primary IDH1 mutant glioma tumors (Figures S1A, S1B) (Wakimoto et al., 2014). To assess the effect of 2-HG reduction on these IDH1 mutant TICs, we exposed cells to a well-characterized R132H and R132C IDH1 mutant specific inhibitor (IDH1i) (See Supplementary Methods and Figure S1C for details of compound chemical synthesis, structure and characterization) (Deng et al., 2015; Popovici-Muller et al., 2012). We confirmed potent inhibition of 2-HG formation in vitro in IDH1R132H mutant glioma TICs and an IDH1R132C mutant fibrosarcoma line (HT1080) (Figure 1A). Notably, despite 2-HG reduction, IDH1i did not inhibit in vitro growth of 8 endogenous IDH1 mutant cell lines tested, including 6 glioma TICs with IDH1R132H, HT1080 (IDH1R132C) and an IDH1R132C mutant melanoma (30T) (Figure 1B). We instead observed small but consistent increases in cell proliferation in most lines (Figure 1B, Figure S1A). Our in vitro findings are similar to prior studies in IDH2 leukemia models (Chen et al., 2013; Wang et al., 2013b), chondrosarcomas (Suijker et al., 2015) and colony forming assays of IDH1 mutant glioma lines (Turcan et al., 2013) where slight in vitro proliferation increases were observed with chemical inhibition of mutant IDH.
Figure 1. Effects of mutant-specific IDH1 inhibitor in endogenous IDH1 mutant cancer cells.

(A) R-2-HG levels in IDH1 mutant lines (MGG119 and MGG152 have IDH1R132H, HT1080 harbors IDH1R132C) measured by liquid chromatography-mass spectrometry (LC-MS) +/− mutant-specific IDH1 inhibitor (IDH1i 5 μM, 48 hr) in vitro. (B) Cell viability assay of cell lines after incubation with IDH1i (CellTiter-Glo, normalized to DMSO control). MGG lines were analyzed at day 9, HT1080 and 30T were analyzed at day 3. (C) Concentrations of mouse MGG152 orthotopic xenograft tissue IDH1i (upper panel) and R-2-HG (lower panel) measured by LC-MS. Data were acquired after 5-day oral gavage, 2×/day, of IDH1i (400 mg/kg, n=5) or vehicle (n=5). (D) Kaplan-Meier estimates of survival by treatment. SCID mice bearing MGG152 orthotopic xenografts were randomly assigned to oral gavage of IDH1i (red, 400 mg/kg 2×/day, n=6) or vehicle (black, 2×/day, n=6). Median survival with IDH1i, 46 days (95% CI; 42–50) and with vehicle, 46 days (95% CI; 45–48, p=0.79). (E) Western blot analysis of histone methylation marks in MGG152 (IDH1R132H) cells +/− IDH1i for 2 days or 10 months. MGG8, IDH1/2 wild-type. Actin, loading control. (F) Methylation-specific PCR (MSP) showing MGMT gene promoter methylation in MGG152 +/− IDH1i. P, passage number; U, unmethylated; M, methylated. (G) Genome-wide distribution of DNA methylation in MGG152 cells treated with DMSO or IDH1i (5 μM) for 2 days (R=0.996, p<0.001, left) and 10 months (R=0.989, p<0.001, right). (H) Cell viability assay (CellTiter-Glo) of MGG152 treated with DMSO or IDH1i (5 μM) for 12 months in vitro. Data were obtained at day 3 and normalized to DMSO control at day 0. (I) Kaplan-Meier estimates of survival after intracerebral implantation of pre-treated MGG152. Cells were incubated +/− IDH1i in vitro for the indicated cell passage number and then 1×105 cells were implanted into SCID mouse brains. Animals were then monitored for survival. In all panels: bars, +/− SEM; *, p<0.05. See also Figure S1.
We then tested the in vivo effect of 2-HG depletion in an orthotopic glioma xenograft model that was derived from an IDH1 mutant recurrent glioblastoma (MGG152) (Wakimoto et al., 2014). Treatment of mice bearing orthothopic MGG152 tumors with IDH1i (400 mg/kg twice a day) for 5 days resulted in IDHi accumulation and near-complete elimination of 2-HG within the brain tumors (Figure 1C). However, continuous daily IDH1i treatment of MGG152 orthotopic tumor bearing mice did not affect the survival of mice compared to vehicle treatment (Figure 1D). At necropsy, we observed no difference between IDH1i- and vehicle-treated animals in the size of orthotopic tumors or expression of mutant IDH1, Ki-67, GFAP or nestin within the tumors. Furthermore, no difference in MGMT promoter DNA methylation or H3K9 di- or tri-methylation was observed in tumor tissue (Figures S1D, S1E, S1F).
We investigated whether epigenetic alterations in endogenous mutant IDH1 glioma cells could be reversed in vitro by depletion of 2-HG. Because many cycles of cell division are required to alter the epigenome after introduction of mutant IDH1 (Turcan et al., 2012), we cultured two glioma TICs in IDH1i at concentrations sufficient to abrogate 2-HG production for up to 12 months. After 12-months, cells retained both the wild-type and mutant IDH1 alleles and expression of the IDH1R132H mutant enzyme (Figures S1G, S1H). Prolonged IDH1i exposure did not detectably alter the global trimethylation or dimethylation of histone 3 lysine 9 (H3K9), global trimethylation of H3K27, genomewide distribution of DNA methylation, or the methylation of O-6-methylguanine-DNA methyltransferase (MGMT) gene promoter (Figure 1E, 1F, 1G). Again, we observed a persistent acceleration of cell proliferation with long-term IDH1i incubation (Figure 1H). In addition, long-term 2-HG depletion did not delay the time-to-tumor-formation or frequency of tumor initiation after implantation of IDH1 mutant glioma TICs into mouse brains (Figure 1I). Surprisingly, long-term exposure of MGG152 to IDH1i prior to intracerebral implantation resulted in significantly shorter animal survival (Figures 1I, S1I).
We then investigated whether DNA demethylation alone or in combination with IDH1i could impact growth of IDH1 mutant cells. We found that decitabine, but not azacytidine, decreased cell viability of MGG152 (Figure S1J). However, there was no additive or synergistic effect on cell viability with the combination of IDH1i and either decitabine or azacytidine (Figure S1K). Taken together, these data demonstrate that in some cell contexts, 2-HG levels can be uncoupled from IDH1 mutant cancer cell proliferation, and that the cancer cell transformation induced in these endogenous IDH1 mutant cancers is not easily reversed by depletion of 2-HG, even after many cell divisions.
Metabolic Profiling of Endogenous IDH1 Mutant Cancer Cells After IDH1 Inhibition Reveals a Susceptibility to Depletion of the Canonical Metabolite NAD+
Cells harboring mutant IDH1 have altered levels of metabolites in addition to 2-HG (Grassian et al., 2014; Metallo et al., 2012; Reitman et al., 2014; Reitman et al., 2011; Seltzer et al., 2010; Zhao et al., 2009). To identify candidate metabolic therapeutic targets other than 2-HG, we performed unbiased metabolite profiling using liquid chromatography–mass spectrometry (LC-MS) in MGG152 after short-term and long-term IDH1i incubation in vitro (Figure 2A, Table S1). As expected, we observed significant 2-HG reduction with both short-term and long-term IDH1i incubation. We targeted as therapeutic candidates those metabolites altered in consistent patterns in both short-term and long-term IDH1i-exposed cultures, reasoning that stable metabolic alterations may require multiple cell divisions. Other than 2-HG, only 3 metabolites were increased or reduced by 50% or greater in both short- and long-term IDH1i exposure: NADH, citrate, and glycerol-3-phosphate (G3P) (Figure 2A, Table S1).
Figure 2. Metabolic profiling after IDH1 inhibition reveals NAD+ level alterations in IDH1 mutant cancers.

(A) Heat map representation of metabolites in MGG152 cells quantified by LC-MS. Analytes are represented as relative change with IDH1i for 2 days (upper row) or 6 months (lower row) compared to DMSO control, with increases in red and decreases in green. (B) NADH levels in MGG152 cells treated with DMSO or IDH1i for 6 months. (C) NAD+ levels in endogenous IDH1 mutant cancer cells +/− IDH1i exposure for the indicated times. (D) NADP+ and NADPH levels in MGG152 +/− IDH1i for 6 months. Bars, +/− SEM, * p<0.05. See also Table S1
Because G3P is a component of the NAD+/NADH shuttle system and NAD+ depletion has been investigated as a therapeutic strategy in cancer (Galli et al., 2013), we further focused on NAD+ and NADH. Using an independent method, we confirmed that IDH1i increased NADH levels in MGG152 (Figure 2B). We then discovered that IDH1i increased NAD+ in MGG152 as well as in other IDH1 mutant lines (Figure 2C). We did not detect a significant change in NADP+ and we observed a decrease in NADPH, the substrate of the mutant IDH1/2 enzyme, after long-term IDH1i incubation (Figure 2D).
We then investigated NAD+ as a potential therapeutic metabolic target in IDH mutant cancer cells. We exposed cells to two chemically dissimilar, highly specific inhibitors of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of the NAD+ salvage pathway; FK866 (Wosikowski et al., 2002; Hasmann and Schemainda, 2003) and GMX1778 (Hjarnaa et al., 1999; Watson et al., 2009). Both NAMPT inhibitors potently inhibited cell viability of 6 endogenous IDH1/2 mutant cancer cell lines, including 3 IDH1R132H mutant GBM TICs (MGG119, MGG152 and BT142), the IDH1R132C mutant lines HT1080 and 30T, and SW1353 chondrosarcoma (IDH2R172S) (Figure 3A), with IC50 values ranging between 1–25 nM (Figures 3A). In contrast, 8 IDH1/2 wild-type cancer lines derived from similar tissue types and normal human astrocytes (NHA) were resistant at doses up to 10 μM (Figures 3A), indicative of a wide therapeutic index.
Figure 3. NAMPT inhibitors selectively inhibit IDH1 mutant cancers in vitro by depleting NAD+.

(A) Cell viability assay (CellTiter-Glo) after 72 hr treatment with FK866 (left panel) and GMX1778 (right panel). IDH status: IDH1R132H: MGG119, MGG152, BT142 (gliomas); IDH1R132C: HT1080 (fibrosarcoma), 30T (melanoma); IDH2R172S, SW1353 (chondrosarcoma); IDH1/2 wild-type: MGG18, MGG23, MGG85, MGG91, MGG102, MGG171, (gliomas), U87 (glioblastoma), UACC257 (melanoma) and NHA (normal human astrocytes). (B) NAMPT inhibitor effect on NAD+ and NADH levels in IDH1 mutant cancer cells. Cells were treated with FK866 (12.5 nM, gray) or GMX1778 (12.5 nM, black) for 24 hr prior to measurement. Data are represented as %DMSO control. (C) R-2-HG levels measured by LC-MS in IDH1 mutant cells +/− incubation with FK866 (12.5 nM, 48 hr, gray) or GMX1778 (12.5nM, 48 hr, black). (D) and (E) Cell viability assay of IDH1 mutant lines incubated with FK866 (12.5nM, 72 hr) or GMX1778 (12.5nM, 72 hr) and increasing concentrations of exogenous NAD+ (72 hr, D) or nicotinamide mononucleotide (NMN, 72 hr, E). *, p<0.05 for difference between addition of NAD+ or NMN vs. FK866 alone; **, p<0.05 for difference between addition of NAD+ or NMN vs. GMX1778 alone. (F) Relative NAD+ levels in IDH1 mutant cancer cells 24 hours after treatment with FK866 (12.5 nM), NMN (1 mM), or the combination of FK866 (12.5 nM) and NMN (1 mM) vs. DMSO (24 hr) control. (G) Cell viability assay of IDH1 mutant cancer cells incubated with FK866 (12.5 nM, 72 hr) or GMX1778 (12.5 nM, 72 hr) and increasing concentrations of exogenous nicotinic acid (NA, 72 hr). Bars, +/− SEM, * p<0.05.
NAMPT inhibition also resulted in significant reduction of intracellular NAD+ and NADH in the IDH1 mutant cancer cells (Figure 3B). NAMPT inhibition also decreased the production of intracellular 2-HG in IDH1 mutant cancer cells (Figure 3C). We next tested whether the effects of NAMPT inhibitors were specifically due to reduction of NAD+ levels and on-target inhibition of NAMPT. We co-incubated IDH1 mutant cancer cells with FK866 or GMX1778 and increasing doses of NAD+ or nicotinoamide mononucleotide (NMN), a precursor of NAD+, which bypasses NAMPT. Both NAD+ and NMN rescued IDH1 mutant cells from the effects of FK866 and GMX1778 (Figures 3D, 3E). In addition, exogenous NMN reversed intracellular NAD+ levels, which was depleted by NAMPT inhibitor (Figure 3F). In contrast, supplementation with nicotinic acid (NA), a precursor for an alternate NAD+ salvage pathway rate-limited by nicotinic acid phosphoribosyltransferase (Naprt1) (Cerna et al., 2012; Chiarugi et al., 2012; Watson et al., 2009), could not rescue IDH1 mutant cells from the effects of NAMPT inhibition (Figure 3G).
Mutant IDH1 Reduces NAD+ Levels and Sensitizes to NAD+ Depletion
We measured basal intracellular NAD+ levels in our cell panel and found NAD+ to be lower in IDH1 mutant compared to IDH1/2 wild-type cells (Figure 4A). The relative decrease of NAD+ levels after treatment with NAMPT inhibitor was not significantly different between IDH1 mutant and IDH1/2 wild-type cells (Figure 4B), suggesting that the lower steady-state NAD+ pool may render IDH1 mutant cells less able to compensate for NAMPT inhibition. We therefore hypothesized that IDH1 mutant cells alter the dynamic steady-state supply of NAD+.
Figure 4. IDH1 mutation reprograms NAD+ metabolism.

(A) Scatter plots of absolute NAD+ (left panel) and NADH (right panel) levels in a panel of glioma tumorsphere lines. (B) Relative NAD+ and NADH decrease after FK866 (12.5 nM, 24 hr) treatment of the lines in panel A compared to DMSO control (24 hr). (C) Western blot analysis of IDH1R132H, NAMPT and Naprt1. Actin, loading control. (D) and (E) Western blot analysis of MGG18 (IDH1/2 wild-type) engineered with a tetracycline (tet)-inducible IDH1R132H gene (MGG18-IDH1-R132H). MGG18-IDH1-R132H was incubated +/− tet for 2 months in vitro, then harvested for analysis. Actin, Vinculin, loading controls. (F) NAD+ levels of MGG18-IDH1-R132H cells +/− tet induction for 2 months. (G) Cell viability assay of MGG18-IDH1-R132H cells +/− tet induction for 2 months followed by +/− FK866 (12.5nM, 5 days). (H) Western blot analysis of MGG152 infected with lentivirus carrying no (MGG152-control) or Naprt1 cDNA (MGG152-Naprt1). Vinculin, loading control. (I) Relative NAD+ change after FK866 treatment (12.5 nM, 24 hr) in MGG152-control and MGG152-Naprt1 compared to DMSO control (24 hr). (J) Cell viability assay of MGG152-control and MGG152-Naprt1 after 48 hr incubation with FK866 or GMX1778. (K) Heatmap of NAPRT1 promoter DNA methylation status in IDH1 mutant (MGG119, MGG152 and HT1080) and IDH1/2 wild-type (MGG18, MGG23, MGG85 and MGG91) lines measured by Infinium arrays. β values represent the fraction of methylated CpG site. Data were acquired by Infinium HumanMethylation 450 BeadChip array. Bars, +/− SEM, * p<0.05. See also Figure S2.
NAMPT and Naprt1 are the rate-limiting enzymes of distinct NAD+ salvage pathways that replenish the intracellular NAD+ pool. We found that expression of NAMPT did not correlate with sensitivity to NAD+ depletion, however all IDH1 mutant cell lines examined had low or undetectable Naprt1 expression (Figure 4C). We therefore asked whether mutant IDH1 was sufficient to alter Naprt1 expression, lower NAD+ levels, and sensitize to NAD+ depletion. We engineered MGG18, an IDH1/2 wild-type, NAMPT inhibitor-resistant glioma TIC (Figure 3A), with a tetracycline-inducible IDH1R132H mutant transgene (MGG18-IDH1-R132H) (Figures 4D, S2A). Induction of IDH1R132H expression resulted in marked elevation of 2-HG levels (Figure S2B) as well as significantly decreased Naprt1 expression (Figures 4E), reduced intracellular NAD+ level (Figure 4F), and remarkably, induced sensitivity to NAD+ depletion (Figure 4G, S2C). We confirmed that mutant IDH1 reduces Naprt1 expression in an isogenic system where heterozygous IDH1 mutation has been engineered in HCT116 cells (Figure S2D). We then tested whether over-expressing Naprt1 could rescue IDH1 mutant cells from the deleterious effects of NAD+ depletion. Lentivirus constructs carrying either NAPRT1 driven by a constitutively active promoter (MGG152-Naprt1) or control vector (MGG152-Control) were used to infect MGG152. After stable lentiviral integration (Figure 4H), the relative NAD+ level decrease with NAMPT inhibition was mild in MGG152-Naprt1 cells but marked in MGG152-control cells (Figure 4I). In addition, this enforced NAD+ increase rescued MGG152-Naprt1 from the anti-proliferative effects of NAMPT inhibition (Figure 4J). Together, these data suggest mutant IDH1 regulation of Naprt1 critically impacts the intracellular NAD+ pool and alters vulnerability to NAMPT inhibitors.
In cancer cells, low Naprt1 expression may be mediated by NAPRT1 promoter methylation (Shames et al., 2013). In keeping with these prior findings, we found that the NAPRT1 promoter CpG island was hypermethylated in our cell lines with very low Naprt1 expression, including all IDH1 mutant lines (Figure 4K). Importantly, not all lines with low Naprt1 expression were sensitive to NAMPT inhibitors, indicating that other genetic and epigenetic factors may mediate sensitivity. As such, we observed NAMPT inhibitor efficacy in several IDH1/2 wild-type cell lines (Figure S2E), consistent with prior reports of NAMPT inhibitor cytotoxicity in IDH1/2 wild-type cancer cell lines (Chiarugi et al., 2012; Galli et al., 2013; Watson et al., 2009).
To assess whether alterations in observed NAD+ levels were due to changes in redox status and utilization of reducing equivalents in IDH1 mutant cells, we calculated NAD+/NADH and NADP+/NADPH ratios after long-term IDH1i treatment, NAMPT inhibitor treatment and mutant IDH1 induction. These various conditions did not significantly or consistently alter NAD+/NADH and NADP+/NADPH ratios (Figures S2F-S2J). These results indicate that the effects observed with mutant IDH1 and NAMPT inhibitors were unlikely to be mediated by changes in net redox status.
NAD+ Depletion Disrupts TCA Cycle Metabolism in IDH1 Mutant Cancer Cells
Because oncogenic IDH1 mutations increase flux through the oxidative TCA cycle (Grassian et al., 2014; Cuyas et al., 2015), we investigated the effect of NAMPT inhibitors on three NAD+-requiring TCA cycle enzymes (alpha-ketoglutarate dehydrogenase, malate dehydrogenase, and isocitrate dehydrogenase) in IDH1 mutant cells. Using LC-MS, we found that short term incubation of MGG152 with FK866 resulted in significant accumulation of TCA cycle precursors upstream of the NAD+-requiring enzymes, including citric acid, fumarate and malic acid (Figure 5). In addition, the products of NAD+-requiring enzymes in the TCA cycle, succinate and oxaloacetate, were significantly decreased. These data indicate that NAD+ depletion may compromise the TCA cycle, upon which IDH1 mutant cells have increased dependence.
Figure 5. NAMPT inhibitor effect on NAD+-utilizing pathways in IDH mutant cancers.

Metabolites of the tricarboxylic acid (TCA) cycle in MGG152 cells were quantified by LC-MS after treatment with DMSO (36 hr, white bars) or FK866 (12.5 nM, 36 hr, black bars). Bar graphs represent the changes in FK866-treated cells relative to DMSO control.
NAD+ Depletion Induces the Intracellular Energy and Nutrient Sensor AMPK and Initiates Autophagy
Since the cell viability assay we used (CellTiter-Glo) quantitates intracellular ATP as a surrogate for cell number, we confirmed that the effects on cell viability represented cell death by counting the number of viable cells that excluded trypan blue. We found that time dependent reduction of cell number with NAMPT inhibitor exposure correlated with ATP-based cell viability assessment (Figure 6A). Cytotoxicity was not primarily mediated by apoptosis, as Annexin V, double staining for Annexin V and propidium iodide (PI), and activated caspases-3/7 did not significantly increase (Figures S3A, S3B). However, LC3-phosphatidylethanolamine conjugate (LC3-II) increased with NAMPT inhibitor exposure (Figure 6B), consistent with initiation of the metabolic-sensing pathway of autophagy, and indeed, the effects of NAD+ depletion on cell viability were rescued with the autophagy inhibitor 3-methyladenine (3-MA, Figure 6C).
Figure 6. NAD+ depletion triggers AMPK-initiated autophagy and cell death in IDH1 mutant cancer cells.

(A) Relative cell viability of cells treated with FK866 (upper row) and GMX1778 (lower row) measured by CellTiter-Glo (white bars) and viable cell count (Trypan blue exclusion assay, black bars) compared to DMSO controls. (B) Western blot analysis of LC3-I and LC3-II expression in MGG152 cells after treatment with FK866 (12.5 nM) and GMX1778 (12.5 nM) compared to DMSO control (72 hr). Vinculin, loading control. (C) Cell viability assay (CellTiter-Glo) of MGG152 cells treated for 72 hr with FK866 (12.5 nM, left), GMX1778 (12.5 nM, right), or DMSO +/− autophagy inhibitor 3-methyladenine (3-MA). Bars, +/− SEM, * p<0.05. (D) Western blot analysis of MGG152 cells treated with DMSO (48 hr), FK866 (12.5 nM, 48 hr), GMX1778 (12.5 nM, 48 hr) and NMN (100 μM, 48 hr) alone and in combinations as indicated. Actin, loading control. See also Figure S3.
We determined that autophagy was initiated by AMP activated protein kinase (AMPK), a key regulator of the metabolic checkpoint which is activated in response to nutrient- and energy-poor conditions (Hardie et al., 2007). Depletion of NAD+ in MGG152 was associated with significant phosphorylation and activation of AMPK (Figure 6D). Concomitantly, we observed activating phosphorylation of the autophagy initiating complex component ULK1 (Ser555), which is catalyzed by AMPK (Egan et al., 2011), and activation of Atg13, another component of the autophagy initiating complex. Additionally, we observed AMPK-mediated phosphorylation of raptor and potent suppression of mTOR signaling, as illustrated by dephosphorylation of S6 ribosomal protein (S6) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1). The autophagy-inhibiting activity of mTOR was therefore decreased, as noted by dephosphorylation of ULK1 Ser757, an mTORC1 substrate (Kim et al., 2011). These findings were partially reversed by NMN supplementation, indicating autophagy initiation was an on-target effect of NAMPT inhibition. Together, these data implicate AMPK activation as an important metabolic state sensor driving the induction of autophagy upon NAD+ depletion.
NAD+ Depletion Inhibits IDH1 Mutant Tumor Growth in vivo
To assess the in vivo effect of NAD+ depletion in endogenous IDH1 mutant xenografts, we implanted HT1080 cells into the flank of SCID mice and treated with either FK866 or vehicle intraperitoneally when xenograft tumor diameters reached 5 mm. After 17 days of treatment, xenograft growth of FK866-treated animals remained near-completely inhibited while xenografts of vehicle-treated animals grew significantly (median tumor volume increase 3.2% ± 36.0% vs 415.6% ± 101.9% p=.003) (Figure 7A). There were significantly less Ki-67 positive cells in FK866-treated versus control tumors (Figure 7B).
Figure 7. NAMPT inhibitors inhibit growth of IDH1 mutant xenograft tumors.

(A) SCID mice implanted with HT1080 (IDH1R132C) cells in the flank were randomly assigned to intraperitoneal injections (4 days/week) of FK866 (30 mg/kg, n=6) or vehicle (n=6). Day 0 (treatment initiation) was the baseline. (B) Hematoxylin and eosin (H&E) and Ki-67 staining of HT1080 xenograft tissues. Ki-67 staining index: 63.5 ± 10.3 % (vehicle, left) vs 7.7 ± 4.8% (FK866, right) p=.001. Scale bars, 100μm. (C) Kaplan-Meier estimates of survival by treatment. SCID mice intracerebrally implanted with 2×105 MGG152 cells were randomly assigned to oral gavage (1/week) of either GMX1778 (250 mg/kg, n=6) or vehicle (n=6). (D) NAD+ levels in MGG152 orthotopic xenografts measured 24 hr after one dose of GMX1778 (250 mg/kg, black bar) or vehicle (white bar). Bars, +/− SEM, * p<0.05. See also Figure S4.
We further tested the effect of NAMPT inhibitors on mice harboring intracerebral MGG152 glioma xenografts. One week after intracerebral implantation, animals began weekly oral dosing of either GMX1778 or vehicle. GMX1778 treatment resulted in a marked extension of survival among MGG152-bearing animals (p=.0005, log rank test) (Figure 7C). We also quantified NAD+ levels in MGG152 orthotopic gliomas 24 hours after a single dose of GMX1778, and observed near-complete elimination of intratumoral NAD+ (Figure 7D). All mice maintained good general health status and stable body weight during treatment (Figures S4A, S4B).
DISCUSSION
Genetic evidence indicates that mutant IDH1 is a driver of tumorigenesis and a rational therapeutic target. Targeted inhibitors of mutant IDH have shown promise in IDH-mutant acute leukemia patients (Cancer Discovery, 2015). However, we found that 2-HG depletion using a direct mutant IDH1 inhibitor was not sufficient to inhibit the growth of several IDH1 mutant solid cancer types. Other genetic and/or epigenetic factors may modulate the effect of direct mutant IDH1 inhibition, and activity may be observed in only a subset of IDH1-mutant patients. We therefore used the direct mutant IDH1 inhibitor in an unbiased screen to identify other metabolic vulnerabilities and discovered an unanticipated and marked susceptibility of IDH1 mutant cancers to depletion of the coenzyme NAD+, which is a critical component of intracellular signaling pathways implicated in cancer cell growth (Chiarugi et al., 2012). Depletion of NAD+ through the use of small molecule inhibitors targeting the salvage NAD+ synthesis enzyme NAMPT resulted in strikingly selective cytotoxicity in IDH mutant cancer cells.
Our findings indicate that mutant IDH1 promotes this selective vulnerability by altering NAD+ supply. Introduction of mutant IDH1 reduces expression of Naprt1, a rate-limiting enzyme within the NAD+ salvage system, and results in lower basal NAD+ levels. Exposure to NAMPT inhibitors thus effectively inhibits both NAD+ salvage pathways (NAMPT and Naprt1) in IDH1 mutant cells, resulting in a metabolic crisis that activates the energy sensor AMPK and initiation of autophagy. In contrast, IDH wild-type cells have either increased expression of Naprt1 to maintain an essential supply of NAD+, or access to alternative metabolic pathways during metabolic stress (Wise et al., 2011; Mullen et al., 2012; Le et al., 2012), neither of which appear functionally-available in IDH mutant cancers (Leonardi et al., 2012; Grassian et al., 2014). It is also possible that upregulated NAD+-utilizing/consuming pathways partially account for the vulnerability of IDH1 mutant cells to NAD+ depletion. Indeed, others have demonstrated that IDH1 mutant cells have a greater dependence on the NAD+-requiring TCA cycle (Grassian et al., 2014; Cuyas et al., 2015). Although the effects of mutant IDH1 on NAD+-consuming pathways warrants further investigation, we find that reduced NAD+ salvage plays a major role in the mechanism of NAMPT inhibitor hypersensitivity.
The marked accumulation of 2-HG by mutant IDH induces global epigenetic changes (CIMP) and blocks differentiation, mediating a tumorigenic effect (Saha et al., 2014; Wang et al., 2013b). However, we did not detect epigenetic state changes nor growth inhibition of IDH1 mutant glioma TICs with pharmacologic reduction of 2-HG. In several other model systems, only minimal alterations in CIMP and moderate inhibition of IDH1 mutant tumor growth have been reported despite near complete abrogation of 2-HG production (Borodovsky et al., 2013; Grassian et al., 2014; Popovici-Muller et al., 2012; Rohle et al., 2013; Turcan et al., 2013). Cellular, genetic, and organ-level context likely impacts 2-HG effect and may explain the contrasting results between our models and in AML, where direct inhibition of mutant IDH2 induces differentiation of leukemia blast cells (Wang et al., 2013b). These differences suggest that IDH1/2 mutation may have additional contributions to tumorigenesis.
Based on the available evidence, we hypothesize that IDH mutations induce a fertile metabolic environment upon which subsequent oncogenic alterations arise, analogous to the selection of oncogenic KRAS and BRAF mutations in hypoglycemic conditions (Yun et al., 2009). In gliomas, IDH1 mutation occurs at the earliest stages of tumorigenesis, yet expression of IDH1 R132H is stable in TICs after multiple passages in vitro and in vivo (Borodovsky et al., 2013; Wakimoto et al., 2014) and clinically-manifest IDH1 mutant gliomas invariably maintain the mutant IDH1 allele despite acquiring additional malignant driver mutations (Johnson et al., 2014; Wakimoto et al., 2014). Progressive IDH1 mutant gliomas may therefore develop and maintain vulnerability to metabolic perturbations through this context-specific development of later mutations. Exploiting such vulnerabilities could theoretically be effective against all similarly-early mutant variants of IDH1 or IDH2, and may potentially be further enhanced by combination with the cellular stress induced by traditional chemotherapeutics or targeted radiation (Cairncross et al., 2014).
In conclusion, we have discovered that IDH1 mutation can render cancer cells “addicted” to a specific metabolite, NAD+. Importantly, our findings may have rapid translational impact as NAMPT inhibitors have entered clinical development. Our study demonstrates that the paradigm of metabolic targeting can be a highly selective and potentially effective strategy for IDH1 mutant cancers.
EXPERIMENTAL PROCEDURES
Cell culture, creation of cell lines, genotyping and cell line fingerprinting
Glioblastoma TICs and xenografts were generated from patient tumors as previously described (Wakimoto et al., 2009; Wakimoto et al., 2014). IDH1R132H overexpressing GBM TIC (MGG18-IDH1-R132H) was generated using pLenti3.3/TR or pLenti6.3/TO/V5 containing IDH1R132H, pCMV-dr8.2-dvpr and pCMV-VSVG (ViraPower HiPerform T-Rex Gateway Expression System, Invitrogen). The Naprt1 overexpressing IDH1 mutant GBM TIC (MGG152-Naprt1) was generated using CCSB-Broad LentiORF-NAPRT Clone (GE Dharmacon), pCMV-dr8.2-dvpr and pCMV-VSVG. Cell lines were obtained from ATCC (BT142, HT1080, U87, SW1353), Sigma-Aldrich (A431), Horizon Discovery (HCT116, MCF10A) and ScienCell (NHA). 30T and UACC257 were provided by Y.S. Chemicals were purchased from Sigma-Aldrich (FK866, GMX1778, NMN, NAD+, NA, decitabine, azacytidine, and 3-MA), or Cayman (GMX1778). For long-term IDH1i exposure, IDH1i (5 μM) or DMSO (0.1%) was added to the media 2–3 times per week.
Western Blot analyses and Immunohistochemistry
Primary antibodies for blotting: IDH1R132H (Dianova), H3K9me2, H3K9me3 (Abcam), H3K4me3, H3K27me3, p-AMPK, p-Raptor, p-mTOR, p-S6, p-4E-BP1, p-ULK1 (Ser555), p-ULK1 (Ser757), LC3B, GAPDH and Actin (Cell Signaling); NAMPT (Bethyl Labs), Naprt1 (Sigma-Aldrich), p-Atg13 (Rockland) and Vinculin (Thermo Scientific). Primary antibodies for IHC: anti-Ki-67 (1:125, Wako), Nestin (1:400, Santa Cruz), GFAP (1:400, Sigma-Aldrich), IDH1R132H (1:200), H3K9me3 (1:400) and H3K9me2 (1:200).
Measurement of cell viability, cytotoxicity, autophagy and apoptosis
Cell viability was measured using CellTiter-Glo (Promega) and cytotoxicity was determined by trypan blue exclusion assay. Apoptosis was measured by staining with propidium iodide (PI) and APC-conjugated Annexin V (eBioscience) and analysis by flow cytometry (Accuri) and BD CSampler software (BD Biosciences) and by Caspase-Glo 3/7 Assay (Promega).
Animal Efficacy Studies
2×106 HT1080 cells were subcutaneously implanted into the right flank of 7–10-week female SCID mice. Mice were randomized to daily intraperitoneal injections of FK866 (30 mg/kg, n=6) or normal saline (n=6) for 17 days. 1–2×105 MGG152 cells were implanted into the right striatum of SCID mice. Animals were randomly assigned for oral gavage treatment with IDH1i (400 mg/kg, 2×/day), GMX1778 (250 mg/kg, 1×/week) or vehicle (20% captisol (CyDex) and 5% dextrose in distilled water). For measurements of IDH1i in tumor tissues, animals were treated for 5 days and then sacrificed. To evaluate tumorigenicity, MGG152 was cultured in DMSO or IDH1i (5 μM) for the indicated passage number then 1×105 cells were implanted intracerebrally. All mouse experiments were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital and were performed in accordance with institutional and national guidelines and regulations.
Liquid Chromatography-Mass Spectroscopy Data, Measurement of NAD+/NADH, NADP+/NADPH and DNA Methylation Analyses
For detailed methods of metabolite quantification, metabolite profiling, 2-HG measurement and IDHi measurement see Supp. Exp. Procedures. Genome-wide methylation analysis was performed using the Illumina Infinium HumanMethylation450 BeadChip array. Methylation values for each site are expressed as a β value, representing a continuous measurement from 0 (completely unmethylated) to 1 (completely methylated). Methylation-specific PCR was performed in a standard two-step approach.
Statistical analyses
Statistical analysis was performed with JMP10 software. For parametric analyses, 2-tailed t tests were used, and for analysis of frequencies of nominal data, 2-tailed Fisher exact test was used. Correlation was assessed using the Pearson’s correlation coefficient. Data were expressed as mean ± SE. Survival analysis was performed using the Kaplan-Meier method and the log-rank test was used to compare survival differences between treatment arms. p<0.05 was considered statistically significant.
For complete details of all experimental procedures, see Supp. Exp. Procedures.
Supplementary Material
Significance.
Recently-discovered tumorigenic mutations in canonical metabolic enzymes highlight the importance of altered metabolism in cancer pathogenesis. IDH mutant cancers are hallmarked by aberrant accumulation of the metabolite 2-HG, which has been found to mediate many of the pathogenic phenotypes observed in these cancers. However, we found that proliferation of IDH1 mutant solid cancers can be decoupled from 2-HG levels in some contexts. Furthermore, by systematically investigating for other metabolic vulnerabilities, we discovered that mutant IDH1 cancers are exquisitely susceptible to depletion of the canonical coenzyme NAD+. Thus, we identify a potential therapeutic metabolic target specifically for IDH1 mutant cancers.
Acknowledgments
A.C. is supported by a Richard B. Simches Scholars Award and the MGH Glioblastoma Research Fund in memory of David M. King, Kirit N. Patel, Sarah E. Rippetoe and Eric W. Schwartz. D.C is supported by a Burroughs Wellcome Fund Career Award. A.C., D.C., H.W., T.B. are supported by NIH P50CA165962-01A1 (T.B., PI). T.B. is supported by NIH K24 CA125440-06. K.T. is supported by a Society of Nuclear Medicine and Molecular Imaging Wagner-Torizuka Fellowship, the Japan Brain Foundation, and the KANAE foundation for the Promotion of Medical Science. R.G. is supported by an American Heart Association Established Investigator Award. J.-R.Y. is supported by NIH R01CA140188 and the MGH Cardiology Hassenfeld Scholar Award. D.W., O.B., G.D., B.Z. and T.H. are full-time employees of Sanofi. We thank NovaBioAssays LLC for 2-HG quantitation.
Footnotes
Accession Number
The genome-wide DNA methylation data have been submitted to the GEO public database at http://www.ncbi.nlm.nih.gov/geo/ (accession number GSE73270).
AUTHOR CONTRIBUTIONS
K.T. and H.W. designed and performed experiments, interpreted data and wrote the paper. H.W., S.T., K.T., A.C. and W.C. generated cell lines. N.L., M.K. and S.S. sequenced samples. D.W., O.B., G.D., B.Z. and T.H. synthesized and characterized IDHi and performed measurements of 2-HG and IDHi. X.S., R.G., Y.Z. and J.-R.Y profiled metabolites. F.L., D.Z., F.N., Q.H., E.R. and L.K performed experiments. A.I., K.F. D.E.F., Y.S. and T.B. designed experiments and interpreted data. A.C and D.C. conceptualized the study, designed experiments, interpreted data and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116:597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- Borodovsky A, Salmasi V, Turcan S, Fabius AW, Baia GS, Eberhart CG, Weingart JD, Gallia GL, Baylin SB, Chan TA, Riggins GJ. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013;4:1737–1747. doi: 10.18632/oncotarget.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, Buckner JC, Fink KL, Souhami L, Laperriere NJ, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32:783–790. doi: 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–1903. doi: 10.1182/blood-2011-11-391748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Discovery. IDH1 inhibitor shows promising early results. Cancer Discov. 2015;5:4. doi: 10.1158/2159-8290.CD-NB2014-178. [DOI] [PubMed] [Google Scholar]
- Capper D, Weissert S, Balss J, Habel A, Meyer J, Jager D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H, et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010;20:245–254. doi: 10.1111/j.1750-3639.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerna D, Li H, Flaherty S, Takebe N, Coleman CN, Yoo SS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J Biol Chem. 2012;287:22408–22417. doi: 10.1074/jbc.M112.357301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Lu C, Cross JR, Morris JPt, Shroff AS, Ward PS, Bradner JE, Thompson C, Lowe SW. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013;27:1974–1985. doi: 10.1101/gad.226613.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome--a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–752. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuyas E, Fernandez-Arroyo S, Corominas-Faja B, Rodriguez-Gallego E, Bosch-Barrera J, Martin-Castillo B, De Llorens R, Joven J, Menendez JA. Oncometabolic mutation IDH1 R132H confers a metformin-hypersensitive phenotype. Oncotarget. 2015;6:12279–12296. doi: 10.18632/oncotarget.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G, Shen J, Yin M, McManus J, Mathieu M, Gee P, He T, Shi C, Bedel O, McLean LR, et al. Selective Inhibition of Mutant Isocitrate Dehydrogenase 1 (IDH1) via Disruption of a Metal Binding Network by an Allosteric Small Molecule. J Biol Chem. 2015;290:762–774. doi: 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli U, Travelli C, Massarotti A, Fakhfouri G, Rahimian R, Tron GC, Genazzani AA. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J Med Chem. 2013;56:6279–6296. doi: 10.1021/jm4001049. [DOI] [PubMed] [Google Scholar]
- Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers C, et al. IDH1 Mutations Alter Citric Acid Cycle Metabolism and Increase Dependence on Oxidative Mitochondrial Metabolism. Cancer Res. 2014;74:3317–3331. doi: 10.1158/0008-5472.CAN-14-0772-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118:469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- Hjarnaa PJ, Jonsson E, Latini S, Dhar S, Larsson R, Bramm E, Skov T, Binderup L. CHS 828, a novel pyridyl cyanoguanidine with potent antitumor activity in vitro and in vivo. Cancer Res. 1999;59:5751–5757. [PubMed] [Google Scholar]
- Izquierdo-Garcia JL, Cai LM, Chaumeil MM, Eriksson P, Robinson AE, Pieper RO, Phillips JJ, Ronen SM. Glioma Cells with the IDH1 Mutation Modulate Metabolic Fractional Flux through Pyruvate Carboxylase. PLoS One. 2014;9:e108289. doi: 10.1371/journal.pone.0108289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell. 2009;16:180–182. doi: 10.1016/j.ccr.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipp BR, Voss JS, Kerr SE, Barr Fritcher EG, Graham RP, Zhang L, Highsmith WE, Zhang J, Roberts LR, Gores GJ, Halling KC. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol. 2012;43:1552–1558. doi: 10.1016/j.humpath.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem. 2012;287:14615–14620. doi: 10.1074/jbc.C112.353946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons D, Jones S, Zhang X, Lin J, Leary R, Angenendt P, Mankoo P, Carter H, Siu I, Gallia G, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F, Gross S, Dang L, Yen KE, Yang H, et al. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med Chem Lett. 2012;3:850–855. doi: 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman ZJ, Duncan CG, Poteet E, Winters A, Yan LJ, Gooden DM, Spasojevic I, Boros LG, Yang SH, Yan H. Cancer-associated Isocitrate Dehydrogenase 1 (IDH1) R132H Mutation and D-2-hydroxyglutarate Stimulate Glutamine Metabolism under Hypoxia. J Biol Chem. 2014;289:23318–23328. doi: 10.1074/jbc.M114.575183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, Yan H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108:3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–114. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shames DS, Elkins K, Walter K, Holcomb T, Du P, Mohl D, Xiao Y, Pham T, Haverty PM, Liederer B, et al. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin Cancer Res. 2013;19:6912–6923. doi: 10.1158/1078-0432.CCR-13-1186. [DOI] [PubMed] [Google Scholar]
- Suijker J, Oosting J, Koornneef A, Struys EA, Salomons GS, Schaap FG, Waaijer CJ, Wijers-Koster PM, Briaire-de Bruijn IH, Haazen L, et al. Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget. 2015;6:12505–12519. doi: 10.18632/oncotarget.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, Viale A, Riggins GJ, Chan TA. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. 2013;4:1729–1736. doi: 10.18632/oncotarget.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto H, Kesari S, Farrell C, Curry WJ, Zaupa C, Aghi M, Kuroda T, Stemmer-Rachamimov A, Shah K, Liu T, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, et al. Targetable Signaling Pathway Mutations Are Associated with Malignant Phenotype in IDH-Mutant Gliomas. Clin Cancer Res. 2014;20:2898–2909. doi: 10.1158/1078-0432.CCR-13-3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013a;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013b;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, Weinstock DM, Sharp KA, Thompson CB. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene. 2012;31:2491–2498. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174:1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, Bernier C, Branchaud S, Chan H, Dairi K, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol. 2009;29:5872–5888. doi: 10.1128/MCB.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosikowski K, Mattern K, Schemainda I, Hasmann M, Rattel B, Loser R. WK175, a novel antitumor agent, decreases the intracellular nicotinamide adenine dinucleotide concentration and induces the apoptotic cascade in human leukemia cells. Cancer Res. 2002;62:1057–1062. [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons D, Jin G, McLendon R, Rasheed B, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.