

Abstract
Background
Pulmonary pleomorphic carcinoma (PPC) follows an aggressive clinical course and outcomes are disappointing. Due to its rarity, however, the clinicopathological and molecular characteristics of this disease remain unclear.
Methods
We retrospectively evaluated the efficacy of chemotherapy and molecular targeted therapy in 16 patients with PPC who received chemotherapy or EGFR-TKI. We also investigated the status of EGFR mutation, KRAS mutation and ALK expression.
Results
On histologic review of the malignant epithelial component, adenocarcinoma was identified in seven cases (43.8 %), large cell carcinoma in four (25.0 %), and squamous cell carcinoma in two (12.5 %). For the sarcomatoid component, 14 cases (87.5 %) had both spindle cell tumor and giant cell and 2 (12.5 %) had giant cell. Eleven patients received cytotoxic chemotherapy as first-line but did not achieve an objective response, although one patient who received docetaxel as second-line achieved a partial response. We also found that one patient achieved long stable disease of about 9 years without progression after receiving cisplatin and gemcitabine treatment. EGFR mutation, KRAS mutation and ALK expression were investigated in 14 patients whose tumor specimens were available. EGFR mutation was observed in 2 (14.3 %) and KRAS mutation in 3 (21.4 %), while no patient was positive for ALK expression. One patient harboring EGFR exon 19 deletion was treated with gefitinib after postoperative recurrence and achieved a complete response of about 35 months.
Conclusions
Although advanced PPC showed a poor response to chemotherapy, one patient with EGFR mutation achieved an extended complete response. We therefore recommend the evaluation of driver gene alteration such as EGFR in the treatment of advanced PPC.
Keywords: Pleomorphic carcinoma, Chemotherapy, Gefitinib, EGFR mutations
Background
Pulmonary pleomorphic carcinoma (PPC) is rare, with an incidence of 0.1–0.4 % of all non-small lung cancers (NSCLC) [1–5]. According to the World Health Organization classification report in 2004 [4], pulmonary pleomorphic carcinoma is defined as poorly differentiated adenocarcinoma, squamous cell carcinoma or large cell carcinoma, containing a component of spindle or giant cells with a sarcomatoid tumor component of at least 10 % [4]. Although clinical outcome is stage-dependent, it follows a more aggressive clinical course and has a worse prognosis than other histological types of NSCLC [6–10]. Furthermore, some recent reports have noted that PPC is often refractory to chemotherapy regimens which provide active treatment for NSCLC [8–12]. Due to its rarity, no optimal treatment for PPC has yet been established.
In NSCLC, the discovery that somatic alterations of driver gene, including epidermal growth factor receptor (EGFR) and anaplastic large kinase (ALK) gene, are found in a subset of lung adenocarcinomas and are associated with sensitivity to molecular target therapy has provided a rationale for the development of therapies in NSCLC [13–15]. Several reports noted that EGFR mutations were recognized in 15–20 % of patients with PPC but that the response to EGFR tyrosine kinase receptor inhibitor (TKI) was weak and transient as a consequence of tumor heterogeneity [8, 10, 11, 16–18].
Here, we retrospectively analyzed the efficacy of chemotherapy and molecular targeted therapy in patients with advanced or metastatic PPC, and characterized their somatic alteration status, particularly for EGFR mutation, K-ras mutation, and ALK immunohistochemistry (IHC).
Patients and methods
Patient selection
PPC was diagnosed according to the 2004 World Health Organization classification [4]. Diagnoses were based on light microscopy findings and confirmed by IHC examination. The histological diagnosis was reviewed by one of the authors (K.T.). From January 1998 to April 2010, 65 patients were histologically diagnosed with PPC by surgical resection, transbronchial lung biopsy, or computed tomography (CT) guided needle biopsy at our institution. Of these 65, 13 had received chemotherapy and 3 had received concurrent chemoradiotherapy, giving a total of 16 consecutive patients for final enrollment as subjects of this study. The protocol was approved by the institutional review board of National Cancer Centre Hospital and we reviewed the medical records of all of these patients.
EGFR mutation, KRAS mutation, and ALK-IHC analysis
Activating EGFR mutations (i.e., exon 19 in-frame deletion and exon 21 L858 R missense mutations) and KRAS mutation in exon 2 (codon 12 and codon 13) were examined in paraffin-embedded tumor specimens by high-resolution melting assay using LCGreen (Idaho Technology) on a LightCycler (Roche Diagnostics), as previously described [19]. These PCR products were denatured at 95 °C for 10 min and cooled to 40 °C to promote the formation of heteroduplexes. The LightCycler capillary was transferred to an HR-1 (Idaho Technology), an high-resolution melting assay instrument, and heated at a transition rate of 0.3 °C/s. Data were acquired and analyzed using the accompanying software (Idaho Technology). After normalization and temperature-adjustment steps, melting curve shapes from 78.5 to 85.5 °C were compared between the tumor samples and control samples. Human Genomic DNA (Roche Diagnostics) was used as the negative control sample with wild-type EGFR. Samples revealing skewed or left-shifted curves as compared with the control samples were judged to have mutations without positive controls.
ALK gene fusions were analyzed by immunohistochemistry. Four-micrometer-thick sections were deparaffinized. Heat-induced epitope retrieval was performed with targeted retrieval solution (pH 9) (Dako, Carpinteria, CA). The slides were then incubated with primary antibodies against ALK protein (1:40, 5A4; Abcam, Cambridge, UK) for 30 min at room temperature. Immunoreactions were detected using the EnVision-FLEX and LINKER (Dako). The reactions were visualized with 3,3′-diaminobenzidine, followed by counterstaining with hematoxylin.
To evaluate the genetic heterogeneity of PPC, we also investigated EGFR IHC in two different histological types. For immunohistochemical staining, formalin-fixed, paraffin-embedded tissues were cut into 4-μm-thick sections and deparaffinized, then subject to heat-induced epitope retrieval with Target Retrieval Solution (Dako, Carpinteria, CA, USA). The primary antibody used was a rabbit monoclonal antibody against human EGFR with the DEL (E746-A750del) mutation (1:100, clone 6B6, Cell Signaling Technology, Danvers, MA, USA) and a rabbit monoclonal antibody against human EGFR with the L858R mutation (1:200, clone 43B2, Cell Signaling Technology) [20]. Antibodies were diluted in SignalStain (Cell Signaling Technology) and the slides were incubated with each primary antibody for 1 h at room temperature. Immunoreactions were detected using the EnVision Plus system (Dako) and 3, 3′-diaminobenzidine, followed by counterstaining with hematoxylin.
Assessment and analysis
Clinical information was obtained from medical records. Clinical disease staging was reassessed according to the latest International Union Against Cancer staging criteria [21]. Response to chemotherapy and survival were assessed retrospectively and classified according to the Response Evaluation Criteria for Solid Tumors, version 1.1 [22]. Progression-free survival (PFS) was defined as the time from the first day of chemotherapy to detection of the earliest signs of disease progression or death from any cause. Overall survival (OS) was defined as the time from the first day of chemotherapy to the last day on which the patient was confirmed to be alive or dead from any cause. Survival was estimated using the Kaplan–Meier method. Fisher’s exact and χ2 test was used to examine the association of two categorical variables. Differences with probability values of <0.05 were considered statistically significant. STATA version 12 (StataCorp LP, College Station, TX, USA) was used for all analyses.
Results
Patient characteristics
Clinical characteristics of the 16 patients who were finally enrolled are listed in Table 1. Median age was 61 years (range 43–77 years). Ten patients (62.5 %) were male and six (37.5 %) were female. Twelve patients (75.0 %) were ex-smokers and their median pack-years was 45.5 (range 17–124). Ten patients (62.5 %) had relapsed after curative surgery, four (25.0 %) were initially diagnosed with advanced disease at stage IV, and two (12.5 %) were diagnosed with locally advanced disease at stage IIIA. Among the relapsed patients who had undergone curative surgery, none had received adjuvant chemotherapy. Two patients with stage IIIA disease and one who had relapse of mediastinal and supraclavicular lymph node metastases received concurrent chemoradiotherapy. Thirteen patients (81.3 %) received systemic chemotherapy, including cytotoxic agents or molecular targeted agents. Ten patients were diagnosed by the resected primary tumor; of the other six, three were diagnosed by CT guided needle biopsy and three by autopsy.
Table 1.
Patient characteristics
| Characteristic | Number of patients (n = 16) |
|---|---|
| Sex | |
| Male | 10 (62.5 %) |
| Female | 6 (37.5 %) |
| Age (years) | |
| Median (range) | 61 (43–77) |
| ECOG-PS | |
| 0 | 3 (18.8 %) |
| 1 | 10 (62.5 %) |
| 2 | 3 (18.8 %) |
| Smoking status | |
| Never smoker | 4 (25.0 %) |
| Ex-smoker | 12 (75.0 %) |
| Pack-yearsa | |
| Median (range) | 45.5 (17–124) |
| Clinical stage | |
| IIIA | 2 (12.5 %) |
| IV | 4 (25.0 %) |
| Recurrence | 10 (62.5 %) |
| Diagnostic procedure | |
| Surgery | 10 (62.5 %) |
| CT-NB | 3 (18.8 %) |
| Autopsy | 3 (18.8 %) |
| Pathological features | |
| Epithelial component | |
| Adenocarcinoma | 7 (43.8 %) |
| Squamous cell carcinoma | 2 (12.5 %) |
| Adenosquamous carcinoma | 1 (6.3 %) |
| Large cell carcinoma | 4 (25.0 %) |
| NOS | 2 (12.5 %) |
| Sarcomatoid component | |
| Spindle cell | 0 (0.0 %) |
| Giant cell | 2 (12.5 %) |
| Spindle cell and giant cell | 14 (62.5 %) |
CT-NB CT-guided needle biopsy, NOS not otherwise specified
aEx-smoker (n = 12)
Pathological features
On histologic review of the malignant epithelial component, adenocarcinoma was identified in seven cases (43.8 %), large cell carcinoma in four (25.0 %), squamous cell carcinoma in two (12.5 %), NSCLC in two (12.5 %), and adenosquamous carcinoma in one (6.3 %). For the sarcomatoid component, two cases (12.5 %) had giant cell tumors. Of the 14 cases (87.5 %) who had both spindle cell tumors and giant cell tumors, the spindle cell tumor was dominant in nine (56.3 %) while the giant cell was dominant in five (31.3 %).
Molecular profile
EGFR mutation, KRAS mutation and ALK-IHC were investigated in the 14 patients whose tumor specimens were available (Table 2). EGFR mutation was observed in 2 (14.3 %) of these 14 patients. The histological features of the patient harboring EGFR exon 19 deletion was adenosquamous carcinoma with spindle cell and giant cell tumor and that of the patient harboring EGFR L858R mutation in exon 21 was large cell carcinoma with spindle cell and giant cell tumor. KRAS mutation was observed in three patients (21.4 %). Two patients with EGFR mutation were never smoker, while all patients with KRAS mutation were ex-smokers. No patient was positive for ALK-IHC.
Table 2.
Association between pathological findings and molecular profile
| No. | Sex | Diagnostic procedure | Smoking index | EGFR mutation | KRAS mutation | ALK IHC | Pathological features | |
|---|---|---|---|---|---|---|---|---|
| Epithelial component | Sarcomatoid component | |||||||
| 1 | M | Surgery | 2100 | Wild | Mutated | Negative | Adeno | Giant cell > Spindle cell |
| 2 | F | Surgery | 0 | Exon 19 del | Wild | Negative | Adenosquamous | Spindle cell > Giant cell |
| 3 | M | Surgery | 1200 | Wild | Wild | Negative | Adeno | Spindle cell > Giant cell |
| 4 | F | Surgery | 0 | Wild | Wild | Negative | Adeno | Spindle cell > Giant cell |
| 5 | M | Surgery | 400 | Wild | Wild | Negative | Adeno | Spindle cell > Giant cell |
| 6 | M | Surgery | 1800 | Wild | Wild | Negative | Squamous | Spindle cell > Giant cell |
| 7 | F | Surgery | 600 | Wild | Wild | Negative | Adeno | Spindle cell > Giant cell |
| 8 | F | Surgery | 345 | Wild | Mutated | Negative | Large | Giant cell > Spindle cell |
| 9 | M | Autopsy | 495 | NA | NA | NA | NOS | Giant cell > Spindle cell |
| 10 | M | Biopsy | 2480 | Wild | Wild | Negative | NOS | Giant cell |
| 11 | M | Autopsy | 480 | Wild | Wild | Negative | Adeno | Giant cell |
| 12 | M | Biopsy | 1600 | Wild | Mutated | Negative | Squamous | Spindle cell > Giant cell |
| 13 | M | Surgery | 660 | Wild | Wild | Negative | Large | Giant cell > Spindle cell |
| 14 | M | Autopsy | 1160 | Wild | Wild | Negative | Adeno | Giant cell > Spindle cell |
| 15 | F | Surgery | 0 | Exon 21 L858R | Wild | Negative | Large | Spindle cell > Giant cell |
| 16 | F | Biopsy | 0 | NA | NA | NA | Large | Spindle cell > Giant cell |
IHC immunohistochemistry, NOS not otherwise specified, Adeno adenocarcinoma, Squamous squamous cell carcinoma, Large large cell carcinoma, Adenosquamous adenosquamous carcinoma
Next, regarding the two cases harboring EGFR mutation, we evaluated the mutated EGFR protein expression using a mutation-specific antibody against EGFR mutations in both the epithelial and sarcomatoid components. In patient No. 2, we found that IHCs by mutation-specific antibody were positive in adenocarcinoma (Fig. 1a), squamous cell carcinoma (Fig. 1b), and sarcomatoid components (Fig. 1c). And patient No. 15 progressed after concurrent chemoradiotherapy and so we could not check the mutated EGFR protein expression in both the epithelial and sarcomatoid components.
Fig. 1.
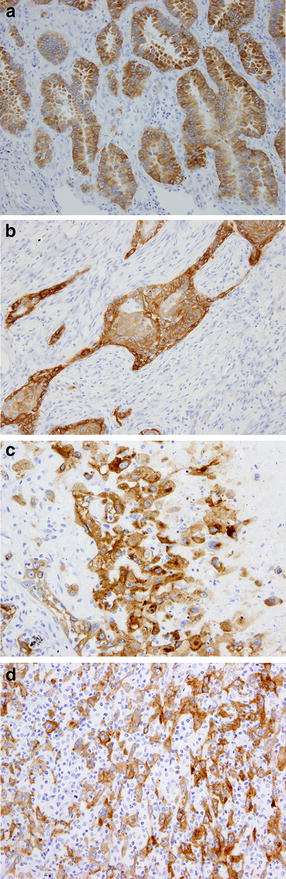
Two representative cases harboring EGFR mutation by mutation-specific antibody against EGFR mutations in both epithelial and sarcomatoid components. a adenocarcinoma component in patient No. 2, ×20; b squamous cell carcinoma component in patient No. 2, ×20; c sarcomatoid component in patient No. 2, ×20; d sarcomatoid component in patient No. 15, ×20
Treatment and efficacy
Of the 13 patients with advanced stage disease, 11 patients received cytotoxic chemotherapy as first-line chemotherapy. Among them, however, no patient achieved an objective response, except one patient (No. 6) who received docetaxel as second-line chemotherapy and achieved a partial response. In all patients, median PFS for first-line chemotherapy was 1.5 (95 % CI 0.6–2.5) months and OS was 7.2 (95 % CI 1.4–10.3) months (Table 3). We also identified one patient (No. 13) who achieved stable disease of about 9 years’ duration without progression after undergoing small intestinal resection for ileus due to a postoperative recurrence lesion in the small intestine and then receiving four cycles of cisplatin and gemcitabine treatment for hilar lymph node and bone metastases. One patient (No. 2) harboring EGFR exon 19 deletion was treated with gefitinib after postoperative recurrence of mediastinal lymph node and achieved a complete response of about 35 months (Fig. 2). Two months after she declined further treatment with gefitinib because of hemoptysis, she developed progressive disease in a mediastinal lymph node and thoracic spine metastases. All three patients who received concurrent chemoradiotherapy with cisplatin plus vinorelbine achieved a partial response (Table 3). Although two patients had early recurrence, the third is alive without progression more than 5 years after completing chemoradiotherapy. One patient (No. 15) harboring EGFR exon 21 point mutation developed multiple brain metastases and carcinomatous meningitis immediately after chemoradiotherapy and was then treated with best supportive care without EGFR-TKI because of poor general condition.
Table 3.
Treatment results by systemic chemotherapy and chemoradiotherapy
| No. | Sex | Age | Stage | PS | EGFR mutation status | Initial treatment | OR | PFS (mos) | OS (mos) | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 55 | Relapsed | 1 | Wild | DTX | PD | 0.7 | 7.3 | Died |
| 2 | F | 69 | Relapsed | 1 | Exon 19 del | Gefitinib | CR | 35.1 | 53.9 | Died |
| 3 | M | 60 | Relapsed | 1 | Wild | CBDCA + PTX | PD | 0.9 | 3.7 | Died |
| 4 | F | 45 | Relapsed | 0 | Wild | Gefitinib | PD | 1.7 | 10.7 | Died |
| 5 | M | 63 | Relapsed | 1 | Wild | CBDCA + PTX | PD | 0.6 | 12.7 | Died |
| 6 | M | 62 | Relapsed | 1 | Wild | CDDP + GEM | PD | 2.5 | 18.7 | Died |
| 7 | F | 53 | Relapsed | 2 | Wild | CBDCA + PTX | PD | 0.4 | 1 | Died |
| 8 | F | 55 | Relapsed | 1 | Wild | DTX | PD | 0.7 | 1.4 | Died |
| 9 | M | 56 | Advanced | 1 | NA | CBDCA + PTX | PD | 0.6 | 1.0 | Died |
| 10 | M | 77 | Advanced | 2 | Wild | CBDCA + ETP | PD | 1.5 | 7.2 | Died |
| 11 | M | 43 | Advanced | 1 | Wild | CBDCA + PTX | SD | 4.5 | 10.3 | Died |
| 12 | M | 62 | Advanced | 2 | Wild | CBDCA + PTX | NE | 0.4 | 0.4 | Died |
| 13 | M | 56 | Relapsed | 0 | Wild | CDDP + GEM | NE | 109.2 | 109.2 | Alive |
| 14 | M | 65 | Stage IIIA | 1 | Wild | CDDP + VNR + TRT | PR | 5.0 | 6.6 | Died |
| 15 | F | 66 | Relapsed | 1 | Exon 21 L858R | CDDP + VNR + TRT | PR | 3.9 | 6.9 | Died |
| 16 | F | 66 | Stage IIIA | 0 | NA | CDDP + VNR + TRT | PR | 65.4 | 65.4 | Alive |
PS performance status, OR overall response, PFS progression-free survival, OS overall survival, DTX docetaxel, CBDCA carboplatin, PTX paclitaxel, CDDP cisplatin, GEM gemcitabine, ETP etoposide, VNR vinorelbine, TRT thoracic radiotherapy, SD stable disease, CR complete response, PD progressive disease, NE not evaluable, PR partial response
Fig. 2.
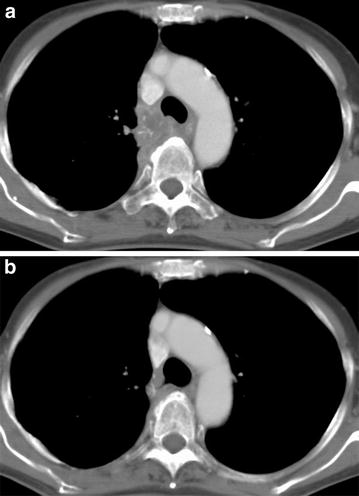
Chest computed tomography images of mediastinal lymph nodes (#4R) before (a) and 6 months after gefitinib treatment (b)
Discussion
Previous retrospective studies and case reports suggested that PPC is often refractory to the chemotherapy regimens used in active treatment for NSCLC [8–11]. This retrospective study also found that PPC is an aggressive tumor associated with high cell proliferation and that palliative chemotherapy is associated with a poor response in advanced PPC, with a few notable exceptions.
With regard to chemoradiotherapy for locally advanced PPC, all three patients achieved a partial response in our study. Although two patients had early recurrence, the third is alive without progression more than 5 years after completing chemoradiotherapy. Some reports revealed that PPC treated with chemoradiotherapy achieved an objective response without recurrence [10, 11]. These results if confirmed suggest that chemoradiotherapy commonly used for NSCLC should be administered in unresectable stage III disease with pleomorphic carcinoma.
Several recent reports have described cases of PPC that responded to cytotoxic chemotherapy containing platinum and gemcitabine [8–12, 23]. Tamiya et al. mentioned that high expression of human equilibrative nucleoside transporter 1 (hENT1), the major transporter of gemcitabine, might be associated with high sensitivity to gemcitabine in PPC [12]. We also experienced one patient who achieved long-term stable disease without progression after receiving cisplatin and gemcitabine treatment. We propose that cisplatin and gemcitabine treatment may be an active chemotherapy for PPC.
In this study, we also investigated EGFR mutation, KRAS mutation and ALK-IHC in the 14 patients with PPC whose tumor specimens were available. We found that two (14.3 %) of these patients harbored EGFR mutation. One patient treated with gefitinib achieved a complete response, which was moreover sustained for about 35 months. Previous reports suggested that the incidence of EGFR mutation was about 15–20 % and that the response to gefitinib was weak and transient [8, 10, 11, 16–18]. Kaira et al. independently investigated EGFR mutations in both the adenocarcinomatous and sarcomatoid components of three patients with EGFR mutation and detected EGFR mutations in the adenocarcinomatous component but not in the sarcomatoid component in all cases. Ushiki et al. revealed a case of PPC whose adenocarcinoma cells had an exon 19 deletion and whose sarcomatous cells had both the deletion 19 and 20 T790 M EGFR mutations [16]. In our present study, we could not clearly separate epithelial tumor cells from sarcomatoid tumor cells because tumor tissue consisted of a mixture of both components. We then investigated IHC by mutated EGFR-specific antibody in the two different histological types to evaluate the genetic heterogeneity of PPC. We found that mutated EGFR protein expression was positive in both components. We consider that our results are unlikely to be false positives, because mutation-specific antibodies against EGFR mutation have high specificity in spite of low sensitivity [20]. Tumorigenesis in lung cancer is known to be a multistage process by which monoclonal cancer cells gradually become heterogeneous owing to clonal evolution and genetic/epigenetic instability [24–27]. Applying computational analysis to the deep sequencing data of NSCLC samples, Govindan et al. suggested that EGFR mutation might be acquired at the very initial phase of tumorigenesis [28]. We consider that the difference in the EGFR mutational heterogeneity of PPC between the epithelial and sarcomatoid components represents the point at which clonal evolution occurred. Additionally, our results also suggest that driver gene alteration such as EGFR should be investigated in the treatment of advanced PPC.
One limitation of our study is that the sample size was small, due to the rarity of this condition. Additionally, 10 of 16 patients diagnosed from the resected primary tumor did not undergone rebiopsy at the start of chemotherapy. For this reason, we did not attempt to identify which component of PPC relapsed.
Conclusions
All the patients who received concurrent chemoradiotherapy achieved at least a partial response and if confirmed, chemoradiotherapy should be considered an effective modality in locally advanced disease. Although this study demonstrated that advanced PPC responds poorly to chemotherapy, further clinical trials are needed to investigate the role of palliative chemotherapy for PPC, including platinum and gemcitabine treatment. Additionally, one patient with EGFR mutation achieved a long-term complete response. We therefore recommend evaluating driver gene alteration, such as EGFR, in the treatment of advanced PPC.
Authors’ contributions
YT, YF, NY, and HN designed the research, and YT, NY, HN, HH, SK, YO promoted the research. YT, YF, and HN analyzed the data, and YT, YF, EK, HN, SK, KT, and YO wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We greatly appreciate the patients who participated in this study and their families. We also appreciate the assistance of the staff of the Department of Thoracic Oncology, National Cancer Center Hospital. The study sponsor had no involvement in the study design, collection, analysis and interpretation of data; in the writing of the manuscript; or in the decision to submit the manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- PPC
pulmonary pleomorphic carcinoma
- NSCLC
non-small cell lung cancer
- EGFR
epidermal growth factor receptor
- ALK
anaplastic large kinase
- TKI
tyrosine kinase receptor inhibitor
- IHC
immunohistochemistry
- CT
computed tomography
- PFS
progression free survival
- OS
overall survival
Contributor Information
Yosuke Tamura, Email: in1285@poh.osaka-med.ac.jp.
Yutaka Fujiwara, Phone: +81-3-3542-2511, Email: fu_ji_@yahoo.co.jp, Email: yutakafu@ncc.go.jp.
Noboru Yamamoto, Email: nbryamam@ncc.go.jp.
Hiroshi Nokihara, Email: hnokihar@ncc.go.jp.
Hidehito Horinouchi, Email: hhorinou@ncc.go.jp.
Shintaro Kanda, Email: skanda@ncc.go.jp.
Yasushi Goto, Email: ygoto@ncc.go.jp.
Emi Kubo, Email: ekubo@ncc.go.jp.
Shinsuke Kitahara, Email: skitahar@ncc.go.jp.
Kenjiro Tsuruoka, Email: ktsuruok@ncc.go.jp.
Koji Tsuta, Email: tsutakoj@hirakata.kmu.ac.jp.
Yuichiro Ohe, Email: yohe@ncc.go.jp.
References
- 1.Fishback NF, Travis WD, Moran CA, Guinee DG, Jr, McCarthy WF, Koss MN. Pleomorphic (spindle/giant cell) carcinoma of the lung. A clinicopathologic correlation of 78 cases. Cancer. 1994;73(12):2936–2945. doi: 10.1002/1097-0142(19940615)73:12<2936::AID-CNCR2820731210>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 2.Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059–1068. doi: 10.1183/09031936.01.00275301. [DOI] [PubMed] [Google Scholar]
- 3.Travis WD. Pathology of lung cancer. Clin Chest Med. 2002;23(1):65–81. [DOI] [PubMed]
- 4.Travis WD, Brambilla E, Muller-Hermelink HK, Harris CC. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon: International Agency for Research on Cancer Press; 2004.
- 5.Beasley MB, Brambilla E, Travis WD. The 2004 World Health Organization classification of lung tumors. Semin Roentgenol. 2005;40(2):90–97. doi: 10.1053/j.ro.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer. 2001;34(1):91–97. doi: 10.1016/S0169-5002(01)00224-0. [DOI] [PubMed] [Google Scholar]
- 7.Mochizuki T, Ishii G, Nagai K, et al. Pleomorphic carcinoma of the lung: clinicopathologic characteristics of 70 cases. Am J Surg Pathol. 2008;32(11):1727–1735. doi: 10.1097/PAS.0b013e3181804302. [DOI] [PubMed] [Google Scholar]
- 8.Bae HM, Min HS, Lee SH, et al. Palliative chemotherapy for pulmonary pleomorphic carcinoma. Lung Cancer. 2007;58(1):112–115. doi: 10.1016/j.lungcan.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Hong JY, Choi MK, Uhm JE, et al. The role of palliative chemotherapy for advanced pulmonary pleomorphic carcinoma. Med Oncol. 2009;26(3):287–291. doi: 10.1007/s12032-008-9117-4. [DOI] [PubMed] [Google Scholar]
- 10.Ito K, Oizumi S, Fukumoto S, et al. Clinical characteristics of pleomorphic carcinoma of the lung. Lung Cancer. 2010;68(2):204–210. doi: 10.1016/j.lungcan.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Kaira K, Horie Y, Ayabe E, et al. Pulmonary pleomorphic carcinoma: a clinicopathological study including EGFR mutation analysis. J Thorac Oncol. 2010;5(4):460–465. doi: 10.1097/JTO.0b013e3181ce3e3c. [DOI] [PubMed] [Google Scholar]
- 12.Tamiya A, Asami K, Shimizu S, et al. Long-term complete response in a patient with disseminated pulmonary pleomorphic carcinoma induced by cisplatin and gemcitabine. Intern Med. 2014;53(22):2625–2628. doi: 10.2169/internalmedicine.53.2637. [DOI] [PubMed] [Google Scholar]
- 13.Azzoli CG, Temin S, Aliff T, et al. 2011 Focused Update of 2009 American Society of Clinical Oncology clinical practice guideline update on chemotherapy for stage IV non-small-cell lung cancer. J Clin Oncol. 2011;29(28):3825–3831. doi: 10.1200/JCO.2010.34.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reck M, Popat S, Reinmuth N et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25 Suppl 3:iii27–39. [DOI] [PubMed]
- 15.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 16.Ushiki A, Koizumi T, Kobayashi N, et al. Genetic heterogeneity of EGFR mutation in pleomorphic carcinoma of the lung: response to gefitinib and clinical outcome. Jpn J Clin Oncol. 2009;39(4):267–270. doi: 10.1093/jjco/hyn155. [DOI] [PubMed] [Google Scholar]
- 17.Saitoh M, Niijima M, Takiguchi Y, et al. An early event of EGFR mutation in pleomorphic carcinoma of the lung. Int J Clin Oncol. 2011;16(6):770–773. doi: 10.1007/s10147-011-0247-x. [DOI] [PubMed] [Google Scholar]
- 18.Lee S, Kim Y, Sun JM, et al. Molecular profiles of EGFR, K-ras, c-met, and FGFR in pulmonary pleomorphic carcinoma, a rare lung malignancy. J Cancer Res Clin Oncol. 2011;137(8):1203–1211. doi: 10.1007/s00432-011-0986-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukui T, Ohe Y, Tsuta K, et al. Prospective study of the accuracy of EGFR mutational analysis by high-resolution melting analysis in small samples obtained from patients with non-small cell lung cancer. Clin Cancer Res. 2008;14(15):4751–4757. doi: 10.1158/1078-0432.CCR-07-5207. [DOI] [PubMed] [Google Scholar]
- 20.Bondgaard AL, Hogdall E, Mellemgaard A, Skov BG. High specificity but low sensitivity of mutation-specific antibodies against EGFR mutations in non-small-cell lung cancer. Mod Pathol. 2014;27(12):1590–8. [DOI] [PubMed]
- 21.Sobin LH, Gospodarowicz M, Wittekind C, editors. TNM classification of malignant tumors. 7th ed. Oxford: Wiley; 2009.
- 22.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 23.Yamanashi K, Marumo S, Miura K, Kawashima M. Long-term survival in a case of pleomorphic carcinoma with a brain metastasis. Case Rep Oncol. 2014;7(3):799–803. doi: 10.1159/000368186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowell PC. Mechanisms of tumor progression. Cancer Res. 1986;46(5):2203–2207. [PubMed] [Google Scholar]
- 25.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Bruin EC, McGranahan N, Mitter R, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346(6206):251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Fujimoto J, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346(6206):256–259. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150(6):1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]