

Abstract
Genetic variation arising from single nucleotide polymorphisms (SNPs) is ubiquitously found among human populations. While disease-causing variants are known in some cases, identifying functional or causative variants for most human diseases remains a challenging task. Rare SNPs, rather than common ones, are thought to be more important in the pathology of most human diseases. We propose that rare SNPs should be divided into two categories dependent on whether the minor alleles are derived or ancestral. Derived alleles are less likely to have been purified by evolutionary processes and may be more likely to induce deleterious effects. We therefore hypothesized that the rare SNPs with derived minor alleles would be more important for human diseases and predicted that these variants would have larger functional or structural consequences relative to the rare variants for which the minor alleles are ancestral. We systematically investigated the consequences of the exonic SNPs on protein function, mRNA structure, and translation. We found that the functional and structural consequences are more significant for the rare exonic variants for which the minor alleles are derived. However, this pattern is reversed when the minor alleles are ancestral. Thus, the rare exonic SNPs with derived minor alleles are more likely to be deleterious. Age estimation of rare SNPs confirms that these potentially deleterious SNPs are recently evolved in the human population. These results have important implications for understanding the function of genetic variations in human exonic regions and for prioritizing functional SNPs in genome-wide association studies of human diseases.
Keywords: single nucleotide polymorphisms, purifying selection, positive selection, ancestral allele, translational selection, RNA structure
Introduction
What genetic variants are most functionally relevant for human diseases? With rapid advances in next-generation DNA sequencing technologies, whole exome, or genome sequencing have been widely applied in genetic association studies to identify the causative genetic variations of human diseases or traits (Goldstein et al. 2013; Soon et al. 2013). A majority of currently known disease-associated variants are common single nucleotide polymorphisms (SNPs), yet these SNPs can only explain a small proportion of genetic variance in diseases (Manolio et al. 2009). One possibility is that rare SNPs, rather than common ones, are more important in the pathology of most human diseases (Cirulli and Goldstein 2010; Gibson 2010, 2012).
Several lines of evidences support this “common disease-rare variants” hypothesis (Iyengar and Elston 2007). First, many studies have related rare genetic variants to complex human diseases, such as type 2 diabetes, hypertriglyceridemia, sick sinus syndrome (McClellan et al. 2007; Nejentsev et al. 2009; Johansen et al. 2010; Holm et al. 2011; Rivas et al. 2011; Need et al. 2012; Lohmueller et al. 2013). Second, disease-causing variants should be selectively unfavorable (i.e., should decrease fitness). Therefore, purifying selection may be common in the evolution of SNPs, preventing deleterious alleles from drifting to a higher frequency in the population (Bulmer 1971). Third, theoretical population genetics suggests that recent explosion of human population size may allow the accumulation of rare alleles with deleterious effects (Lynch 2010; Simons et al. 2014). Finally, accumulating human polymorphism data from 1000 Genomes Project (1000 Genomes Project Consortium 2012), Personal NHLBI Exome Sequencing Project (Tennessen et al. 2012; Fu et al. 2013), and International HapMap Project (International HapMap 3 Consortium 2010) exemplify that the majority of human SNPs are rare. Rare SNPs are enriched in genomic regions that are evolutionarily conserved (Zhu et al. 2011; 1000 Genomes Project Consortium 2012) or functionally important (Wray 2007; Montgomery et al. 2011; Maurano et al. 2012). Therefore, rare SNPs should be considered as a high priority when identifying causative genetic variants in genetic association studies and medical genome sequencing (Pabinger et al. 2014).
In general, SNPs with the minor allele frequency (MAF) less than 1% are defined to be rare variants (Frazer et al. 2009). Among rare variants, some are evolutionarily conserved, suggesting that if they are deleterious, they have small functional effects or are maintained in the genome by various mechanisms (e.g., compensatory or epistatic effects or antagonistic pleiotropy [Davis et al. 2009]). These SNPs may be less strongly associated with phenotypic changes. Newly arisen variants in the genome, however, have had less time to be purged by selection or other evolutionary processes, and may harbor more deleterious effects. Therefore, we hypothesized that rare SNPs that have arisen recently should be given priority when determining what genetic variants are functionally relevant for human diseases. We predicted that 1) MAF would be negatively associated with deleteriousness only for derived alleles, and 2) the derived allele frequency (DAF) would better predict phenotypic effects of SNPs.
The potential deleteriousness of a variant is related to the strength of its functional effects (Fisher 1930). Previous efforts have primarily focused on nonsynonymous variants occurring in protein coding regions (International HapMap 3 Consortium 2010; Zhu et al. 2011; 1000 Genomes Project Consortium 2012; Tennessen et al. 2012; Fu et al. 2013). However, little is known about the functional consequences of synonymous SNPs or SNPs outside coding regions. Several previous studies have used conservation scores, which are derived from evolutionary comparisons, in estimating the functional importance of such SNPs (Chorley et al. 2008; Goode et al. 2010; Montgomery et al. 2011; Maurano et al. 2012; Kircher et al. 2014) because it is difficult to quantify the functional consequences of noncoding or synonymous SNPs. Recent high-throughput RNA structure profiling data and bioinformatics analyses (Li et al. 2012; Mortimer et al. 2014) have revealed important regulatory roles performed by RNA structures in different mRNA regions, such as mRNA translation initiation regions (Gu et al. 2010), microRNA target site regions (Gu et al. 2012), 5′-untranslated regions (5′-UTRs) near the cap site (Gu et al. 2014b), and RNA splicing regions (Li et al. 2013). Single nucleotide variations, including several synonymous variants, have been proposed to cause diseases by altering mRNA structure (Sauna and Kimchi-Sarfaty 2011; Salari et al. 2013). Hence, a systematic analysis of structural effects caused by exonic variants, including synonymous SNPs, nonsynonymous SNPs, SNPs in 5′-UTRs, and SNPs in 3′-UTRs will help provide insights into the functional and structural effects of rare exonic SNPs in the human genome.
Here, we performed a genome-wide analysis of functional and structural changes caused by all exonic SNPs in the human genome. We calculated changes to mRNA structure made by each exonic SNP in silico. We also calculated the effects on mRNA translation for synonymous SNPs, and alterations on protein function for nonsynonymous SNPs. For the SNPs for which the minor alleles is derived, functional changes made by rare exonic variants are stronger than those made by common variants. This pattern suggests purifying selection, rather than positive or balancing selection, as a major driver in shaping exonic variation in human populations (Zhu et al. 2011). Notably, this pattern is reversed when we focus only on those variants for which the minor alleles are ancestral. Exonic variants with DAF larger than 99% have the least functional consequences. Therefore, rare variants for which the minor alleles are ancestral are less likely to be deleterious (Gorlova et al. 2012). We propose that these variants can be excluded when prioritizing causative variants in genetic association studies. In addition, we observed a significant enrichment of rare variants, which have the biggest functional and structural effects, in exonic regions of human genome. Age distribution analysis of these rare variants confirmed a recent explosion of deleterious SNPs in the human population (1000 Genomes Project Consortium 2012; Fu et al. 2013).
Materials and Methods
SNP and Random Mutation Data
We obtained exonic SNPs within protein-coding genes from dbSNP (May 21, 2014, release; NCBI dbSNP Human Build 141) (Sherry et al. 2001). The ancestral state of each SNP was obtained from Ensembl (Flicek et al. 2014), which was based on a six-way primate genome-wide alignment (Paten et al. 2008). Only the SNPs with annotated ancestral allele and global MAF were included. We excluded the SNPs with more than two alleles. The SNPs without unique gene annotation were also excluded. We matched each SNP to the corresponding transcripts defined by Ensembl (Flicek et al. 2014). Only the longest transcript for each SNP was saved for further analysis. In total, we collected 720,480 exonic SNPs, including 46,157 SNPs located in 5′-UTR, 261,697 SNPs located in 3′-UTR, 240,813 nonsynonymous SNPs, and 171,813 synonymous SNPs. We also randomly put 100,000 random point mutations into the transcripts, which guaranteed the same transition/transversion ratio in 5′-UTR, 3′-UTR, and coding region. In total, we obtained 53,307 and 46,693 random point mutations in coding and UTRs, respectively.
PhyloP, GERP, CADD, and SIFT Scores
We downloaded CADD (Kircher et al. 2014) score with annotations for all possible human (version hg19) SNPs from CADD website (http://cadd.gs.washington.edu/download, last accessed October 12, 2015), and parsed PhyloP (Siepel et al. 2006), GERP (Cooper et al. 2010), CADD (Kircher et al. 2014), and SIFT (Ng and Henikoff 2003) scores for each exonic human SNP.
Amino Acid Chemical Distance, Hydrophobicity Index, and BLOSUM62 Score
We used the amino acid chemical distance, change in amino acid hydrophobicity index (|ΔH|), and BLOSUM62 score to evaluate the potential functional effect caused by nonsynonymous SNPs. The definition of amino acid chemical distance was obtained from a previous study (Grantham 1974). The table of amino acid hydrophobicity index was obtained from Argos et al’s study (Argos et al. 1982). BLOSUM62 is a weighting matrix for amino acid substitution (Henikoff S and Henikoff JG 1992). A BLOSUM62 score of zero indicates that the frequency of the given amino acid substitution does not differ from that expected by chance, while a positive score indicates that the substitution occurs more frequently than by chance, while negative score indicates that the substitution is found less often than by chance.
Evolutionary Rate
Nonsynonymous substitutions rate (dN) and synonymous substitutions rate (dS) of each human transcript were obtained from BioMart of Ensembl (Flicek et al. 2014). Both mouse and chimp orthologs were used in this study.
Prediction of mRNA Secondary Structure
Two software, remuRNA (Salari et al. 2013) and RNAsnp (Sabarinathan et al. 2013), were used to predict the effect of single-point substitution on local mRNA secondary structure. remuRNA computes the structural relative entropy between the Boltzmann ensembles of the native and a mutant structure (Salari et al. 2013). As suggested by Salari et al. (2013), we used the local version of remuRNA with a window size of 150 nt to compute the mRNA local structural entropy caused by SNPs or random mutations, using the default settings. RNAsnp focuses on the local regions of maximal structural change between the native and a mutant mRNA. Structural distance between wild-type and mutant sequences was calculated from base pairing probability matrices (Sabarinathan et al. 2013). We applied “Mode 2” in RNAsnp with default settings to compute the structural distance caused by SNPs or random mutations.
Codon Optimality
We used the method presented in our previous study (Zhou et al. 2009) to compute codon optimality. We first obtained the expression data for human genes from Su et al. (2014). We measured expression level as the geometric mean of expression among different tissues. Next, we compared the codon usage pattern between the gene groups showing the lowest 20% and highest 20% expression level. Codon optimality was defined as the odds ratio of codon usage between highly and lowly expressed groups, calculated separately for each codon (supplementary table S1, Supplementary Material online).
Translation Initiation Sites and Splicing Sites
We defined translation initiation sites (TISs) as the 5′-UTR regions of 50 bp upstream of the start codon. Splicing sites (SSs) were defined as exonic regions of 3 bp from exon–intron boundary.
Gene Categories
First, we stratified human genes into subgroups according to their functional roles as essential or nonessential genes. The definition of functionally essential genes was obtained from Liao and Zhang (2008). Next, human genes were categorized according to their involvement in Mendelian diseases as Mendelian or non-Mendelian genes. The genes involved in human Mendelian genetic disorders were obtained from OMIM.org (Amberger et al. 2015).
SNP Age
We downloaded the age information for protein-coding SNPs from the NHLBI Exome Sequencing Project (Tennessen et al. 2012; Fu et al. 2013), which was estimated based upon the demographic models with recent accelerated population growth. The SNP age data are available for both European American and African American. In this study, we mainly focused on the age data in European American.
Statistical Analysis
All the statistical analyses were performed within the R platform. Spearman’s rank correlation test, t-test, and Wilcoxon rank sum test were conducted using “cor.test,” “t.test,” and “wilcox.test” functions, respectively. Cumulative distributions were computed by “ecdf” function, which is a step function with jumps i/n at observation values. Here, i is the number of tied observations whereas n is the total number of observations.
Results
Selective Constraint Is Not Necessarily a Function of MAF
Rare SNPs are thought to be more deleterious and more likely to be associated with human genetic disorders (Kryukov et al. 2007; Zhu et al. 2011; Subramanian 2012). However, these SNPs can be stratified into two groups depending on whether the minor allele is derived or ancestral. Because there is a potential difference in the history of selection on these two categories of SNPs (Zhu et al. 2011), we examined each category separately.
First, PhyloP (Goode et al. 2010) and GERP (Cooper et al. 2005) scores were used to evaluate the conservation of and selective constraints on each variant site, respectively. Larger PhyloP scores suggest higher degree of conservation, while larger GERP scores suggest stronger evolutionary constraints. Consistent with previous findings (Cooper et al. 2010; Zhu et al. 2011), MAF is negatively correlated (P < 10−10 by Spearman’s rank correlation test) with PhyloP and GERP scores for exonic SNPs for which the minor alleles are derived (fig. 1A and B). However, MAF was positively correlated (Spearman’s rank correlation test: P < 10−10) with PhyloP and GERP scores for exonic SNPs for which the minor alleles are ancestral (fig. 1A and B).
Fig. 1.—

Correlation between MAF of exonic SNPs and corresponding conservation/pathogenicity score. (A) Relationship between MAF and PhyloP score; (B) Relationship between MAF and GERP score; (C) Relationship between MAF and CADD score. Each point represents the mean of the corresponding category. Error bars indicate the standard error of the mean.
Next, we measured the pathogenicity of exonic SNPs using the CADD (Kircher et al. 2014) score. A higher CADD score implies a more deleterious effect caused by the SNP. The relationship between MAF and CADD score mirrors the pattern we found for PhyloP and GERP scores (fig. 1C). MAF and CADD scores are negatively correlated (Spearman’s rank correlation test: P < 10−10) for SNPs for which the minor alleles are derived, but positively correlated (Spearman’s rank correlation test: P < 10−10) for SNPs for which the minor alleles are ancestral. The positive correlation between MAF and CADD score of SNPs for which the minor alleles are ancestral may be interpreted by purifying selection: only the alleles with the weakest deleterious effect can drift to a high frequency (a low ancestral allele frequency) in human populations. Also, some rare SNPs with ancestral minor alleles may experience positive selection in human evolution. Therefore, rare SNPs with ancestral minor alleles tend to have the weakest pathogenicity and the lowest CADD score (fig. 1C), which can be reflected by the positive correlation between MAF and CADD score.
Taken together, these results provide evidence that rare exonic SNPs should be categorized into two distinct groups—those with derived minor alleles and those with ancestral minor alleles. Selective constraint or pathogenicity is not necessarily a function of MAF. Instead, they are a function of DAF (supplementary fig. S1, Supplementary Material online). DAF may, by itself, serve as a predictor of functional consequence of exonic SNPs (Kryukov et al. 2007). Although we observed an enormous excess of rare exonic SNPs (MAF < 0.01) at the both extremes of the DAF (supplementary fig. S2, Supplementary Material online), the minor alleles of most rare SNPs (99.1%) are derived. In this study, in total, there are 577,956 exonic SNPs with DAF < 0.01, but only 5,500 exonic SNPs with DAF > 0.99. From this point on, we only focus on DAF when examining the functional and structural consequences of rare exonic SNPs.
DAF Is Modulated by Functional Effects of Nonsynonymous SNPs
Nonsynonymous sites in coding sequences are thought to be under very strong purifying selection. In this study, 33.5% of the exonic SNPs are nonsynonymous. Here, we assessed the relationship between DAF and the functional properties of nonsynonymous SNPs. Nonsynonymous SNPs were divided into eight bins according to their DAF. The SNP-induced functional alteration were measured by amino acid chemical distance (Grantham 1974), change in amino acid hydrophobicity index (|ΔH|) (Argos et al. 1982), BLOSUM62 score (Henikoff S and Henikoff JG 1992), and SIFT score (Ng and Henikoff 2003). Random point mutations at nonsynonymous sites were used as a background.
Chemical distance and |ΔH| induced by nonsynonymous SNPs decreased monotonically with increasing DAF (Spearman’s rank correlation test: P < 10−10 for chemical distance and P = 0.002 for |ΔH|) (fig. 2A and B). The physicochemical alterations caused by random nonsynonymous mutation are significantly higher (t-test: P < 10−10) than that of nonsynonymous SNPs (fig. 2A and B).
Fig. 2.—

Functional effects caused by nonsynonymous SNPs as a function of DAF. (A) Amino acid chemical distance caused by nonsynonymous SNPs as a function of DAF; (B) Change in amino acid hydrophobicity index (|ΔH|) as a function of DAF; (C) BLOSUM62 scores as a function of DAF; (D) SIFT scores as a function of DAF; (E) Comparison of the functional effect caused by nonsynonymous SNPs between the genes undergoing stronger and weaker purifying selection. The median of ω (calculated using mouse orthologs) is used as the cutoff. Each point represents the mean of the corresponding category. Error bars indicate the standard error of the mean.
BLOSUM62 scores measures the likelihood of substitution between two amino acids. Larger BLOSUM62 scores indicate a higher likelihood of substitution at a specific site. SIFT scores predicts whether an amino acid substitution affects protein function (Ng and Henikoff 2003). Lower SIFT scores imply more deleterious effect caused by nonsynonymous substitution. Figure 2C and D reveal that BLOSUM62 and SIFT scores are positively correlated with DAF (Spearman’s rank correlation test: P < 10−10). The BLOSUM62 and SIFT scores of random nonsynonymous mutations are significantly lower (t-test: P < 10−10) than that of nonsynonymous SNPs in our study (fig. 2C and D).
We used the ω value, ratio of nonsynonymous substitutions rate (dN) to synonymous substitutions rate (dS), to evaluate the selective pressure acting on coding sequences. When ω < 1, this is indicative of purifying selection (Kryazhimskiy and Plotkin 2008). We first focused on the ω data generated between human and mouse. Interestingly, we found that the functional effect caused by nonsynonymous SNPs of genes undergoing stronger purifying selection is significantly weaker (t-test: P < 10−10) than that of genes under weaker purifying selection (fig. 2E and supplementary fig. S3, Supplementary Material online). A similar pattern was observed when human transcripts were compared with chimp orthologs (supplementary fig. S4, Supplementary Material online). All together, these results suggest that purifying selection prevents deleterious derived alleles at nonsynonymous sites from drifting to a higher frequency in human populations.
DAF Is a Function of Alterations to mRNA Structure Caused by Exonic SNPs
Selection against mutations that significantly change mRNA structure is widespread (Chamary and Hurst 2005; Kudla et al. 2009; Gu et al. 2010, 2012, 2014a, 2014b; Zhou and Wilke 2011). Here, we examined the relationship between DAF of exonic SNPs and corresponding alteration in mRNA secondary structure. Two independent programs, remuRNA (Salari et al. 2013) and RNAsnp (Sabarinathan et al. 2013), were used to predict the effect of single-point mutation on local mRNA secondary structure. remuRNA computes the structural entropy in Boltzmann ensembles between wild-type and mutant structure (Salari et al. 2013). Larger structural entropy indicates a more substantial alteration in mRNA secondary structure. RNAsnp focuses on the local regions of maximal structural change caused by mutation (Sabarinathan et al. 2013). Structural distance between wild-type and mutant sequences was calculated from base pairing probability matrices (Sabarinathan et al. 2013). Larger structural distance indicates higher dissimilarity in mRNA secondary structure. Figure 3A indicates that the structural entropy of random point mutations is significantly higher (t-test: P < 10−10) than that of exonic SNPs. Together, the structural entropy of the SNPs in bins 7 (0.99 < DAF ≤ 0.999) and 8 (DAF > 0.999) is significantly lower (t-test: P < 10−10) than the structural entropy of the other bins (bin 1–6).
Fig. 3.—

The mRNA structural effect caused by exonic SNPs as a function of DAF. (A) Structural entropy caused by exonic SNPs as a function of DAF; (B) MFE gap between two alleles (|ΔΔGMFE|) as a function of DAF; (C) Structural distance between two alleles as a function of DAF; (D) Comparison of the structural entropy between SNPs located in 5′- and 3′-UTR; (E) Comparison of the structural entropy between nonsynonymous and synonymous SNPs. Each point represents the mean of the corresponding category. Error bars indicate the standard error of the mean.
Similar patterns were observed when we looked into the mRNA structure of minimum free energy (MFE) (fig. 3B). The folding energy gap of MFE structure (|ΔΔGMFE|) in bin 7 and 8 is much lower (t-test: P < 10−6) than that in the other bins (fig. 3B). In addition, as shown in figure 3C, the structural distance predicted by RNAsnp largely mirrors the pattern in figure 3A and B. These results demonstrate that DAF is closely related to the structural effects of rare exonic SNPS, and also suggest strong selection against mutations that significantly change mRNA structure. Derived alleles with less structural effect are more likely to eventually become fixed in human population.
A comparison of SNPs located in 5′- and 3′-UTR revealed that the structural effect of the SNPs in 5′-UTR is significantly higher (t-test: P < 10−10) than those in 3′-UTR (fig. 3D). This finding suggests that the 3′-UTR undergoes stronger purifying selection against mRNA structural alterations compared with the 5′-UTR. Furthermore, the structural effect caused by nonsynonymous SNPs is significantly higher (t-test: P < 10−10) than the effect of synonymous SNPs (fig. 3E). Thus, synonymous sites seem to be subject to stronger selection in keeping RNA secondary structures compared with nonsynonymous sites in coding sequences.
Selection on Translational Efficiency in Synonymous SNPs
In this study, 23.8% of the exonic SNPs are synonymous. Selection on synonymous sites for translational efficiency has been observed among a diversity of prokaryotes and eukaryotes, including for humans (Drummond and Wilke 2008; Zhou et al. 2009; Lee et al. 2010). In general, translational optimal codons are the ones that are efficiently and rapidly translated because their cognate tRNAs are highly abundant (Zhou et al. 2010). Optimal codons may confer a selective advantage to genes for which high translational speed and accuracy is required. However, there is also evidence that nonoptimal codons are selected at specific coding sites to facilitate cotranslational protein folding (Thanaraj and Argos 1996; Komar et al. 1999; Zhang et al. 2009). Here, we investigated the relationship between DAF and selection on translational efficiency in synonymous variants. We followed the method presented in one previous study (Zhou et al. 2009) to define codon optimality (see Materials and Methods for details). Change in codon optimality (|ΔOcodon|) was calculated for each synonymous SNPs. A significant negative correlation (Spearman’s rank correlation test: P < 10−10) was found between DAF and |ΔOcodon| (fig. 4). Also, |ΔOcodon| of random synonymous mutations is significantly higher (t-test: P < 10−10) than that of synonymous SNPs (fig. 4). These results suggest that purifying selection on synonymous sites, which may substantially affect translational efficiency or cotranslational folding, prevents those derived alleles from drifting to a higher frequency in human populations.
Fig. 4.—
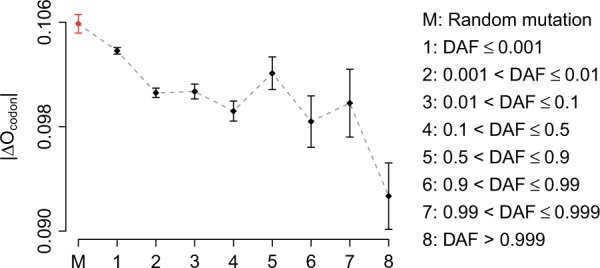
Change in codon optimality (|ΔOcodon|) as a function of DAF. Each point represents the mean of the corresponding category. Error bars indicate the standard error of the mean.
Regional Difference in DAF
All of the above observations indicate that DAF is modulated by the functional and structural consequences of exonic SNPs. For this reason, DAF may serve as a predictor of functional significance of exonic SNPs. Therefore, comparison of DAF among different mRNA sites may help us understand the patterns of selection acting on exonic SNPs.
We found that the DAF of nonsynonymous SNPs is significantly lower (Wilcoxon rank sum test: P < 10−10) than the DAF of synonymous ones (fig. 5A). Also, the DAF of SNPs in 3′-UTR is significantly lower (Wilcoxon rank sum test: P < 10−10) than the DAF of SNPs in 5′-UTR (fig. 5A). Together, these results suggest that the genomic positions of nonsynonymous SNPs are under the least evolutionarily labile while the positions of SNPs in 5′-UTR are undergoing the weakest selection.
Fig. 5.—

Cumulative distribution of DAF. (A) Comparison of DAF between SNPs located in 5′-UTR, 3′-UTR, and coding regions; (B) Comparison of DAF between 5′-UTR SNPs within and outside of TIS; (C) Comparison of DAF between 5′-UTR SNPs located within and outside of splice site (SS); (D) Comparison of DAF between coding SNPs located within and outside of SS.
Although 5′-UTR may have been under weaker purifying selection, the 5′-UTR is important for the regulation of translation in eukaryotes. For example, the TISs, located in the 5′-UTR immediately upstream of the start codon, are under strong selection to facilitate translation initiation (Dvir et al. 2013; Gu et al. 2014a). Here, we define TISs as the regions of 50 bp upstream of the start codon. Figure 5B indicates that the DAF of the SNPs within TIS (n = 8,565) is significantly lower (Wilcoxon rank sum test: P < 10−10) than the DAF of non-TIS variants, which is consistent with the action of purifying selection.
Splice sites (SSs) in mRNA are also thought to be under strong purifying selection (Orban and Olah 2001; Parmley et al. 2006; Lawrie et al. 2013). Here, SSs are defined as exonic regions of 3 bp from exon–intron boundary. We found that, in 5′-UTR, the DAF of the SNPs within SS (n = 744) is significantly lower (Wilcoxon rank sum test: P < 10−10) than the DAF of non-SS variants (fig. 5C). Also, in coding region, the DAF of the SNPs within SS (n = 4,480 for synonymous SNPs and n = 6,418 for nonsynonymous SNPs) is significantly lower (Wilcoxon rank sum test: P = 1.8 × 10−5 for synonymous SNPs and P < 10−10 for nonsynonymous SNPs) than the DAF of non-SS variants (fig. 5D). However, we did not find significant difference in DAF between SS and non-SS SNPs in 3′-UTR (Wilcoxon rank sum test: P = 0.681), which may be due to the limited number of SS variants in 3′-UTR (n = 107).
Difference in DAF between Gene Categories
We investigated the difference in DAF among different gene categories. First, we categorized the SNPs according to the functional essentiality of their host gene. We found that the DAF of SNPs in essential genes (n = 5,504) is significantly lower (Wilcoxon rank sum test: P = 5.4 × 10−3) than the DAF of SNPs in non-essential genes (supplementary fig. S5A, Supplementary Material online). Next, we checked the difference between Mendelian and non-Mendelian genes. We found that the DAF of SNPs in Mendelian genes (n = 522,696) is significantly lower (Wilcoxon rank sum test: P < 10−10) than that in non-Mendelian genes (supplementary fig. S5B, Supplementary Material online).
DAF and SNP Age
SNP age was previously reported to be negatively correlated with SNP-induced deleterious effect (Fu et al. 2013). We collected the age information for protein-coding SNPs from the NHLBI Exome Sequencing Project (Tennessen et al. 2012; Fu et al. 2013). The SNP age data are available for both European American and African American. Here, we focused on the age data in European American first.
Not surprisingly, we found a strong positive correlation (Spearman’s rank correlation test: P < 10−10) between SNP age and DAF (fig. 6A). A negative relationship (Spearman’s rank correlation test: P < 10−10) between age and PhyloP, GERP, and CADD scores was also observed (supplementary fig. S6, Supplementary Material online). However, figure 6A indicates that there are several outliers aggregated at the top left (age < 300 kiloyears and DAF > 0.8) and bottom right (age > 700 kiloyears and DAF < 0.2) corners of the figure. The SNPs at the top left are those in which derived allele became major allele quickly (“quick-running” SNP). The SNPs at the bottom right corner are old variants with relatively low DAF (“slow-running” SNP). Further investigation reveals that the deleterious effect caused by both quick-running and slow-running SNPs is significantly weaker (t-test: P < 10−2) than that of the SNPs aggregated at the top right (age > 700 kiloyears and DAF > 0.8) and bottom left (age < 300 kiloyears and DAF < 0.2) corners (supplementary fig. S7, Supplementary Material online and fig. 6B–D), which suggests that, even with similar age, the SNPs at the two DAF extremes differ in the strength of purifying selection. Pathway enrichment analysis was conducted for the genes of quick-running SNPs based on KEGG database (Kanehisa et al. 2014). Only the genes of SNPs with DAF > 0.8 were used as the background of enrichment study. We observed that “Osteoclast differentiation,” “Bladder cancer,” “Butanoate metabolism,” and “Olfactory transduction” are among the top pathways enriched in the genes of quick-running SNPs (supplementary fig. S8A, Supplementary Material online). We also conducted pathway analysis for the genes of slow-running SNPs. Here, the genes of SNPs with DAF < 0.2 were used as the background of enrichment study. Interestingly, Olfactory transduction is the most significant pathway enriched in the genes of slow-running SNPs (supplementary fig. S8B), Supplementary Material online. The above findings can be largely reproduced when the age data in African American are used (supplementary figs. S9–S12, Supplementary Material online).
Fig. 6.—

Relationship between SNP age and DAF. (A) DAF increases with SNP age. Outliers are aggregated at the top left (red points with age < 300 kiloyears and DAF > 0.8) and bottom right (blue points with age > 700 kiloyears and DAF < 0.2) corners. SNPs located in the top right (green points with age > 700 kiloyears and DAF > 0.8) and bottom left corners (orange points with age < 300 kiloyears and DAF < 0.2) are highlighted. (B) Comparison of PhyloP score among SNPs located in the four corners in panel A. (C) Comparison of GERP score among SNPs located in the four corners in panel A. (D) Comparison of CADD score among SNPs located in the four corners in panel A.
Discussion
All our analyses demonstrated a robust and consistent pattern of functional and structural changes with regard to the frequency of derived allele (figs. 2–4). The changes on protein structure and/or function, RNA local structure, and translation efficiency are larger for SNPs at lower DAF in the human population. Compared to artificially generated, randomized mutations in the human genome, naturally observed human SNPs, even those SNPs with very small DAFs, have smaller functional/structural changes (figs. 2–4). Several previous studies have shown that rare SNPs with derived minor alleles are more likely to be located in evolutionary conserved region (Zhu et al. 2011; Tennessen et al. 2012; Fu et al. 2013, 2014). In particular, Zhu et al. (2011) sequenced whole genomes of 29 individuals of European origin and assessed allele frequencies. Using SNP information in this small-size population, they found that genomic positions with low-DAF SNPs are more conserved than positions corresponding to high-DAF SNPs and rare SNPs with derived minor alleles are more likely to be located in functional genomic regions (Zhu et al. 2011). Furthermore, Fu et al. (2014) analyzed human coding SNPs from 6,515 individuals of European and African origin and identified putative deleterious SNPs in this population based on PhyloP conservation score (Fu et al. 2014). They also showed that derived minor alleles were more deleterious than ancestral minor alleles. In comparison to these previous studies, we performed a genome-wide and systematic analysis of the functional and structural consequences of all known human exonic SNPs. Other than coding nonsynonymous SNPs, we also analyzed coding synonymous SNPs and noncoding SNPs in 5′- and 3′-UTRs. In addition to evolutionary conservation, we focused on the consequences of human exonic SNPs at three biological levels. First, nonsynonymous SNPs in protein coding regions change encoded amino acids, which in turn will have effects on the structure and function of the final protein products (Bromberg and Rost 2007). We computed several measures for each nonsynonymous SNP in estimating its effect on protein structure and function, including amino acid chemical distance (Grantham 1974), change in amino acid hydrophobicity index (|ΔH|) (Argos et al. 1982), BLOSUM62 score (Henikoff S and Henikoff JG 1992), and SIFT score (Ng and Henikoff 2003). Second, RNA structures are very important in several biological processes, such as RNA splicing (Parmley et al. 2006), RNA translation initiation (Gu et al. 2010), and microRNA-mediated gene regulation (Gu et al. 2014b). Human exonic mutations can affect those biological processes by altering local mRNA structures. We computed the change of RNA structure for each exonic SNP with several different measures (Sabarinathan et al. 2013; Salari et al. 2013) and estimated its effect on local RNA structure through these calculations. Third, synonymous SNPs in the protein coding region can regulate translational process of messenger RNA (Gingold and Pilpel 2011). Biased usages of synonymous codons are related to translation efficiency and accuracy, and thus cotranslational protein folding process (Hershberg and Petrov 2008). We investigated the effect on mRNA translation made by synonymous SNPs in the human genome by calculating the change in codon optimality (|ΔOcodon|) (Zhou et al. 2009). Our results showed that derived, young alleles had higher disruptive effects than ancestral alleles at all three biological levels, which explained why derived minor alleles are the most deleterious. This observation complemented previous findings (Zhu et al. 2011; Gorlova et al. 2012; Fu et al. 2014), which taken together suggested that derived rare alleles in human genomes had higher probability in causing human diseases than ancestral rare alleles.
Minor allele can be separated into two groups according to ancestral state. Our results show that these SNPs have completely different functional consequences on protein structure/function, RNA translation, and mRNA secondary structure (figs. 1–4). Variants with the DAF less than 1% have the biggest functional changes, whereas variants with DAF larger than 99% have the smallest functional consequences. Furthermore, we observed more than 100-fold enrichment of rare variants with DAF less than 0.1% in the human genome, but only 2- to 10-fold enrichment for variants with DAF larger than 99.9%. Genetic variants in the human population are enriched with rare variants, especially those with highest functional changes. These observations suggest that the causative variants for most human complex diseases may be rare derived variants with large functional effects, such as those with DAF less than 0.1%. This supports the common disease-rare variants hypothesis. Human SNPs with DAF larger than 99.9% are less likely to be deleterious and therefore less likely to be causally related to human diseases. When prioritizing variants in genetic association studies or whole exome/genome sequencing, we should consider whether a variant is derived in addition to its frequency.
Many studies have observed that synonymous codons are selected for loose or tight RNA structure to facilitate RNA splicing, RNA translation initiation, and microRNA binding (Chamary and Hurst 2005; Chamary et al. 2006; Parmley et al. 2006; Gu et al. 2010, 2012, 2014a, 2014b; Mortimer et al. 2014). When filtering functional synonymous SNPs that may be related to human diseases, secondary structure alteration is an important feature to estimate its pathogenicity (Buske et al. 2013). RNA secondary structure change caused by nonsynonymous SNPs is rarely considered in functional annotation of nonsynonymous SNPs (Frousios et al. 2013). Recent studies have implicated the important role of RNA structure in gene regulation (Mortimer et al. 2014; Yang et al. 2014; Wang et al. 2015), and nonsynonymous mutations in human exonic region should be selected for functional RNA structures (Gu et al. 2014a). Our results show that both nonsynonymous and synonymous SNPs altered local RNA structure, and the structural entropy difference caused by nonsynonymous SNPs is higher than synonymous SNPs (fig. 3E). Hence, we propose that nonsynonymous SNPs could also cause abnormal gene expression or function by altering RNA secondary structure, which should be considered in functional annotation of nonsynonymous SNPs.
In addition to synonymous and nonsynonymous SNPs in protein coding region, human SNPs in 5′-UTR and 3′-UTR have substantial effects on local RNA secondary structure. Previous studies have found important biological roles performed by RNA structure in 5′-UTR and 3′-UTR. RNA structure near the cap site is crucial for microRNA-mediated gene regulation in humans (Gu et al. 2014b). RNA secondary structure upstream of the start codon is also important in efficient translation initiation(Goodman et al. 2013). microRNA target sites are located in 3′-UTR of most human genes, implicating these sites as an important target of epigenetic regulation (Bartel 2009). Many studies have confirmed that secondary structure of microRNA targets and their flank region is related to microRNA target accessibility and microRNA binding. In our results, we observed that structural effects caused by SNPs in 5′- or 3′-UTR are similar to those caused by SNPs in protein coding region (fig. 3D). This finding hinted that altering RNA structure is an important mechanism by which exonic SNPs can cause aberrant gene expression and human disease.
Interestingly, human nonsynonymous SNPs in genes under different selective constraints also show significant differences in functional consequences. We observed that chemical distance (Grantham 1974), change in amino acid hydrophobicity index (|ΔH|) (Argos et al. 1982), and BLOSUM62 (Henikoff S and Henikoff JG 1992) score are consistently higher for SNPs located in genes with higher ω value (fig. 2E and supplementary figs. S3 and S4, Supplementary Material online). Higher ω value suggests a relaxed selective constraint on protein coding sequence evolution and that SNPs in genes with weaker selective constraints have larger functional changes. This suggests purifying selection on rare SNPs is a major factor shaping genetic variation in the human genome.
We also investigated the DAF of human exonic SNPs in different genomic regions. We observed that the cumulative distribution of DAF is significantly different for SNPs in different genome regions (fig. 5A). The DAF of SNPs in coding region is significantly lower than the DAF of SNPs in 5′- and 3′-UTR s (fig. 5A). Also, we observed lower DAF for SNPs in TIS (fig. 5B) and SS (fig, 5C and D) compared with non-TIS and non-SS regions, respectively. In addition, we found that the DAF of SNPs in functionally essential genes (supplementary fig. S5A, Supplementary Material online) and Mendelian genes (supplementary fig. S5B, Supplementary Material online) is significantly lower than that in nonessential genes and non-Mendelian genes, respectively. The regional difference in DAF confirms the role of purifying selection in human genome evolution.
The excessive number of evolutionarily young SNPs with a low DAF (bottom left of fig. 6A) is consistent with recent explosion of human SNPs, as well as with the recent demographic history of human population (Gravel et al. 2011; Fu et al. 2013). Younger SNPs have lower DAF, which may have substantial functional and structural consequences. We also showed that younger SNPs are more likely to be found in conserved regions (fig. 6B and supplementary figs. S6A and S7A, Supplementary Material online). These results are compatible with previous findings that recent human SNPs are more likely to be rare and deleterious (Gravel et al. 2011; Fu et al. 2013).
Finally, we want to draw attention to the distinction between slow-running and quick-running SNPs in the human genome (fig. 6A). For the quick-running SNPs, the derived allele may have very high selective advantage over the ancestral allele, and can be fixed very quickly in the population. The PhyloP score of quick-running SNPs is significantly less than zero, which means those quick-running SNPs experienced positive selection. Curiously, those SNPs are enriched in genes performing olfactory transduction function (supplementary fig. S8, Supplementary Material online), which are known to be under positive selection in human evolution (Young and Trask 2002). Several other quick-running SNPs are located in genes coding Zinc-finger proteins, such as ZNF33B and ZNF460. Previous studies have also shown zinc-finger proteins have experienced lineage-specific positive selection in humans (Schmidt and Durrett 2004). Interestingly, slow-running SNPs have similar conservation scores as quick-running SNPs, and pathway analysis suggests that both these categories of SNPs are enriched in olfactory related genes (supplementary fig. S8, Supplementary Material online). The reason for these similar selective constraints is unknown. The ancestral alleles of human SNPs were inferred by the comparison of human genome to six-way primate genome alignment (Paten et al. 2008), which may lead to incorrect ancestral allele definition of some human SNPs. The misdefinition of ancestral allele of human SNPs could partially explain the signal of positive selection observed in slow-running SNPs. Furthermore, our results suggest that positive selection and genetic hitchhiking (Fay and Wu 2000) may account for the quick fixation of some “fast-running” SNPs. However, other possible mechanisms, such as weak background selection or chance alone, could also explain fast-running for some human SNPs. Hence, more thorough analysis will be needed to explore the exact mechanisms for the fast-running and slow-running SNPs.
In conclusion, our systematic analysis of functional and structural consequences of human exonic SNPs suggested that purifying selection, rather than positive or balancing selection, shapes much of the genetic variation in human populations. Rare exonic SNPs with DAF less than 1%, which appeared recently in the human population, cause the largest functional and structural changes, whereas rare exonic SNPs with DAF larger than 99% have the smallest functional consequences. This will have important implications for understanding the molecular underpinnings of complex diseases.
Supplementary Material
Supplementary table S1 and figures S1–S12 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors would like to thank Michael F. Hammer, Hugo Gruson, Dawn Higginson, Gavin Leighton, Erin Morrison, and Georgy Semenov for comments on the development of this manuscript. This work was supported by National Basic Research Program of China (2012CB316501 to W.G.), National High Technology Research and Development Program of China (2012AA020401 to W.G.), National Natural Science Foundation of China (61171143, 61372164, 61471112 and 61571109 to W.G.), and Tsinghua National Laboratory for Information Science and Technology (TNList) Cross-Discipline Foundation. This work was also supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-2011-0016587 to J.H.K.).
Literature Cited
- 1000 Genomes Project Consortium, 2012.1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. 2015. OMIM.org: online mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 43:D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos P, Rao JK, Hargrave PA. 1982. Structural prediction of membrane-bound proteins. Eur J Biochem. 128:565–575. [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. 2007. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 35:3823–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer MG. 1971. The effect of selection on genetic variability. Am Nat. 105:201–211. [Google Scholar]
- Buske OJ, Manickaraj A, Mital S, Ray PN, Brudno M. 2013. Identification of deleterious synonymous variants in human genomes. Bioinformatics 29:1843–1850. [DOI] [PubMed] [Google Scholar]
- Chamary JV, Hurst LD. 2005. Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol. 6:R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary JV, Parmley JL, Hurst LD. 2006. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet. 7:98–108. [DOI] [PubMed] [Google Scholar]
- Chorley BN, et al. 2008. Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: current and developing technologies. Mutat Res. 659:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Goldstein DB. 2010. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 11:415–425. [DOI] [PubMed] [Google Scholar]
- Cooper GM, et al. 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15:901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, et al. 2010. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods. 7:250–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BH, Poon AFY, Whitlock MC. 2009. Compensatory mutations are repeatable and clustered within proteins. Proc Biol Sci. 276:1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir S, et al. 2013. Deciphering the rules by which 5’-UTR sequences affect protein expression in yeast. Proc Natl Acad Sci U S A. 110:E2792–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu CI. 2000. Hitchhiking Under Positive Darwinian Selection. Genetics 155:1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. 1930. The genetical theory of natural selection. Oxford: Clarendon Press. [Google Scholar]
- Flicek P, et al. 2014. Ensembl 2014. Nucleic Acids Res. 42:D749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Murray SS, Schork NJ, Topol EJ. 2009. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 10:241–251. [DOI] [PubMed] [Google Scholar]
- Frousios K, Iliopoulos CS, Schlitt T, Simpson MA. 2013. Predicting the functional consequences of non-synonymous DNA sequence variants—evaluation of bioinformatics tools and development of a consensus strategy. Genomics 102:223–228. [DOI] [PubMed] [Google Scholar]
- Fu W, et al. 2013. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Gittelman RM, Bamshad MJ, Akey JM. 2014. Characteristics of neutral and deleterious protein-coding variation among individuals and populations. Am J Hum Genet. 95:421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. 2010. Hints of hidden heritability in GWAS. Nat Genet. 42:558–560. [DOI] [PubMed] [Google Scholar]
- Gibson G. 2012. Rare and common variants: twenty arguments. Nat Rev Genet. 13:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y. 2011. Determinants of translation efficiency and accuracy. Mol Syst Biol. 7:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB, et al. 2013. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 14:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode DL, et al. 2010. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res. 20:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman DB, Church GM, Kosuri S. 2013. Causes and effects of N-terminal codon bias in bacterial genes. Science 342:475–479. [DOI] [PubMed] [Google Scholar]
- Gorlova OY, et al. 2012. Derived SNP alleles are used more frequently than ancestral alleles as risk-associated variants in common human diseases. J Bioinform Comput Biol. 10:1241008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R. 1974. Amino acid difference formula to help explain protein evolution. Science 185:862–864. [DOI] [PubMed] [Google Scholar]
- Gravel S, et al. 2011. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A. 108:11983–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Li M, et al. 2014. The impact of RNA structure on coding sequence evolution in both bacteria and eukaryotes. BMC Evol Biol. 14:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Xu Y, et al. 2014. The role of RNA structure at 5’ untranslated region in microRNA-mediated gene regulation. RNA 20:1369–1375. [Google Scholar]
- Gu W, Wang X, Zhai C, Xie X, Zhou T. 2012. Selection on synonymous sites for increased accessibility around miRNA binding sites in plants. Mol Biol Evol. 29:3037–3044. [DOI] [PubMed] [Google Scholar]
- Gu W, Zhou T, Wilke CO. 2010. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol. 6:e1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. 2008. Selection on codon bias. Annu Rev Genet. 42:287–299. [DOI] [PubMed] [Google Scholar]
- Holm H, et al. 2011. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 43:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap 3 Consortium. 2010. Integrating common and rare genetic variation in diverse human populations. Nature 467:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar S, Elston R. 2007. The genetic basis of complex traits. In: Collins A, editor. Linkage disequilibrium and association mapping. Totowa: Humana Press; p. 71–84. [Google Scholar]
- Johansen CT, et al. 2010. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 42:684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, et al. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42:D199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, et al. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar AA, Lesnik T, Reiss C. 1999. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 462:387–391. [DOI] [PubMed] [Google Scholar]
- Kryazhimskiy S, Plotkin JB. 2008. The population genetics of dN/dS. PLoS Genet. 4:e1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. 2007. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 80:727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrie DS, Messer PW, Hershberg R, Petrov DA. 2013. Strong purifying selection at synonymous sites in D. melanogaster . PLoS Genet. 9:e1003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Zhou T, Tartaglia GG, Vendruscolo M, Wilke CO. 2010. Translationally optimal codons associate with aggregation-prone sites in proteins. Proteomics 10:4163–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, et al. 2012. Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. Plant Cell 24:4346–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2013. Far upstream element-binding protein 1 and RNA secondary structure both mediate second-step splicing repression. Proc Natl Acad Sci U S A. 110:E2687–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao BY, Zhang J. 2008. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci U S A. 105:6987–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, et al. 2013. Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet. 93:1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 107:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, et al. 2009. Finding the missing heritability of complex diseases. Nature 461:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan JM, Susser E, King MC. 2007. Schizophrenia: a common disease caused by multiple rare alleles. Br J Psychiatry. 190:194–199. [DOI] [PubMed] [Google Scholar]
- Montgomery SB, Lappalainen T, Gutierrez-Arcelus M, Dermitzakis ET. 2011. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 7:e1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer SA, Kidwell MA, Doudna JA. 2014. Insights into RNA structure and function from genome-wide studies. Nat Rev Genet. 15:469–479. [DOI] [PubMed] [Google Scholar]
- Need AC, et al. 2012. Exome sequencing followed by large-scale genotyping suggests a limited role for moderately rare risk factors of strong effect in schizophrenia. Am J Hum Genet. 91:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. 2009. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324:387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2003. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orban TI, Olah E. 2001. Purifying selection on silent sites—a constraint from splicing regulation?. Trends Genet. 17:252–253. [DOI] [PubMed] [Google Scholar]
- Pabinger S, et al. 2014. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 15:256–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmley JL, Chamary JV, Hurst LD. 2006. Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol Biol Evol. 23:301–309. [DOI] [PubMed] [Google Scholar]
- Paten B, Herrero J, Beal K, Fitzgerald S, Birney E. 2008. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 18:1814–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas MA, et al. 2011. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 43:1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabarinathan R, et al. 2013. RNAsnp: efficient detection of local RNA secondary structure changes induced by SNPs. Hum Mutat. 34:546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salari R, Kimchi-Sarfaty C, Gottesman MM, Przytycka TM. 2013. Sensitive measurement of single-nucleotide polymorphism-induced changes of RNA conformation: application to disease studies. Nucleic Acids Res. 41:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauna ZE, Kimchi-Sarfaty C. 2011. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 12:683–691. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Durrett R. 2004. Adaptive evolution drives the diversification of zinc-finger binding domains. Mol Biol Evol. 21:2326–2339. [DOI] [PubMed] [Google Scholar]
- Sherry ST, et al. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Pollard KS, Haussler D. 2006. New methods for detecting lineage-specific selection. Res Comput Mol Biol. 3909:190–205. [Google Scholar]
- Simons YB, Turchin MC, Pritchard JK, Sella G. 2014. The deleterious mutation load is insensitive to recent population history. Nat Genet. 46:220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soon WW, Hariharan M, Snyder MP. 2013. High‐throughput sequencing for biology and medicine. Mol Syst Biol. 9:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, et al. 2012. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 101:6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S. 2012. Quantifying harmful mutations in human populations. Eur J Hum Genet. 20:1320–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, et al. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanaraj TA, Argos P. 1996. Ribosome-mediated translational pause and protein domain organization. Protein Sci. 5:1594–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Zhou T, Saadat L, Garcia JG. 2015. A MYLK variant regulates asthmatic inflammation via alterations in mRNA secondary structure. Eur J Hum Genet. 23:874–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray GA. 2007. The evolutionary significance of cis-regulatory mutations. Nature Rev Genet. 8:206–216. [DOI] [PubMed] [Google Scholar]
- Yang JR, Chen X, Zhang J. 2014. Codon-by-Codon Modulation of Translational Speed and Accuracy Via mRNA Folding. PLoS Biol. 12:e1001910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JM, Trask BJ. 2002. The sense of smell: genomics of vertebrate odorant receptors. Hum Mol Genet. 11:1153–1160. [DOI] [PubMed] [Google Scholar]
- Zhang G, Hubalewska M, Ignatova Z. 2009. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat Struct Mol Biol. 16:274–280. [DOI] [PubMed] [Google Scholar]
- Zhou T, Gu W, Wilke CO. 2010. Detecting positive and purifying selection at synonymous sites in yeast and worm. Mol Biol Evol. 27:1912–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Weems M, Wilke CO. 2009. Translationally optimal codons associate with structurally sensitive sites in proteins. Mol Biol Evol. 26:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Wilke CO. 2011. Reduced stability of mRNA secondary structure near the translation-initiation site in dsDNA viruses. BMC Evol Biol. 11:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, et al. 2011. A genome-wide comparison of the functional properties of rare and common genetic variants in humans. Am J Hum Genet. 88:458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.