

Abstract
Background
Hereditary predisposition is rarely suspected for childhood acute lymphoblastic leukemia (ALL). Recent studies identified germline ETV6 variations associated with marked familial clustering of hematologic malignancies, pointing to this gene as a potentially important genetic determinant for ALL susceptibility. The aims of the current study are to comprehensively identify ALL predisposition variants in ETV6 and to determine the extent to which they contribute to the overall risk of childhood ALL.
Methods
Whole-exome sequencing of an index family with multiple cases of ALL was performed to identify causal variants for ALL predisposition. Targeted sequencing of ETV6 was done in 4,405 children from the Children's Oncology Group (COG) and St. Jude Children's Research Hospital frontline ALL trials. Patients were included in this study on the basis of their enrollment in these clinical trials and the availability of germline DNA. ETV6 variant genotypes were compared with non-ALL controls to define ALL-related germline risk variants. ETV6 variant function was characterized bioinformatically and correlated with clinical and demographic features in 2,021 children with ALL.
Findings
We identified a novel nonsense ETV6 variant (p.R359X) with a high penetrance of familial ALL. Subsequent targeted sequencing of ETV6 in 4,405 childhood ALL cases discovered 31 exonic variants (4 nonsense, 21 missense, 1 splice site, and 5 frame shift variants) that are potentially related to ALL risk in 35 cases (0.79%). Fifteen (48%) of the 31 ALL-related ETV6 variants clustered in the ETS domain and predicted to be highly deleterious. Children with ALL-related ETV6 variants were significantly older at leukemia diagnosis than others (10.2 years [IQR 5.3-13.8] vs 4.7 years [IQR 3.0-8.7], P=0.017). The hyperdiploid leukemia karyotype was strikingly overrepresented in ALL cases harboring germline ETV6 risk variants compared to the wildtype group (9 of 14 cases [64.3%] vs 538 of 2,007 cases [26.8%]; P=0.0050).
Interpretation
Our findings indicated germline ETV6 variations as the basis of a novel genetic syndrome associated with predisposition to childhood ALL.
Funding
This study was supported by the National Institutes of Health and by the American Lebanese Syrian Associated Charities.
Research in context
Evidence before this study
As the most common cancer in children, acute lymphoblastic leukemia (ALL) is generally considered as driven by acquired genomic abnormalities and hereditary predisposition is rarely considered in clinical practice. Recent studies identified ETV6 germline variants associated with rare hereditary thrombocytopenia and a plausible increased susceptibility to hematologic malignancies, including ALL.
Added value of this study
Combining whole-exome discovery in familial ALLs and targeted sequencing in a large frontline national clinical trials (N=4,405), we comprehensively identified a panel of 31 ETV6 germline variants that were likely associated with ALL risk. Children with ALL-relate ETV6 variants had distinct clinical features, suggesting unique leukemia etiology. The substantial proportion of childhood ALL cases carrying ETV6 germline variants of potential pathologic significance highlights the importance of ETV6 as a leukemia predisposition gene and also indicate that inherited susceptibility to ALL may be much more substantial than currently believed.
Implications of all the available evidence
Germline ETV6 variations are the basis for a novel genetic syndrome associated with strong predisposition to hematologic malignancies (particularly ALL), implying potential benefit of clinical identification and subsequent surveillance of the at risk individuals.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer in children and a prototype of cancer that can be cured by chemotherapy alone with risk-adapted chemotherapeutic regimens.1-4 However, the etiology of this aggressive cancer remains largely unknown. The contribution of environmental factors to the development of ALL is debated, although there is growing evidence in support of the influence of exposure to infectious agents early in life.5-7 In a Swedish-Finnish population-based study of ∼4,000 patients with childhood ALL, children with affected siblings had a 2- to 4-time increased risk to develop ALL and this risk increased up to 163-time for monozygotic twins,8, 9 pointing to a potential genetic basis of susceptibility to ALL. In fact, common germline genetic polymorphisms affecting genes involved in lymphoid development and tumor suppression (e.g., ARID5B,10, 11 IKZF1,10, 11 CEBPE,10 GATA3,12, 13 CDKN2A,14, 15 BMI1-PIP4K2A,16 TP6317) have been associated with the risk of developing ALL, although with mostly modest effects. Only a small fraction of ALL cases are thought to be related to congenital genetic disorders (e.g., Down syndrome18, 19, Robertsonian translocation20), and hereditary predisposition is rarely considered in clinical practice. Recent studies of familial ALL have identified rare germline mutations in PAX5 and SH2B3 with high penetrance.21, 22 More strikingly, germline TP53 mutations were found in ∼50% of children with low-hypodiploid ALL, suggesting that this subtype of ALL may be a manifestation of Li Fraumeni syndrome.23 Together, these data raise the possibility that the proportion of ALL cases attributable to inherited genetic mutations may be much higher than currently proposed.
ETV6 is a transcriptional repressor that belongs to the ETS family and is essential for hematopoiesis, particularly thrombopoiesis.24, 25 Somatic ETV6 mutations have been associated with myelodysplastic syndromes and T-cell leukemias.26, 27 In childhood ALL, the ETV6-RUNX1 fusion is the most common somatic genetic aberration and is associated with a good outcome with modern therapy.28 Germline ETV6 variations have recently been reported in rare families with hereditary thrombocytopenia and susceptibility to hematologic malignancies (particularly ALL).29-31 These highly damaging variants result in loss of functional ETV6 protein in a dominant negative fashion,29-31 although the exact biological mechanism of excessive ALL risk in these subjects remains unclear. These recent observations raise the questions as to whether additional ALL predisposition variants in ETV6 exist and to what extent they contribute to ALL risk in general.
In the present study, we identified a novel damaging ETV6 germline variant driving predisposition to familial ALL, described a population-based screen of ETV6 germline variation in 4,405 children with ALL, and evaluated the potential functions of ALL-related ETV6 variants and their association with clinical features of ALL.
Methods
Subjects and samples
A family of European ancestry with multiple ALL cases was identified at St. Jude Children's Research Hospital, Memphis USA (Fig. 1). ALL cases were treated on the St. Jude frontline ALL protocols Total Therapy XIIIA32, XIIIB33 and XV.34
Figure 1. A novel nonsense germline variant in ETV6 (p.R359X) associated with familial ALL.

Filled symbols represented individuals with ALL. “WT” and “p.R359X” indicate the wildtype and heterozygous genotype at this ETV6 variant, respectively.
The ETV6 targeted-sequencing cohort comprised 4,405 children with newly-diagnosed ALL (3,807 with B-ALL and 93 with T-ALL) who were treated on the Children's Oncology Group (COG) AALL0232, P9904, P9905, P9906 protocols35, 36 and St. Jude Total Therapy XIIIA, XIIIB and XV studies. Individuals in the NHLBI GO Exome Sequencing Project cohort37 (ESP, http://evs.gs.washington.edu/EVS/) served as the primary non-ALL control cohort because the prevalence of childhood ALL is extremely low in the general population. In addition, we utilized the Broad Institute Exome Aggregation Consortium cohort [ExAc, http://exac.broadinstitute.org/] as a secondary non-ALL cohort, as it has a considerably larger sample size with greater diversity in ancestry.
Germline DNA was extracted from bone marrow samples or peripheral blood of children with ALL obtained during remission. Most of the ALL cases had been previously genotyped with genome-wide SNP arrays and genetic ancestry (European, African, Native American, and Asian) was estimated using STRUCTURE38 on the basis of genotypes at 30,000 randomly selected SNPs.12, 39, 40 This study was approved by the appropriate institutional review boards and informed consent was obtained from parents, guardians, or patients, as appropriate.
Exome sequencing and analyses of the pedigree with familial ALL
Germline genomic DNA was subjected to exome capture (62 Mb) with the Illumina TruSeq kit. Exome sequencing (pair-end 101 bp) was performed on the Illumina HiSeq 2500 platform with a coverage of 10× for > 84% ∼ 94% of target regions. Sequencing reads were mapped using the Burrow-Wheller Aligner and variant calls in the gene coding regions were made by bambino41 (with parameters: -min-quality 20, -min-flanking-quality 20, -min-alt-allele-count 3, -min-minor-frequency 0, -broad-min-quality 10, -mmf-max-hq-mismatches 4, -mmf-max-hq-mismatches-xt-u 10, -mmf-min-quality 15, -mmf-max-any-mismatches 6, -unique-filter-coverage 2). Sequencing results were also analyzed using the GATK pipeline (version 3.1)42 for calling single nucleotide variants and insertions and deletions. All germline variants were subjected to rigorous quality control including checking for paralogs, repeats, and low variant allele frequency. Using an in-house integrated variant prioritization algorithm, variants were classified into multiple tiers of differing importance on the basis of allele function prediction (e.g., polyphen2, SIFT, mutation assessor, protein truncating, etc), gene function (e.g., known cancer genes in COSMIC43, ACMG disease predisposition genes44), prior biological evidence for pathologic effects (e.g., HGMD45, ClinVar46), etc. Variants were evaluated for co-segregation with ALL based on an autosomal dominant mode of inheritance: we hypothesized that the risk variant should be present in subjects with ALL (I-2, II-2, and II-3) and absent in I-1; Because it is not uncommon for ALL to be diagnosed in older children and adolescents, we reason that there is a distinct possibility that II-1 would eventually develop ALL. As a result, we did not require risk variant to be absent in II-1 (appendix p 7). Variants were then filtered based on frequency in non-ALL controls and known gene function in cancer and/or hematopoiesis to identify the final candidates related to cancer risk (appendix p 2, 7).
Targeted ETV6 sequencing and function prediction
Illumina dual-indexed libraries were created from the germline DNA of 4,405 children with ALL, and pooled in sets of 96 prior to hybridization with customized Roche NimbleGene SeqCap EZ probes to capture the ETV6 genomic region. Quantitative PCR was used to determine the appropriate capture product titer necessary to efficiently populate an Illumina HiSeq 2000 flowcell for paired-end 2×101 bp sequencing. A coverage of >20× depth was achieved across >90% of the targeted regions for nearly all samples. Sequence reads in FASTQ format were mapped and aligned using the Burrow-Wheller Aligner, and genetic variants were called using the GATK pipeline (version 3.1), as previously described.14, 42
Potential damaging effects of ETV6 variants were predicted using the combined annotation dependent depletion (CADD, v1.0)47 and each variant was assigned a CADD phred-like score. A CADD phred-like score of 10, 20, or 30 represents the top 10%, 1%, and 0.1% of the most deleterious variants, respectively.
Each ETV6 variant identified in the ALL cohort was curated manually and classified as “ALL-related” or “common” to indicate its potential role in predisposition to ALL, on the basis of variant frequency in ancestry-matched ALL vs. non-ALL cohorts (appendix p 8). For example, variants that were observed only in the ALL cohort were most likely to be related to ALL risk, whereas variants common in non-ALL cohorts were less likely to confer predisposition to this cancer.
Statistical analyses
The St. Jude Total Therapy protocols and the COG P9900 studies (P9904, P9905, and P9906 protocols) are frontline clinical trials for newly-diagnosed ALL across diverse risk groups and demographics.32-35 As such, they were used in population-based analyses of the association of ETV6 genotype with ALL clinical features. Patients were first classified as “with ALL-related ETV6 variants” or “without”. The association of germline ETV6 status with categorical clinical features (tumor translocation [ETV6-RUNX1, E2A-PBX1, BCR-ABL1, MLL rearrangements], hyperdiploidy [DNA index ≥1.16], and leukocyte count [< or ≥ 50×109/l]) was determined by using Fisher's exact test. Age at diagnosis (as a continuous variable) was compared between two ETV6 groups by using the non-parametric Wilcoxon rank sum test because of age did not follow normal distribution. Association of ETV6 status and genetic ancestry was tested using a multivariate Firth logistic regression model with European, African, and Asian ancestry as covariates. Relapse was treated as a time-dependent variable and its relationship with the status of germline ETV6 risk variant was evaluated by using the Fine and Gray hazard regression model48. Early treatment response was measured as minimal residual disease at the end of remission induction therapy and positivity was defined as ≥ 0.01%33-35, and its association with germline ETV6 status was evaluated by using the Fisher's exact test. Over-representation of ALL-related ETV6 variants in the ETS domain was evaluated by comparing the observed frequency with what would be expected if variants were randomly distributed in this gene, and the statistical significance was determined by using Fisher's exact test. R (version 3.1) was used for all statistical analyses unless otherwise indicated.
Role of the funding source
The funding source was not directly involved in the design of the study, the collection, analysis, and interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript. TM, GW, MQ, WY, CC, XC, SR, SW, ME, HW, EM, RF, CM, JZ, MVR, JJY had access to the raw data. The corresponding author had full access to all of the data and the final responsibility to submit for publication.
Results
We identified a family of European descent with an unusual clustering of childhood ALL at St. Jude Children's Research Hospital in the United States (Fig. 1): the mother (I-2) and 2 of her 3 daughters (II-2 and II-3) developed ALL at the ages of 9, 3, and 2, respectively. All 3 ALL cases were of B-lineage, although with different leukemia molecular subtypes. Mild thrombocytopenia was noted for I-2 and II-3 (appendix p 3), II-2 was diagnosed with Turner syndrome and mild intellectual disability, and II-3 was diagnosed with a learning disability (Table 1). The family history did not reveal other hematologic malignancies within the extended kindred.
Table 1. Clinical features of individuals in the studied pedigree with familial ALL.
| Individual | ETV6 genotype | ALL status | Age at ALL diagnosis (years) | Leukemia karyotype | Cytopenias | Additional Features |
|---|---|---|---|---|---|---|
| I-1 | Wild Type | None | NA | NA | None | None |
| I-2 | p.R359X | pre-B ALL | 9 | 56,XX,+X,+4,+6,+10,+11,+14, +del(17)(p11.2),+18,+21,+21 | Thrombocytopenia | None |
| II-1 | p.R359X | None* | NA* | NA | None | None |
| II-2 | p.R359X | pre-B ALL | 3 | 46,X,i(X)(q10)c | None | Turner syndrome |
| II-3 | p.R359X | pre-B ALL | 2 | 46,X,i(X)(q10),del(6)(q13q21) | Thrombocytopenia | Learning disability |
Abbreviations: ALL, acute lymphoblastic leukemia
This individual was 11 and did not have leukemia at the time when this manuscript was written
We hypothesized that predisposition to ALL in this family was driven by a rare but highly penetrant germline genetic variation. Whole exome sequencing of all 5 family members was performed and variants were first prioritized on the basis of bioinformatic function annotation (e.g., damaging effects, gene function). We postulated that the causal variant should follow an autosomal dominant mode of inheritance and thus would be recurrent in ALL cases but absent in the unaffected father. We identified a total of 9 nonsilent variants that followed this pattern of segregation with ALL and were also rare in non-ALL controls (minor allele frequency [MAF] <0.01%), of which ETV6 was the most likely candidate because of its known involvement in ALL pathogenesis (appendix p 2, 7). This nonsense variant in ETV6 (c.1075C>T, p.R359X) was predicted to create a stop codon within the ETS domain and result in a truncated protein without DNA-binding function and was shared by all 3 ALL cases. Interestingly, it was also present in the healthy daughter (II-1), suggesting incomplete penetrance. However, it is formally possible that she was still at risk of ALL given her young age of 11. All carriers in the index family were heterozygous for this ETV6 variant. Consistent with recent reports of recurring germline ETV6 mutations associated with familial thrombocytopenia and overrepresentation of hematologic malignancies,29-31 this p.R359X variant in ETV6 is likely to be responsible for the ALL predisposition in this family.
To determine the overall prevalence of ETV6-related predisposition to childhood ALL, we performed targeted sequencing of ETV6 in germline DNA from 4,405 children enrolled on St. Jude and the COG frontline clinical trials for newly diagnosed ALL. In this unselected cohort, we identified 43 exonic ETV6 variants (Fig. 2A and appendix p 4-6). Of these, 12 ETV6 variants were recurrent (e.g., MAF≥0.01%) in ancestry-matched non-ALL populations (the NHLBI GO Exome Sequencing Project cohort [ESP, N=6,503] and/or in the Exome Aggregation Consortium cohort [ExAC, N=60,706]) and thus less likely to be related to ALL risk. In contrast, 31 ETV6 variants were observed only in children with ALL or were exceedingly rare in non-ALL populations (MAF<0.01%) and thus were considered as “ALL-related”. All but 5 ALL-related ETV6 variants were singletons. In total, 35 (0.79%) of 4,405 children in this cohort had rare ETV6 variants that were potentially related to ALL predisposition, including 4 nonsense, 21 missense, 1 splice, and 5 frameshift variants.
Figure 2. ETV6 variants identified by targeted sequencing in 4,405 unselected ALL cases.
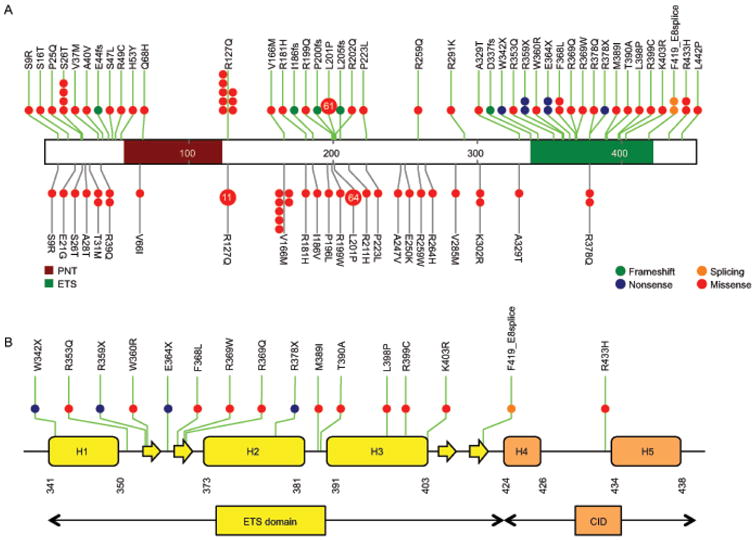
(A) ETV6 was sequenced using Illumina HiSeq platform following capture-based enrichment of this genomic region in 4,405 unselected ALL cases. Variants in non-ALL controls were based on publicly available data from the NHLBI Exome Sequencing Project cohort (N=6,503). Exnoic variants are classified as frameshift, nonsense, missense, and splicing (green, blue, red, or orange solid circles, respectively) for ALL cases (green vertical lines) and non-ALL controls (gray vertical lines). Functional domains are indicated by color based on Pfam annotation. Each circle represents a unique individual carrying the indicated variant (heterozygous or homozygous) except variants recurring in more than 10 individuals for which the number in the circle indicates the exact frequency. (B) ALL-related ETV6 variants are highly enriched in the ETS domain which consists of 3 helices (yellow boxes) and 4 β-sheets (yellow arrows). H3 is responsible for direct binding with DNA, which is negatively regulated by the CID domain (H4 and H5, orange boxes) at the C-terminal. Variant function is denoted by the color of each line.
Fifteen of the 31 ALL-related ETV6 variants (48.4%) were clustered within the ETS DNA-binding domain (Fig. 2B), while only 6 were expected if mutations were randomly distributed in ETV6 (P=3.4×10-4). These 15 included all 4 nonsense variants (p.W342X, p.R359X, p.E364X, and p.R378X), 10 missense variants (p.R353Q, p.W360R, p.F368L, p.R369W, p.R369Q, p.M389I, p.T390A, p.L398P, p.R399C, and p.K403R), and 1 splice variant (F419_E8splice). Using the combined annotation dependent depletion (CADD) algorithm, we predicted 18 of the ALL-related ETV6 variants to be highly deleterious (CADD phred-like score> 20). The p.D337fs variant had the highest CADD phred-like score (99), followed by 4 nonsense variants (score 40 for p.W342X, p. R359X, and p.E364X, and 39 for p.R378X). Compared with CADD phred-like scores of exonic ETV6 variants observed in the non-ALL controls (i.e., the ExAC cohort), the ALL-related ETV6 variants were significantly more likely to be damaging (mean CADD phred-like score of 25.6 vs 15.2; p<0.0001. appendix p 9). Interestingly, of the 18 most deleterious ETV6 variants, 10 resided in the ETS domain but none were located in the helix that directly interacts with target DNA.49 Instead, 7 of the 10 variants in the ETS domain were between the first and second helices (Fig. 2B).
We next analyzed the relationship between germline risk variants in ETV6 and clinical features of ALL, in a subset of 2,021 cases that were comprehensively characterized for clinical features and representative of the US childhood ALL population across risk and demographic groups (Table 2). Children with ALL-related ETV6 variants were significantly older at the time of diagnosis than those without these variants (10.2 years [IQR 5.3-13.8] vs 4.7 years [IQR 3.0-8.7], P=0.017). The hyperdiploid leukemia karyotype was strikingly overrepresented in ALL cases harboring germline risk variants in ETV6 compared to the wildtype group (9 [64.3%] of 14 cases vs 538 [26.8%] of 2,007 cases), P=0.0050). In contrast, the frequency of somatic ETV6-RUNX1 fusion was only 7.1% (1 of 14) in cases with germline ETV6 risk variants, compared to 22.7% (455 or 2,007) in cases with wildtype ETV6 in the host genome. Of note, there was also a trend towards overrepresentation of females in carriers of ALL-related ETV6 variants (71.4% [10 of 14] vs 45.7% [918 of 2,007], respectively), although it did not reach statistical significance. These germline ETV6 risk variants were not associated with genetic ancestry, early treatment response (minimal residual disease at the end of remission induction therapy, P=0.29), or the risk of relapse (P=0.36).
Table 2. Clinical characteristics of ALL cases with or without risk variants in ETV6a.
| COG9904/5 /6 (N=1,445) | SJ T13A/T13 B/T15 (N=576) | Combined (N=2,021) | P value | ||||
|---|---|---|---|---|---|---|---|
| Carriers of ETV6 variants ALL-related | Carriers of ETV6 variants ALL-related | Carriers of TV6 variants ALL-related E | |||||
| Yes N=8 | No N=1,437 | Yes N=6 | No N=570 | Yes N=14 | No N=2,007 | ||
| Age at diagnosis, ears | 12.0 | 4.5 | 7.1 | 5.8 | 10.2 | 4.7 | 0.017* |
| (median [range, IQR]) | (4.6-15.4, 7.0-13.4) | (1.0-20.6, 3.0-7.7) | (2.0-15.4, 3.2-13.0) | (0.1-18.8, 3.3-10.9) | (2.0-15.4, 5.3-13.8) | (0.1-20.6, 3.0-8.7) | |
|
| |||||||
| Leukocyte count 109/l | |||||||
| <50 | 7 (87.5%) | 1,229 (85.5%) | 6 (100%) | 419 (73.5%) | 13 (92.9%) | 1,648 (82.1%) | 0.49** |
| ≥50 | 1 (12.5%) | 205 (14.3%) | 0 (0%) | 151 (26.5%) | 1 (7.1%) | 356 (17.7%) | |
| Unknown | 0 (0%) | 3 (0.2%) | 0 (0%) | 0 (0%) | 0 (0%) | 3 (0.1%) | |
|
| |||||||
| DNA indexb | |||||||
| ≥1.16 | 4 (50%) | 404 (28.1%) | 5 (83.3%) | 134 (23.5%) | 9 (64.3%) | 538 (26.8%) | 0.0050** |
| <1.16 | 4 (50%) | 989 (68.8%) | 1 (16.7%) | 433 (76.0%) | 5 (35.7%) | 1,422 (70.9%) | |
| Unknown | 0 (0%) | 44 (3.1%) | 0 (0%) | 3 (0.5%) | 0 (0%) | 47 (2.3%) | |
|
| |||||||
| Gender | |||||||
| Male | 2 (25%) | 775 (53.9%) | 2 (33.3%) | 311 (54.6%) | 4 (28.6%) | 1,086 (54.1%) | 0.063** |
| Female | 6 (75%) | 659 (45.9%) | 4 (66.7%) | 259 (45.4%) | 10 (71.4%) | 918 (45.7%) | |
| Unknown | 0 (0%) | 3 (0.2%) | 0 (0%) | 0 (0%) | 0 (0%) | 3 (0.1%) | |
|
| |||||||
| Tumor translocation | |||||||
| ETV6-RUNX1 | 1 (12.5%) | 354 (24.6%) | 0 (0%) | 101 (17.7%) | 1 (7.1%) | 455 (22.7%) | 0.21** |
| E2A-PBX1 | 1 (12.5%) | 68 (4.7%) | 0 (0%) | 31 (5.4%) | 1 (7.1%) | 99 (4.9%) | 0.51** |
| MLL rearrangements | 0 (0%) | 15 (1.0%) | 0 (0%) | 8 (1.4%) | 0 (0%) | 23 (1.2%) | 1** |
| BCR-ABL1 | 0 (0%) | 0 (0%) | 0 (0%) | 12 (2.1%) | 0 (0%) | 12 (0.6%) | 1** |
| Others | 6 (75%) | 992 (69%) | 6 (100%) | 418 (73.3%) | 12 (85.7%) | 1,410 (70.3%) | |
| Unknown | 0 (0%) | 8 (0.6%) | 0 (0%) | 0 (0%) | 0 (0%) | 8 (0.4%) | |
|
| |||||||
| Ancestryc (median %) | |||||||
| European | 99.0% | 98.1% | 99.0% | 99.0% | 99.0% | 98.3% | 0.79*** |
| African | 0.1% | 0.3% | 0.1% | 0.2% | 0.1% | 0.3% | |
| Asian | 0.5% | 0.5% | 0.5% | 0.3% | 0.5% | 0.4% | |
| Native American | 0.4% | 0.5% | 0.5% | 0.4% | 0.5% | 0.5% | |
P value was estimated using
the Wilcoxon rank sum test,
the Fisher's exact test and
the Firth logistic regression test.
Data are presented as No. (%) of patients unless otherwise indicate.
DNA index of ≥ 1.16 indicates cases with high hyperdiploidy (>50 chromosomes in ALL blasts).
Genetic ancestry was estimated as European, African, Native American, and Asian using STRUCTURE, on the basis of genotypes at 30,000 randomly selected SNPs, with HapMap samples and indigenous Native American references as ancestral populations. Numbers represent median value for each patient group.
Discussion
In this study, we systematically identified germline variants in the ETV6 gene that are associated with increased risk to childhood ALL, and comprehensively described clinical characteristics unique in children with ALL carrying these risk alleles. Inherited genetic predisposition to ALL refers to an increased likelihood of developing this cancer that is attributable to germline (constitutional) genetic variations. The contribution of germline genetic variants to ALL risk is now well established, particularly through genome-wide association studies.10-16 However, the susceptibility conferred by these common variants is usually modest (1.5-2-time increase in ALL risk for each copy of the variant allele).50 In contrast, a smattering of congenital genetic disorders have been linked to ALL predisposition (e.g., PAX5,21 SH2B322 and TP5323 germline mutations), resulting in a dramatic increase in disease risk. However, these risk variants often have incomplete penetrance and the accompanying non-malignant symptoms can be mild.17 Therefore, it is conceivable that a familial cause will not be routinely suspected and it is challenging to accurately determine the extent to which sporadic ALL is attributable to rare germline predisposition variants. This is exemplified by the wide spectrum of ALL risk alleles identified in ETV6 in our current study: close to 1% of unselected sporadic ALL cases carry likely damaging and potentially highly penetrant germline risk variants in a single gene ETV6. Our findings challenge the current paradigm that ALL is primarily driven by somatic genetic alterations and imply that the inherited genetic predisposition to this cancer may be much greater than previously believed. Thus, these data from us and others29-31 provide a strong rationale for including family history examination as part of the standard approach to the diagnosis and work up of pediatric ALL. For example, the family history should determine the presence of leukemia, the types of leukemia, the ages at leukemia diagnosis, and other hematological abnormalities (e.g., antecedent thrombocytopenia). However, future studies to comprehensively characterize the clinical features and natural course of ALL with inherited predisposition are needed to define clear guidelines for clinical actions appropriate for at-risk patients.
The unusual clustering of ALL-related ETV6 variants within the critical ETS domain suggests that the loss or alteration of DNA-binding function of ETV6 may be critical to the promotion of leukemogenesis. Experimental characterization of other ETV6 variants in the ETS domain indicates that they may function in a dominate-negative manner in that the loss-of-function variant protein still oligomerizes with wildtype ETV6, thus altering subcellular localization and dramatically abrogating transcriptional repression29, 30. We hypothesize that this may be also true for ALL-related ETV6 variants identified in the current study (especially those in the ETS domain), and thus predict a dominant transmission of disease phenotype in individuals carrying these risk alleles. In contrast, the function of ETV6 variants outside of the ETS domain is much less understood and it is possible that they alter gene function in an additive manner with dosage effects on ALL predisposition. Interestingly, the ETS domain is also truncated in the ETV6-RUNX1 fusion protein as a result of the somatic t(12;21) translocation and the remaining copy of wildtype ETV6 is often subsequently deleted in overt leukemia51. In fact, we analyzed 15 ALL cases carrying germline ETV6 risk variants with materials available and identified 2 with acquired copy number loss of ETV6 in ALL blasts (not shown). Although common in childhood ALL in general,28, 52 the ETV6-RUNX1 fusion is underrepresented in children who carry the ETV6 germline risk alleles (Table 2), and this mutual exclusivity might imply possible overlap in the molecular mechanisms by which somatic and germline ETV6 variations influence ALL leukemogenesis. For example, ETV6-RUNX1 fusion arises in utero prenatally as one of the first acquired genomic abnormalities during the development of ALL53, and thus the constitutional germline ETV6 variation may also play a role at the very early stage of leukemogenesis. Because both types of genetic changes negatively impact ETV6 function, it is conceivable that they render vulnerability to subsequent oncogenic events affecting common signaling pathways.
However, it should be noted that a critical function of ETV6-RUNX1 is to disrupt RUNX1-mediated transcription regulation54 which may be distinct from the effects of ETV6 germline variants. ETV6 is indispensable for the survival of adult hematopoietic stem cells but with little effect on their progeny.24, 55 Selective inactivation of ETV6 in lineage committed progenitor cells showed profound defects in terminally differentiated megakaryocytes (reduction of platelet counts),55 consistent with the thrombocytopenia observed in patients with loss-of-function ETV6 germline variants (Table 1).29-31 In contrast, B cell development is unaffected by lineage-specific ETV6 deletion in mice55 and a distinct mechanism may exist to explain the increased risk in B-ALL conferred by germline defects in ETV6.
The possible incomplete penetrance of ETV6 variants in the index family strongly argues that secondary mutations (most likely somatic) are needed for overt leukemogenesis, consistent with the relatively late disease onset in individuals who carry the ETV6 risk variants (Table 2). Paradoxically, the frequency of hyperdiploid ALL (usually more common in young children) was significantly greater in this group despite their older age. One can hypothesize that somatic lesions characteristic of the hyperdiploid karyotype (e.g., mutations in the RAS pathway) may act synergistically with ETV6 germline variants during leukemogenesis, which should be tested experimentally in future studies. Also of interest, there were 8 additional germline variants identified from whole exome seq of the index family (CEP95, CEP250, GUSB, SNTG1, AGBL1, RAB7A, RSPRY1, and PRR23C, appendix p 2) that also co-segregated with ALL and were rare (MAF<0.01%) in non-ALL controls. While there is no obvious link of these genes with leukemogenesis, their functions have been sparsely characterized and it remains unknown whether they also contributed to ALL risk in this family (especially for the few variants predicted to be deleterious by CADD).
In conclusion, we identify germline mutations in the gene encoding the critical hematopoietic transcription factor ETV6 that co-segregate with ALL and thrombocytopenia in a rare leukemia-prone kindred. This observation, as well as similar recent findings29-31 suggests the presence of a novel genetic syndrome characterized by a predisposition to ALL and thrombocytopenia with a common underlying genetic cause (i.e., ETV6 germline variants). We also comprehensively analyzed a large cohort of unselected pediatric ALL patients and identified that close to 1% of patients harbor potential predisposing germline ETV6 variants in ETV6. Future longitudinal family studies are needed to better define the penetrance, age specific leukemia incidence and clinical features characterizing this syndrome. Similarly, experimental characterization is warranted to define the exact function of the potential ALL-related ETV6 variants and to elucidate the molecular pathways by which they influence leukemia formation. These clinical and mechanistic studies are absolutely critical for the development of recommendations for clinical interventions in the future for individuals harboring ALL-related ETV6 variants.
Supplementary Material
Acknowledgments
We thank the patients and parents who participated in the St. Jude and COG protocols included in this study, the clinicians and research staff at St. Jude and COG institutions, and J. Pullen (University of Mississippi at Jackson) and A. Carroll (University of Alabama at Birmingham) for assistance in the classification of patients with ALL. J.J.Y. is supported by the American Society of Hematology Scholar Award and by the Order of St. Francis Foundation, and T.M. is supported by the Study-Abroad Scholarship of Mie Prefecture, Japan. We thank M. Shriver (Pennsylvania State University) for sharing SNP genotype data of the Native American references. This work was supported by the National Institutes of Health (grant numbers CA21765, CA98543, CA114766, CA98413, CA180886, CA180899, and GM92666, GM115279, and GM097119) and the American Lebanese Syrian Associated Charities.
Footnotes
Contributors: TM, MLM, GW, SW, C-HP, CM, MVR, KEN, MLL, and JJY contributed to writing of the manuscript. MLM, CM and JJY designed the study. TM, MLM, RN, MD, EQ, SR, JG-F, ER, EL, PLM, WPB, NW, SW, HX, RF, C-HP, WEE, SPH, MVR, KEN and MLL contributed the data collection. TM, MLM, GW, RN, MQ, WY, CC, XC, ME, HX, RF, WEE and JZ contributed the data analysis. MLM, GW, MD, EQ, SR, YK, SW, EM, C-HP, CM, SPH, MVR, KEN, MLL and JJY interpreted the data. All authors critically reviewed the manuscript and agreed to submit the paper for publication.
Declaration of interests: The authors declared no conflicts of interest relevant to this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(5):551–65. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(14):1663–9. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moorman AV, Enshaei A, Schwab C, Wade R, Chilton L, Elliott A, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124(9):1434–44. doi: 10.1182/blood-2014-03-562918. [DOI] [PubMed] [Google Scholar]
- 4.Stanulla M, Schrappe M. Treatment of childhood acute lymphoblastic leukemia. Seminars in hematology. 2009;46(1):52–63. doi: 10.1053/j.seminhematol.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Kinlen L. Childhood leukaemia, nuclear sites, and population mixing. British journal of cancer. 2011;104(1):12–8. doi: 10.1038/sj.bjc.6605982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swaminathan S, Klemm L, Park E, Papaemmanuil E, Ford A, Kweon SM, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nature immunology. 2015;16(7):766–74. doi: 10.1038/ni.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nature reviews Cancer. 2006;6(3):193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 8.Kharazmi E, da Silva Filho MI, Pukkala E, Sundquist K, Thomsen H, Hemminki K. Familial risks for childhood acute lymphocytic leukaemia in Sweden and Finland: far exceeding the effects of known germline variants. British journal of haematology. 2012;159(5):585–8. doi: 10.1111/bjh.12069. [DOI] [PubMed] [Google Scholar]
- 9.Hemminki K, Jiang Y. Risks among siblings and twins for childhood acute lymphoid leukaemia: results from the Swedish Family-Cancer Database. Leukemia. 2002;16(2):297–8. doi: 10.1038/sj.leu.2402351. [DOI] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nature genetics. 2009;41(9):1006–10. doi: 10.1038/ng.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nature genetics. 2009;41(9):1001–5. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez-Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nature genetics. 2013;45(12):1494–8. doi: 10.1038/ng.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Migliorini G, Fiege B, Hosking FJ, Ma Y, Kumar R, Sherborne AL, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122(19):3298–307. doi: 10.1182/blood-2013-03-491316. [DOI] [PubMed] [Google Scholar]
- 14.Xu H, Zhang H, Yang W, Yadav R, Morrison AC, Qian M, et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nature communications. 2015;6:7553. doi: 10.1038/ncomms8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherborne AL, Hosking FJ, Prasad RB, Kumar R, Koehler R, Vijayakrishnan J, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nature genetics. 2010;42(6):492–4. doi: 10.1038/ng.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu H, Yang W, Perez-Andreu V, Devidas M, Fan Y, Cheng C, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. Journal of the National Cancer Institute. 2013;105(10):733–42. doi: 10.1093/jnci/djt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellinghaus E, Stanulla M, Richter G, Ellinghaus D, te Kronnie G, Cario G, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2012;26(5):902–9. doi: 10.1038/leu.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet. 2000;355(9199):165–9. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- 19.Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nature genetics. 2009;41(11):1243–6. doi: 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508(7494):98–102. doi: 10.1038/nature13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nature genetics. 2013;45(10):1226–31. doi: 10.1038/ng.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez-Garcia A, Ambesi-Impiombato A, Hadler M, Rigo I, LeDuc CA, Kelly K, et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood. 2013;122(14):2425–32. doi: 10.1182/blood-2013-05-500850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nature genetics. 2013;45(3):242–52. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang LC, Swat W, Fujiwara Y, Davidson L, Visvader J, Kuo F, et al. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes & development. 1998;12(15):2392–402. doi: 10.1101/gad.12.15.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kar A, Gutierrez-Hartmann A. Molecular mechanisms of ETS transcription factor-mediated tumorigenesis. Critical reviews in biochemistry and molecular biology. 2013;48(6):522–43. doi: 10.3109/10409238.2013.838202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. The New England journal of medicine. 2011;364(26):2496–506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, et al. ETV6 mutations in early immature human T cell leukemias. The Journal of experimental medicine. 2011;208(13):2571–9. doi: 10.1084/jem.20112239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhojwani D, Pei D, Sandlund JT, Jeha S, Ribeiro RC, Rubnitz JE, et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia. 2012;26(2):265–70. doi: 10.1038/leu.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nature genetics. 2015;47(2):180–5. doi: 10.1038/ng.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nature genetics. 2015;47(5):535–8. doi: 10.1038/ng.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Topka S, Vijai J, Walsh MF, Jacobs L, Maria A, Villano D, et al. Germline ETV6 Mutations Confer Susceptibility to Acute Lymphoblastic Leukemia and Thrombocytopenia. PLoS genetics. 2015;11(6):e1005262. doi: 10.1371/journal.pgen.1005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pui CH, Mahmoud HH, Rivera GK, Hancock ML, Sandlund JT, Behm FG, et al. Early intensification of intrathecal chemotherapy virtually eliminates central nervous system relapse in children with acute lymphoblastic leukemia. Blood. 1998;92(2):411–5. [PubMed] [Google Scholar]
- 33.Pui CH, Sandlund JT, Pei D, Campana D, Rivera GK, Ribeiro RC, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood. 2004;104(9):2690–6. doi: 10.1182/blood-2004-04-1616. [DOI] [PubMed] [Google Scholar]
- 34.Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. The New England journal of medicine. 2009;360(26):2730–41. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borowitz MJ, Devidas M, Hunger SP, Bowman WP, Carroll AJ, Carroll WL, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood. 2008;111(12):5477–85. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borowitz MJ, Wood BL, Devidas M, Loh ML, Raetz EA, Salzer WL, et al. Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children's Oncology Group study AALL0232. Blood. 2015 doi: 10.1182/blood-2015-03-633685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–9. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang JJ, Cheng C, Devidas M, Cao X, Fan Y, Campana D, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nature genetics. 2011;43(3):237–41. doi: 10.1038/ng.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernandez CA, Smith C, Yang W, Mullighan CG, Qu C, Larsen E, et al. Genome-wide analysis links NFATC2 with asparaginase hypersensitivity. Blood. 2015;126(1):69–75. doi: 10.1182/blood-2015-02-628800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edmonson MN, Zhang J, Yan C, Finney RP, Meerzaman DM, Buetow KH. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics. 2011;27(6):865–6. doi: 10.1093/bioinformatics/btr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic acids research. 2015;43(Database issue):D805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2013;15(7):565–74. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human genetics. 2014;133(1):1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic acids research. 2014;42(Database issue):D980–5. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. Journal of the American Statistical Association. 1999;94(446):496–509. [Google Scholar]
- 49.De S, Chan AC, Coyne HJ, 3rd, Bhachech N, Hermsdorf U, Okon M, et al. Steric mechanism of auto-inhibitory regulation of specific and non-specific DNA binding by the ETS transcriptional repressor ETV6. Journal of molecular biology. 2014;426(7):1390–406. doi: 10.1016/j.jmb.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moriyama T, Relling MV, Yang JJ. Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood. 2015;125(26):3988–95. doi: 10.1182/blood-2014-12-580001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nature genetics. 2014;46(2):116–25. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alpar D, Wren D, Ermini L, Mansur MB, van Delft FW, Bateman CM, et al. Clonal origins of ETV6-RUNX1(+) acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia. 2015;29(4):839–46. doi: 10.1038/leu.2014.322. [DOI] [PubMed] [Google Scholar]
- 53.Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nature reviews Cancer. 2003;3(9):639–49. doi: 10.1038/nrc1164. [DOI] [PubMed] [Google Scholar]
- 54.Guidez F, Petrie K, Ford AM, Lu H, Bennett CA, MacGregor A, et al. Recruitment of the nuclear receptor corepressor N-CoR by the TEL moiety of the childhood leukemia-associated TEL-AML1 oncoprotein. Blood. 2000;96(7):2557–61. [PubMed] [Google Scholar]
- 55.Hock H, Meade E, Medeiros S, Schindler JW, Valk PJ, Fujiwara Y, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes & development. 2004;18(19):2336–41. doi: 10.1101/gad.1239604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.