

Abstract
Demand remains for new inhibitors of HIV-1 replication and the inhibition of HIV-1 entry is an extremely attractive therapeutic approach. Using field-based bioisosteric replacements, we have further extended the chemotypes available for development in the HIV-1 entry inhibitor class. Moreover, using field-based disparity analysis of the compounds, 3D structure-activity relationships were derived that will be useful in the further development of these inhibitors towards clinical utility.
Keywords: computer-aided drug design, HIV-1 envelope protein, field-based, bioisosteric replacement, scaffold-hopping, SAR analysis, structure-activity landscape, antiviral
Graphical Abstract
Inhibiting the process of entry of HIV-1 into susceptible cells remains an elusive but extremely attractive intervention point for the development of novel anti-HIV therapies. HIV-1 entry into cells is solely orchestrated by the Env complex on the surface of the virion. The Env complex is organized on the virion surface as trimeric spikes, composed of three gp120 molecules non-covalently linked to three gp41 molecules. Recently, two structures of a soluble, cleaved HIV-1 Env trimer from a clade A founder virus has been solved providing additional and much needed information on the quaternary organization of the Env complex.1, 2,3
HIV-1 infection usually occurs only after two sequential and specific binding steps. The first interaction is between gp120 and CD4 antigen present on CD4+ T cells, monocyte/macrophages, and other immune and nonimmune cells. This interaction results in a series of conformational rearrangements in gp120 that permits the second binding event to occur. This second interaction occurs between gp120 and a member of the chemokine receptor subfamily, within the large G protein–coupled family of receptors, mainly CCR5 and/or CXCR4. This interaction also promotes considerable rearrangement in gp120 and transduction of this conformational signal to gp41. This then elicits the exposure of the fusion peptide within the N-terminus of gp41, which through additional conformational rearrangements in gp41 facilitates fusion between the viral and cellular membranes and release of the viral core into the cell.
Several groups are actively involved in the development of small molecules targeted to gp120 that disrupt the Env molecular machine to stop HIV-1 entry into cells. 4–13 Despite this only one chemotype, developed by Bristol Myers Squibb, has successfully made it to clinical trials. The newest compound in the drug class, BMS-663068, a phosphonooxymethyl prodrug of BMS-626529,14 recently performed favourably in a Phase IIb clinical study, highlighting the potential utility of these Env-directed entry inhibitor class of compounds (presented at the 22nd Conference on Retroviruses and Opportunistic Infections [CROI]).
Our group recently described the computational design of new compounds designed to act through a common binding site to that of the Bristol Myers Squibb piperazine-based entry inhibitors, of which BMS-663068/BMS626529 are members. Our most potent compounds, SC11 and SC26, both contain a dipyrrolodine core scaffold, and specifically inhibit HIV-1JR-CSF at 0.8 and 2 nM, respectively.15
Having successfully demonstrated that scaffold-hopping of the piperazine moeity can be achieved, in this study we sought to extend the core chemotypes available for the entry inhibitor class in the hopes of improving drug-like properties. To accomplish this we performed computationally directed scaffold-hopping studies, coupled to synthesis, antiviral potency analysis and computational 3D Quantitative Structure-Activity Relationship (QSAR).
Due to the lack of structural information on the bioactive conformation of our inhibitors and the BMS piperazine based inhibitors, we first used FieldTemplater (Forge, Cresset)16–26 to determine the most likely 3D conformation adopted by BMS-377806,12 BMS-488043,27 BMS-626529,28 and SC11/SC2615 upon binding to the HIV-1 Env target (Figure 1). This FieldTemplater-derived 3D conformation was then used as input into Spark (Cresset, UK). Spark searches a database of up to 600,000 fragments to find bioisosteres that exhibit similar shape and electronic properties as the region of interest when placed in the context of the final molecule. To maximize the likelihood of identifying interesting potential replacements, we performed bioisosteric searches of the piperazine groups of BMS-377806, BMS-488043, and BMS-626529, in addition to the dipyrrolodine group of compounds SC11/SC26. The results of each search were analyzed and common structures were identified. From this analysis, four different core chemotypes were chosen for investigation based upon diversity and BIF% scores (a factor that indicates how good the replacement is in the context of the conformation of the entire molecule). Compounds containing core pyrrolo-pyrazole, azetidine, tetrahydropyridine, azabicyclo-hexane and diazaspiro-decane groups were then synthesized. First, a common head group to be used in all of the molecules was synthesized, compound 6, according to Supplemental Scheme 1. This was subsequently used in the synthesis of compounds SC12, SC14, SC15, SC27, SC28, and SC45 as outlined in supplemental schemes 1–7.
Figure 1. Overlaid field point representation of compounds BMS-377806, BMS488043, BMS-626529, and SC11 from the derived binding mode (Forge, UK).

Blue field points highlight energy minima for a positively charged probe, red for a negative probe. Yellow spheres represent attractive van der Waals minima for a neutral probe and orange spheres represent hydrophobic centroids. Oxygen atoms are shown in red, nitrogen in blue. The size of the points is related to the strength of a potential interaction (i.e., absolute value of the field strength at that point in space).
After successful synthesis of each of the five novel-scaffold derivatives, we then tested them for specificity and potency against HIV-1. To do this we used the HIV-1 single round infection assay. In this system, recombinant single-round infectious envelope-pseudotyped luciferase-reporter HIV-1 viruses are produced by co-transfection of 293T cells with the envelope-deficient HIV-1 NL4-3 vector (pNL4-3-LucR+E−; a gift of N. R. Landau, New York University),29 which carries the luciferase-reporter gene; and the HIV-1 JR-CSF, HIV-1 B41,30 HIV-1 HxBc2 or amphotropic murine leukemia virus (AMLV) envelope expressing vector. These recombinant viruses are then used to infect U87.CD4.CCR5 (JR-CSF and B41 Envs) target cells or U87.CD4.CXCR4 (HxBc2 Env) and infectivity is quantified by measuring luciferase activity in cell lysates (Luciferase Assay System, Promega) using a microplate luminometer (GloMax, Promega). The effect of the compounds on the infection of target cells by AMLV Env pseudotyped recombinant HIV-1 was used to determine the specificity of the compounds to HIV-1 Env-mediated cell entry. The results of this analysis are shown in Table 1.
Table 1.
Specificity and potency of compounds against HIV-1JR-CSF, HIV-1B41, HIV-1HxBc2 Env, and AMLV pseudotyped HIV-1.
| Compound | IC50 JR-CSF (μM) | IC50 B41 (μM) | IC50 HxBc2 (μM) | IC50 AMLV (μM) |
|---|---|---|---|---|
SC12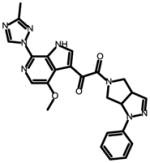
|
0.008 ± 0.002 | 0.006 ± 0.003 | 0.08 ± 0.02 | N.A. |
SC14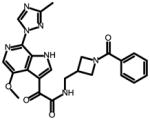
|
2.5 ± 0.4 | 3.8 ± 0.4 | 3.8 ± 1.6 | N.A. |
SC15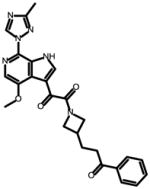
|
0.003 ± 0.001 | 0.007 ± 0.001 | 0.009 ± 0.0011 | N.A. |
SC27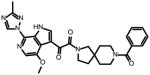
|
13.0 ± 9.0 | 3.9 ± 0.7 | 36 ± 21 | N.A |
SC28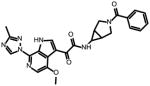
|
0.096 ± 0.019 | 0.085 ± 0.03 | 0.069 ± 0.014 | N.A. |
SC45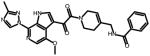
|
0.224 ± 0.017 | 0.35 ± 0.03 | 0.38 ± 0.03 | N.A. |
N.A. = not active; N.D. = not determined.
As can be seen from the results in Table 1, all compounds had specific anti-HIV activity but their half maximum inhibitory concentrations (IC50) showed noticibale variation. To better understand the factors driving activity of these HIV-1 entry inhibitors, including those compounds previously reported by our group, we assessed them computationally using Activity Miner and Activity Atlas (Cresset, UK). Activity Miner provides an interface for the rapid assessment of structure-activity landscapes for a set of aligned compounds in the context of activity cliffs. It achieves this by comparing pairs of molecules to find where small changes in the molecule’s structure or local properties results in a large change in activity. In the software, this notion of disparity is expressed as:
The higher the calculated disparity corresponds to a sharper change in activity between a pair of very similar molecules and hence an activity cliff.
Activity Atlas (Cresset, UK) provides a probabilistic approach to analyzing Structure Activity Relationships (SAR). Like all 3D SAR approaches, comprehensive conformation hunting and correct alignments are critical, thus, alignments were hand checked to reduce noise arising from inadequate alignments. This is a key and necessary step, since, being a probabilistic method, Activity Atlas assumes that the alignments are correct. Activity Atlas helps provide insight into what feature(s) the active compounds in a collection have in common – in essence, it summarizes the Activity Miner analysis to illuminate what activity cliffs reveal about the nature of the SAR.
Compounds SC03, SC04, SC07, SC08, SC11 and SC26 from our previous report,15 and compounds SC12, SC14, SC15, SC27, SC28 and SC45 from this analysis were input into Activity Miner. From the pairwise comparisons (50% field – 50% shape) a Disparity Matrix was generated (Figure 2) which provides an overview of the dataset. For each pair, the comparison was color-coded by color and shade, with red and green meaning a decrease or increase in activity, respectively. The shading corresponds to the disparity: darker shading indicates a higher disparity, and thus, a steeper activity cliff.
Figure 2. Disparity Matrix showing the pairwise comparisons for the compounds reported using IC50 HIV-1JR-CSF as activity.

Red and green boxes correspond to decreases and increases in activity between the pair, respectively. The darker the shading, the sharper the activity cliff (higher calculated Disparity). The matrix is symmetrical – for every red box, there is a corresponding green box for the comparison going in the opposite direction.
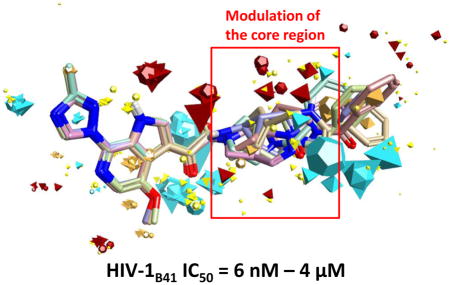
As can be clearly seen in Figure 2, the highest ranked activity cliff is between molecules SC11 (pIC50 = 9.1) and SC27 (pIC50 = 4.890). This pair has a 3D Similarity of 0.844 (quite high), combined with a ΔpIC50 = 4.21. When the field differences are examined, as shown in Figure 3, it can be observed that the electron density spanning the dipyrollidine in SC11 is absent in the central diazaspiro-decane group of SC27. This suggests that electron density in the center of the molecule may be important for activity.
Figure 3. Comparison of field differences for a selection of molecules in which the negative electrostatics over the central moiety appears to be important for activity.

The field difference map displays the surfaces as relative values between pairs of molecules. The regions where the surface is displayed have stronger electrostatic fields than the corresponding regions in the compared molecule. In Panel A, SC27 is compared to SC11; in the Panel B, SC15 is compared to SC14. Again, IC50 refers to IC50 HIV-1JR-CSF.
This observation is conserved in the comparisons of SC15 and SC26 to SC27, with the higher potency compounds having this region of electron density. Furthermore, this observation is even borne out in the comparison of SC15 to SC14. SC14 and SC15 share a core azetidine moiety but differ in its orientation. Again, the reduction of negativity in the core region is seen in the lower potency compound SC14 but maintained in the highly active SC15 compound.
Comparison of SC11 to its parental compound SC04 (IC50 HIV-1JR-CSF = 100 μM), highlights key field differences that could explain the corresponding 5-order of magnitude difference in activity (Figure 4). Most striking are the differences between the “head regions” of the compounds. The acenaphthene head group of SC04 shows an intact π-cloud, as well as positivity associated with the in-plane hydrogens that is not observed in the azaindole group of the more active SC11 (Figure 4, Panel B).
Figure 4. (Panel A) Comparing the field differences between SC11 and SC04.

The π-cloud associated with the fused rings remains intact, whereas in the more active SC11, that electron density has been pulled out of the region. (Panel B) Comparison of the calculated electrostatic surfaces for SC11 and SC04. Notice the intact π-cloud of the acenaphthene, as well as the positive field associated with the in-plane hydrogens, which leaves them available for potential hydrogen bonding.
Evaluating the complex structure-activity relationship for this set of compounds, which have only loosely related 2D structure based on Tanimoto similarity scores (based on Merck Atom Pairs), resulted in a number of comparisons to examine in Activity Miner. The visualization of single-pair comparisons provides plenty of insight for molecule pairs with only minor structural or featural changes resulting in activity cliffs. However, when examining more complex datasets containing multiple chemotypes with multiple simultaneous scaffold modifications, it is more convenient and illuminative to summarize the pair-wise information using Activity Atlas (Cresset, UK).
Activity Atlas analyzes SAR by employing a Bayesian approach to qualitatively attain a global overview of available 3D structure and activity information. Effectively, this approach automates and summarizes the pair comparisons that could be examined and exploited for SAR understanding in Activity Miner. The aim of Activity Atlas is to: determine what electrostatic/hydrophobic/shape features active molecules in the data set have in common; to understand what the activity cliffs suggest about the SAR; and finally, to be able to assess how well the compounds cover available chemical (or feature) space.
Once the Activity Atlas calculated average field contributions to activity for the data set are plotted, one can look at new molecules in the context of these averages to assess suitability for progression.
Examining three of the compounds in the dataset with high, mid, and low potencies in the context of the average field contributions (Figure 5, Panel A), we see that one of the most active compounds (SC26), shows a field pattern that aligns well with the average electrostatics. SC14, which has mid-range activity (IC50 JR-CSF = 2.5 μM), has a field point pattern which aligns acceptably in some areas, but doesn’t meet the negative electrostatic directionality associated with the carbonyl group shown on the right. Finally, SC04, with IC50 of 100 μM, shows reasonable field matching across the core, but the acenaphthene “head region” lacks much of the fields present in the higher potency molecules (Figure 5, Panel B).
Figure 5. (Panel A) Compound SC11 showing the average electrostatics and average hydrophobic regions based on the Activity Atlas summary of active molecules in the dataset. (Panel B) Assessing three compounds in the context of the average electrostatics associated with activity based on the dataset.

The higher active molecules have better correlation with the average fields for actives (shown in Panel A), while the less active compounds miss critical features of the SAR. Red and blue maps depict the average positive and negative electrostatics associated with active molecules, based on the dataset. IC50 refers to IC50 HIV-1JR-CSF. (Panel C) Summary graphic of the activity cliffs found by pairwise comparisons in Activity Miner and summarized in Activity Atlas. The maps in this graphic show the key features that drive activity, as determined from the alignment of the reported compounds. Note that “more negative” can also mean “less positive”, and vice versa.
When the activity cliff information is summarized using Activity Atlas (Figure 5, Panel C), it is seen that there are regions with specific electrostatic conditions, as well as hydrophobicity considerations. In essence, these regions qualitatively express our 3D SAR in a graphical fashion and provide a blueprint for the design of further analogues.
In summary, we have extended our previous work using computational means to diversify the core scaffolds in the entry inhibitor class, identifying 5 new core chemotypes. Computational analysis of these core chemotype diversified inhibitors has provided a blueprint that can be used in the design of next generation entry inhibitors to improve the potency. Taken together, the results from this study validate the combined use of field-based bioisosteric replacement and field-based disparity analysis as a means to further the potential utility of Env-targeted HIV-1 entry inhibitors. Future studies in our group will include assessment of the drug-like properties of these new chemotypes, modulation and optimization of their potencies based upon SAR relationships derived in this study and simultaneously against genetically distinct isolates, and detailed mechanism of action studies, including structural studies.
Supplementary Material
Acknowledgments
This work was supported by NIH/NIAID grant 1R21AI104354-01A1 (Cocklin, PI) and W. W. Smith Charitable Trust grant #A1301 (Cocklin, PI). We’d like to thank Pavel Pugachev and John P. Moore (Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York, NY 10021, USA) for the generous gift of the B41 gp160 expression plasmid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, Wilson IA, Ward AB. Science. 2013;342:1484. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. Science. 2013;342:1477. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GB, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. Nature. 2014 doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Courter JR, Madani N, Sodroski J, Schon A, Freire E, Kwong PD, Hendrickson WA, Chaiken IM, Lalonde JM, Smith AB., 3rd Accounts of chemical research. 2014 doi: 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwon YD, LaLonde JM, Yang Y, Elban MA, Sugawara A, Courter JR, Jones DM, Smith AB, 3rd, Debnath AK, Kwong PD. PloS one. 2014;9:e85940. doi: 10.1371/journal.pone.0085940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z, Zhou N, Sun Y, Ray N, Lataillade M, Hanna GJ, Krystal M. Antimicrobial agents and chemotherapy. 2013;57:4172. doi: 10.1128/AAC.00513-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lalonde JM, Le-Khac M, Jones DM, Courter JR, Park J, Schon A, Princiotto AM, Wu X, Mascola JR, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB., 3rd ACS Med Chem Lett. 2013;4:338. doi: 10.1021/ml300407y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curreli F, Choudhury S, Pyatkin I, Zagorodnikov VP, Bulay AK, Altieri A, Kwon YD, Kwong PD, Debnath AK. Journal of medicinal chemistry. 2012;55:4764. doi: 10.1021/jm3002247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams DH, Adam F, Fenwick DR, Fok-Seang J, Gardner I, Hay D, Jaiessh R, Middleton DS, Mowbray CE, Parkinson T, Perros M, Pickford C, Platts M, Randall A, Siddle D, Stephenson PT, Tran TD, Vuong H. Bioorganic & medicinal chemistry letters. 2009;19:5246. doi: 10.1016/j.bmcl.2009.06.080. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Virology. 2005;339:213. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, 3rd, Sodroski JG. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5036. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:11013. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herschhorn A, Gu C, Espy N, Richard J, Finzi A, Sodroski JG. Nature chemical biology. 2014;10:845. doi: 10.1038/nchembio.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nettles RE, Schurmann D, Zhu L, Stonier M, Huang SP, Chang I, Chien C, Krystal M, Wind-Rotolo M, Ray N, Hanna GJ, Bertz R, Grasela D. J Infect Dis. 2012;206:1002. doi: 10.1093/infdis/jis432. [DOI] [PubMed] [Google Scholar]
- 15.Tuyishime M, Danish M, Princiotto A, Mankowski MK, Lawrence R, Lombart HG, Esikov K, Berniac J, Liang K, Ji J, Ptak RG, Madani N, Cocklin S. Bioorganic & medicinal chemistry letters. 2014;24:5439. doi: 10.1016/j.bmcl.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheeseright T, Mackey M, Rose S, Vinter A. J Chem Inf Model. 2006;46:665. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- 17.Cheeseright T, Mackey M, Phd, Rose S, Phd, Vinter A., Phd Expert Opin Drug Discov. 2007;2:131. doi: 10.1517/17460441.2.1.131. [DOI] [PubMed] [Google Scholar]
- 18.Cheeseright TJ, Holm M, Lehmann F, Luik S, Gottert M, Melville JL, Laufer S. Journal of medicinal chemistry. 2009;52:4200. doi: 10.1021/jm801399r. [DOI] [PubMed] [Google Scholar]
- 19.Cheeseright TJ, Mackey MD, Melville JL, Vinter JG. J Chem Inf Model. 2008;48:2108. doi: 10.1021/ci800110p. [DOI] [PubMed] [Google Scholar]
- 20.Cheeseright TJ, Mackey MD, Scoffin RA. Curr Comput Aided Drug Des. 2011;7:190. doi: 10.2174/157340911796504314. [DOI] [PubMed] [Google Scholar]
- 21.Collins JC, Armstrong A, Chapman KL, Cordingley HC, Jaxa-Chamiec AA, Judd KE, Mann DJ, Scott KA, Tralau-Stewart CJ, Low CMR. Medchemcomm. 2013;4:1148. [Google Scholar]
- 22.Kirpotina LN, Khlebnikov AI, Schepetkin IA, Ye RD, Rabiet MJ, Jutila MA, Quinn MT. Mol Pharmacol. 2010;77:159. doi: 10.1124/mol.109.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Low CM, Buck IM, Cooke T, Cushnir JR, Kalindjian SB, Kotecha A, Pether MJ, Shankley NP, Vinter JG, Wright L. Journal of medicinal chemistry. 2005;48:6790. doi: 10.1021/jm049069y. [DOI] [PubMed] [Google Scholar]
- 24.Low CM, Vinter JG. Journal of medicinal chemistry. 2008;51:565. doi: 10.1021/jm070880t. [DOI] [PubMed] [Google Scholar]
- 25.Mackey MD, Melville JL. J Chem Inf Model. 2009;49:1154. doi: 10.1021/ci8003978. [DOI] [PubMed] [Google Scholar]
- 26.Webster SP, Binnie M, McConnell KM, Sooy K, Ward P, Greaney MF, Vinter A, Pallin TD, Dyke HJ, Gill MI, Warner I, Seckl JR, Walker BR. Bioorganic & medicinal chemistry letters. 2010;20:3265. doi: 10.1016/j.bmcl.2010.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Z, Zadjura LM, Marino AM, D’Arienzo CJ, Malinowski J, Gesenberg C, Lin PF, Colonno RJ, Wang T, Kadow JF, Meanwell NA, Hansel SB. J Pharm Sci. 2010;99:2135. doi: 10.1002/jps.21948. [DOI] [PubMed] [Google Scholar]
- 28.Nowicka-Sans B, Gong YF, McAuliffe B, Dicker I, Ho HT, Zhou N, Eggers B, Lin PF, Ray N, Wind-Rotolo M, Zhu L, Majumdar A, Stock D, Lataillade M, Hanna GJ, Matiskella JD, Ueda Y, Wang T, Kadow JF, Meanwell NA, Krystal M. Antimicrobial agents and chemotherapy. 2012;56:3498. doi: 10.1128/AAC.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connor RI, Chen BK, Choe S, Landau NR. Virology. 1995;206:935. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 30.Pugach P, Ozorowski G, Cupo A, Ringe R, Yasmeen A, de Val N, Derking R, Kim HJ, Korzun J, Golabek M, de Los Reyes K, Ketas TJ, Julien JP, Burton DR, Wilson IA, Sanders RW, Klasse PJ, Ward AB, Moore JP. Journal of virology. 2015;89:3380. doi: 10.1128/JVI.03473-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.