

Abstract
(±)-Tetrabenazine was synthesized in six steps from commercially available compounds. The key cyclization substrate was assembled rapidly via Baylis-Hillman and aza-Michael reactions. Annulation of the final ring was achieved through visible light photocatalysis, wherein carbon-carbon bond formation was driven by the oxidation of a tertiary amine. Solvent played a critical role in the photoredox cyclization outcome–whereas methanol led to a mixed ketal, acetonitrile/water (10:1) gave direct cyclization to (±)-tetrabenazine and occurred more rapidly.
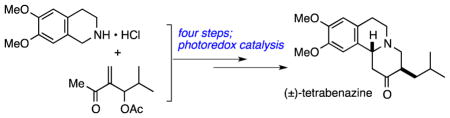
Graphical abstract
First prepared in the 1950’s as an antipsychotic,1–2 tetrabenazine (TBZ, (±)−1, Fig. 1) has reemerged as a therapeutically useful compound for the treatment of hyperkinetic disorders. An important milestone for this benzo[a]quinolizine came in 2008, when the FDA approved the racemic mixture of 1 for the treatment of Huntington’s chorea in the United States.3–5 Metabolically TBZ is rapidly reduced to dihydrotetrabenazine (DTBZ, (±)−2), which is responsible for most of the pharmacological activity. Both TBZ and its metabolite, DTBZ, reversibly bind to vesicular monoamine transporter 2 (VMAT2) with nanomolar affinities.6–7 VMAT2 is responsible for shuttling monoamines into the presynaptic vesicles from the neuronal cytoplasm.8 Inhibition of VMAT2 directly decreases neurotransmitters available for release, and further indirectly depletes monoamine levels, as those stranded in cytoplasm become degraded by monoamine oxidases. The value of these compounds extends beyond the treatment of hyperkinetic disorders. The high VMAT2 affinity of (+)-(2R,3R,11bR)-DTBZ (Ki 0.97 nM) has spurred the development of radiolabeled analogs that are valuable imaging agents for diagnosing neurological conditions.9–15 Furthermore, Parkinsonian tremors can be modeled by dosing rodents with TBZ to induce tremulous jaw movements.16
Figure 1.

Tetrabenazine and Dihydrotetrabenazine.
TBZ and DTBZ’s compelling pharmacology has stimulated the development of several methods for the preparation of these alkaloids,2,17–23 as well as the synthesis of numerous analogs.11,24–27 Our synthetic approach was inspired by a late-stage oxidative aza-Prins cyclization strategy that was reported by Cho, Min, and co-workers (Scheme 1A).21 In that work, stoichiometric DDQ oxidized tertiary amine 3 to an iminium ion, which cyclized with a tethered allylsilane to provide 4. Tetrabenazine and dihydrotetrabenazine were subsequently obtained through additional oxidation state adjustments. A key finding of their work was that the iminium ion cyclization occurred stereoselectively, establishing the correct relative stereochemistry between C-11b and C3. We envisioned that a related approach involving an oxidative Mannich cyclization would offer advantages with respect to step and redox economies (Scheme 1B).28 In principle the cyclization substrate would be available through the conjugate addition of tetrahydroisoquinoline 7 to an appropriately functionalized enone, exemplified by 8a/8b.
Scheme 1.

Prior Work and Retrosynthetic Analysis of Tetrabenazine.
Furthermore, we recognized this as an opportunity to examine a more environmentally benign alternative to stoichiometric oxidants in the context of a target-directed synthesis. A number of aerobic methods have been developed for oxidative Mannich-type couplings of carbon nucleophiles with relevant tertiary amines,29 including those mediated by transition metals, such as Cu30–33 and Fe,34–35 as well as other catalytic radical initiators, such as triarylaminium radical salts,36 I2,37–38 and SO2Cl2.39 Visible light photocatalysis has emerged as a capable and environmentally benign method for driving redox processes.40–46 Specifically, Stephenson, and others, have shown that N-aryltetrahydroisoquinolines are excellent substrates for visible light photoredox catalysis.47–53 We elected to pursue photoredox catalysis for the cyclization of 6 to TBZ based on our own work in this area that demonstrated N-alkylated tetrahydroisoquinolines, as well as other tertiary amines, remain viable substrates for oxidation under these conditions.54 N-Alkylated tetrahydroisoquinolines55 have a slightly more positive redox potentials compared to N-aryl analogs.56–57 This could partially explain why oxidative coupling reactions of N-alkylated tetrahydroisoquinolines are not as developed as their N-aryl counterparts.58 Diverse nucleophiles can be coupled with iminium ions generated under photoredox conditions, including enol ethers and ketones, although the later must be activated through enamine catalysis.59–62 Although intermolecular couplings of N-aryltetrahydroisoquinolines generally require an excess of the nucleophilic partner, we reasoned that the proposed cyclization of 6a/6b to TBZ would be facilitated by the high effective concentration inherent with an intramolecular process.
An important question was whether a ketone or an enol ether would be optimal for the cyclization (Scheme 2). This was addressed through the rapid preparation of 6a in 93% yield through the reaction of tetrahydroisoquinoline 7 and methyl vinyl ketone in the presence of aqueous NaOH. Unfortunately, we were unable to identify conditions for the direct cyclization of 6a to 9 under standard photoredox conditions with Ru(bpy)3Cl2. Cyclization of 6a was also attempted in the presence of proline or pyrrolidine·TFA, conditions that have been effective for oxidative couplings of ketones with tetrahydroisoquinoline,61–62 but annulation did not occur in this case. 1H NMR analysis of the crude reaction mixture confirmed the presence of starting material 6a and a component whereby the benzylic position of tetrahydroisoquinoline ring had been oxidized to give amide. Oxidation of some of this material to an amide was supported by the disappearance of its benzylic CH2 singlet at 3.5 ppm and the downfield shift of an aromatic CH singlet from 6.51 to 7.55 ppm in the 1H NMR spectrum. Our group, as well as others, has observed the oxidation of tetrahydroisoquinolines to amides under photoredox conditions.54,63–65 We reasoned that the low levels of nucleophile, formed through keto/enol tautomerization or via catalytic formation of the enamines through 2° amine catalysis, was insufficient to compete with the undesired oxidation pathway. Thus, we converted 6a to TIPS enol ether 10 and found that it cyclized slowly under photoredox conditions in MeOH to provide a mixed methyl triisopropylsilyl ketal that was immediately hydrolyzed to ketone 9 (28% yield for two steps). Two factors contributed to the poor yield of 9. Conversion for the cyclization of 10 to the mixed ketal was incomplete, even after 4 d, and ketone 9 was difficult to purify by chromatography as it co-eluted with TIPSOH. We attempted the photoredox cyclization of 10 in CH3CN/H2O (10:1) in hopes of avoiding the formation of the mixed ketal and the extra hydrolysis step. Fortunately, 10 cyclized directly to 9 in 51% yield after only 2 d of irradiation in this system. Gagné has noted that water as a co-solvent can improve the rates of photocatalytic reactions and posited that this rate enhancement stems from resolvation of the catalyst after reductive quenching, which decreases the rate of competing back-transfer reactions.66
Scheme 2.

Proof-of-Principle Photoredox Cyclization.
Pleased that the photoredox cyclization would be a viable approach, we turned our attention to the preparation of branched substrate 6b (Scheme 3). In addition to directly providing TBZ, the cyclization of 6b should be more facile compared to 6a due to gauche interactions present within the side chain, which should serve to pre-organize the side chain and decrease the activation energy. The first step to prepare this substrate involved modified conditions for a known Baylis-Hillman reaction between methyl vinyl ketone (8a) and isobutyraldehyde,67 followed by acetylation to provide 12. Treating 12 with commercially available tetrahydroisoquinoline salt 7 in the presence of aqueous hydroxide promoted rapid conjugate addition, which was accompanied by spontaneous β-elimination of acetate, forming enone 14 as a 6:1 mixture of E:Z isomers. Selective reduction of the alkene in 14 was not trivial–several conditions for 1,4-hydride delivery instead predominantly gave the undesired 1,2-reduction product.68–71 Hydrogenation under acidic conditions ultimately proved to be rapid and high yielding process, giving 6b in 79% yield. Addition of TFA was essential in this reaction, as hydrogenolysis of the benzylic amine and other side products were formed in its absence. Standard conditions were then used to convert 6b to TIPS enol ether 15, which could be purified by chromatography.
Scheme 3.

Synthesis of (±)-Tetrabenazine.
Photoredox cyclization of 15 was accomplished using a blue LED light source with the standard Ru(bpy)3Cl2 catalyst. NMR analysis of the resulting mixed ketal 16 was complicated by the presence of multiple diastereomers, so it was immediately hydrolyzed by refluxing in aqueous HCl to provide (±)-tetrabenazine (1) in 33% yield over two steps (5:1 dr). Although this represented a modest improvement in yield compared to the oxidative cyclization of 10 in MeOH, this sequence remained relatively inefficient due to incomplete conversion after 3 d and the need for an additional step to hydrolyze the ketal. Once again, CH3CN/H2O was a superior solvent system providing complete conversion of 15 to (±)-tetrabenazine after only 16 h.66 It is interesting to note that both solvent systems ultimately provided 1 with identical diastereoselectivity. Unfortunately, we were unable to separate these diastereomers and thus could not investigate whether this is a kinetic or thermodynamic ratio. Finally, tetrabenazine (1) was reduced to dihydrotetrabenazine (2) by NaBH4 reduction. The diastereoselectivity for this reduction was consistent with Rishel’s results,18 where 2 was formed in 73% yield as a 95:5 mixture, favoring the shown α-diastereomer.
In summary, (±)-tetrabenazine was prepared in six steps from commercially available materials. Application of the Baylis-Hillman and aza-Michael reactions enabled the rapid preparation of the tetrabenazine framework. Key observations from photoredox cyclization included the identification of silyl enol ether as the optimal nucleophile, and the critical role of solvent. Whereas cyclization of 15 in methanol led to the formation of mixed ketals, the desired ketone was obtained directly by changing the solvent to acetonitrile/water, which also improved the reaction rate. This work demonstrates the viability of photoredox catalysis as a key step for the synthesis of polycyclic alkaloids.
EXPERIMENTAL SECTION
4-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butan-2-one (6a)
Methyl vinyl ketone (1 mL, 11.0 mmol) was added to a solution of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride (1.32 g, 5.74 mmol) in 2 M aqueous NaOH (3.5 mL). After stirring rapidly for 25 min, the reaction was diluted with water (20 mL), and extracted with ether (3 x 15 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure to provide ketone 6a (5.36 g, 93%) as light tan syrup. This material was used immediately without further purification. 1H NMR (CDCl3) δ 6.59 (1H, s), 6.52 (1H, s), 3.845 (3H, s), 3.838 (3H, s), 3.56 (2H, br s), 2.85-2.80 (4H, comp), 2.75-2.71 (4H, comp), 2.21 (3H, s); 13C NMR (CDCl3) δ 207.9 (C), 147.5 (C), 147.2 (C), 126.1 (C), 125.9 (C), 111.2 (CH), 109.3 (CH), 55.9 (CH3), 55.8 (CH3), 55.6 (CH2), 52.4 (CH2), 51.0 (CH2), 41.7 (CH2), 30.2 (CH3), 28.5 (CH2); IR (film) 2912, 2833, 1709, 1516, 1226, 1129, 749 cm−1; HRMS (CI) m/z 264.1604 [C15H22NO3 (M+1) requires 264.1600].
6,7-Dimethoxy-2-(3-((triisopropylsilyl)oxy)but-3-en-1-yl)-1,2,3,4-tetrahydroisoquinoline (10)
Triethylamine (1 mL, 7.17 mmol) and TIPSOTf (1 mL, 3.72 mmol) were added sequentially to a room temperature solution of ketone 6a (648.9 mg, 2.46 mmol) in CH2Cl2 (15 mL). After 1 h the reaction mixture was treated with 2 M aqueous NaOH (50 mL). The resulting layers were separated and the aqueous phase was extracted with CH2Cl2 (3 x 50 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (alumina), eluting with EtOAc/hexanes (10:90, then 15:85) to provide TIPS enol ether 10 (723.6 mg, 70%) as a light yellow oil. 1H NMR (CDCl3) δ 6.59 (1H, s), 6.52 (1H, s), 4.06 (2H, br s), 3.85 (3H, s), 3.84 (3H, s), 3.60 (2H, s), 2.83 (2H, t, J = 5.5 Hz), 2.77-2.73 (4H, comp), 2.38 (2H, AA’XX’), 1.22 (2H, m), 1.10 (18H, d, J = 7.0 Hz), 1.07 (1H, m); 13C NMR (CDCl3) δ 158.0 (C), 147.4 (C), 147.1 (C), 126.6 (C), 126.1 (C), 111.3 (CH), 109.4 (CH), 89.6 (CH2), 56.2 (CH2), 55.9 (CH3 x 2), 55.6 (CH2), 51.1 (CH2), 34.9 (CH2), 28.7 (CH2), 18.0 (CH3 x 6), 12.6 (CH x 3); IR (film) 2944, 2866, 1517, 1216, 748 cm−1; HRMS (CI) m/z 420.2927 [C24H42NO3Si (M+1) requires 420.2934].
(±)-9,10-Dimethoxy-3,4,6,7-tetrahydro-1H-pyrido[2,1-a]isoquinolin-2(11bH)-one (9)
A 20 mL vial was charged with a solution of TIPS enol ether 10 (270 mg, 0.643 mmol) and Ru(bpy)3Cl2 (5.6 mg, 0.0075 mmol) in MeOH (7.6 mL). The vial was sealed with a rubber septa, opened to the air with an 18 gauge needle, and was irradiated using a blue LED strip for 4 d at 45 °C. Although incomplete by TLC, solvent was removed under reduced pressure and the residue was treated with 1 M aqueous HCl (4 mL), MeOH (1 mL) and was refluxed for 1.5 h. The reaction mixture was cooled and treated with 2 M aqueous NaOH (10 mL). The resulting basic solution was extracted with CH2Cl2 (3 x 10 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel), eluting with MeOH/CH2Cl2 (5:95) to provide 9 (47 mg, 28%) as a light yellow, waxy, solid. mp 153–154 °C; 1H NMR (CDCl3) δ 6.63 (1H, s), 6.54 (1H, s), 3.86 (3H, s), 3.84 (3H, s), 3.52 (1H, dd, J = 12.4, 2.3 Hz), 3.29 (1H, m), 3.16-3.08 (2H, comp), 2.91 (1H, app dt, J = 14.7, 2.8 Hz), 2.75-2.69 (3H, comp), 2.61 (1H, app td, J = 12.8, 6.4 Hz), 2.49 (1H, dd, J = 14.2, 12.0 Hz), 2.44 (1H, app dt, J = 11.5, 2.3 Hz); 13C NMR (CDCl3) δ 208.8 (C), 147.7 (C), 147.4 (C), 128.4 (C), 126.1 (C), 111.3 (CH), 107.6 (CH), 61.5 (CH3), 55.9 (CH3), 55.8 (CH), 54.7 (CH2), 50.8 (CH2), 47.6 (CH2), 41.1 (CH2), 29.3 (CH2); IR (AT-IR) 1708, 1692 cm−1; HRMS (CI) m/z 262.1437 [C15H20NO3 (M+1) requires 262.1443].
One-Step Preparation of (±)-9,10-Dimethoxy-3,4,6,7-tetrahydro-1H-pyrido[2,1-a]isoquinolin-2(11bH)-one (9)
A 20 mL vial was charged with a solution of TIPS enol ether 10 (205 mg, 0.49 mmol) and Ru(bpy)3Cl2 (7.8 mg, 0.0104 mmol) in CH3CN (4 mL) and water (0.4 mL). The vial was sealed with a rubber septa, opened to the air with an 18 gauge needle, and was irradiated using a blue LED strip for 2 d. CH2Cl2 (10 mL) and 10% w/w aqueous NaOH (10 mL) were added to the reaction mixture. The layers were separated and the aqueous phase was extracted with CH2Cl2 (4 x 10 mL). The combined organic extracts were dried (Na2SO4) and concentrated under reduced pressure. Filtration through a plug of alumina, trituration with hexanes, provided 9 as an off-white solid. Characterization data was identical for 9 as prepared above through the two-step procedure.
2-Methyl-4-methylene-5-oxohexan-3-yl acetate (12)72
A mixture of methyl vinyl ketone (6 mL, 74 mmol), isobutyraldehyde (8.5 mL, 93 mmol), 1,4-diazbicyclo[2.2.2]octane (1.64 g, 14.6 mmol), and 2,6-di-tert-butyl-4-methylphenol (122.4 mg, 0.55 mmol) was stirred overnight. The reaction mixture was diluted with ether (200 mL) and washed with 2 M aqueous HCl (2 x 50 mL), followed by brine (50 ml). The organic phase was dried (Na2SO4) and concentrated under reduced pressure to give the Baylis-Hillman adduct as an orange-colored oil (8.18 g). This oil was dissolved in CH2Cl2 (150 mL) and treated, in sequence, with pyridine (3 mL, 37.4 mmol) and acetyl chloride (2.4 mL). Once complete, as monitored by GC-MS, the reaction mixture was diluted with CH2Cl2 (100 mL) and washed with 2 M aqueous HCl (100 mL x 3), followed by 2 M aqueous NaOH (100 mL). The organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (silica), eluting with EtOAc/hexanes (15:85 then 2:8) to give enone 12 (5 g, 46%) as a clear, colorless, oil. 1H NMR (CDCl3) δ 6.15 (1H, s), 5.88 (1H, d, J = 0.9 Hz), 5.51 (1H, dd, J = 5.5, 0.9 Hz), 2.35 (3H, s), 2.08 (3H, s), 1.96 (1H, m), 0.9 (3H, d, J = 6.9 Hz), 0.86 (3H, d, J = 6.9 Hz); 13C NMR (CDCl3) δ 197.9 (C), 170.0 (C), 147.5 (C), 125.4 (CH2), 75.3 (CH), 31.3 (CH3), 26.1 (CH3), 21.0 (CH), 19.0 (CH3), 16.9 (CH3); IR (film) 2967, 2876, 1735, 1677, 1367, 1230, 1023 cm−1; HRMS (CI) m/z 185.1179 [C10H17O3 (M+1) requires 185.1178].
3-((6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)methyl)-5-methylhex-3-en-2-one (14)
6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride (7) (2.03 g, 8.84 mmol) and enone 12 (1.93 g, 10.47 mmol) were suspended in 2 M aqueous NaOH (6.5 mL) and stirred at ambient temperature for 1 h. The reaction mixture was treated with aqueous 2 M HCl (125 mL) and then washed with ether (75 mL x 2). The pH of the aqueous layer was adjusted to 11 by addition of 2 M aqueous NaOH (150 mL), and was then extracted with CH2Cl2 (3 x 80 mL). The combined organic layers were washed with brine (75 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (silica), eluting with MeOH/CH2Cl2 (1:9) to give enone 14 (2.336 g, 83%) as a clear, colorless, oil (6:1 mixture of E:Z isomers). Characterization is provided for the major isomer. 1H NMR (CDCl3) δ 6.57 (1H, s), 6.55 (1H, d, J = 10.0 Hz), 6.49 (1H, s), 3.84 (3H, s), 3.83 (3H, s), 3.52 (2H, s), 3.40 (2H, s), 2.88 (1H, dsept, J = 10.0, 6.4 Hz), 2.75 (2H, t, J = 5.5 Hz), 2.66 (2H, t, J = 5.5 Hz), 2.37 (3H, s), 1.07 (6H, d, J = 6.4 Hz); 13C NMR (CDCl3) δ 200.4 (C), 152.5 (CH), 147.3 (C), 147.0 (C), 135.6 (C), 126.7 (C), 126.2 (C), 111.2 (CH), 109.3 (CH), 55.8 (CH3 x 2), 55.4 (CH2), 51.9 (CH2), 50.3 (CH2), 28.7 (CH2), 28.1 (CH), 26.4 (CH3), 22.3 (CH3 x 2); IR (film) 2958, 2868, 2835, 1665, 1517, 1254, 1226, 1126, 909, 727 cm−1; HRMS (CI) m/z 318.2066 [C19H28NO3 (M+1) requires 318.2069],
(±)-3-((6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)methyl)-5-methylhexan-2-one (6b)
A solution of enone 14 (2.47 g, 7.78 mmol) and trifluoroacetic acid (1.19 mL, 15.57 mmol) in MeOH (70 mL) was added to a flask containing 10% w/w Pd/C (0.33 g). Hydrogen was bubbled through the resulting suspension for 20 min. After completion by TLC the reaction mixture was filtered through Celite, treated with 2 M aqueous NaOH (100 mL), and the extracted with CH2Cl2 (3 x 100 mL). The combined organic extractions were dried (Na2SO4) and concentrated under reduced pressure. An analytically pure sample was obtained by flash chromatography (silica), eluting with MeOH/CH2Cl2 (1:9) to give ketone 6b (1.96 g, 79%) as a light yellow solid. mp 62–65.5 °C; 1H NMR (CDCl3) δ 6.56 (1H, s), 6.50 (1H, s), 3.82 (3H, s), 3.81 (3H, s), 3.60 (1H, d, J = 14.6 Hz), 3.46 (1H, d, J = 14.6 Hz), 2.93 (1H, m), 2.82-2.72 (4H, comp), 2.57 (1H, ddd, J = 10.5, 6.9, 5.3 Hz), 2.44 (1H, dd, J = 11.5, 5.7 Hz), 2.15 (3H, s), 1.57-1.47 (2H, comp), 1.23 (1H, m), 0.91 (3H, d, J = 6.0 Hz), 0.88 (3H, d, J = 6.0 Hz); 13C NMR (CDCl3) δ 212.4 (C), 147.3 (C), 147.0 (C), 126.4 (C), 126.1 (C), 111.2 (CH), 109.2 (CH), 60.7 (CH2), 55.9 (CH2), 55.8 (CH3 x 2), 50.9 (CH2), 49.0 (CH), 39.3 (CH2), 28.8 (CH3), 28.6 (CH2), 26.1 (CH), 23.0 (CH3), 22.2 (CH3); IR (film) 2954, 2869, 2836, 1709, 1517, 725 cm−1; HRMS (CI) m/z 320.2226 [C19H30NO3 (M+1) requires 320.2226].
(±)-6,7-Dimethoxy-2-(4-methyl-2-(1-((triisopropylsilyl)oxy)vinyl)pentyl)-1,2,3,4-tetrahydroisoquinoline (15)
Ketone 6a (510 mg, 1.60 mmol) was dissolved in CH2Cl2 (11 mL), followed by the sequential addition of Et3N (0.7 mL, 5.02 mmol), and TIPSOTf (0.65 mL, 2.42 mmol). After 30 min, the reaction mixture was diluted with CH2Cl2 (50 mL) and 2 M aqueous NaOH (50 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 x 50 mL). All organic extractions were combined, then dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (alumina), eluting with EtOAc/hexanes (1:9) to give TIPS enol ether 15 (610 mg, 80%) as a clear, colorless, oil. 1H NMR (CDCl3) δ 6.58 (1H, s), 6.52 (1H, s), 4.00 (1H, br s), 3.98 (1H, br s), 3.84 (3H, s), 3.83 (3H, s), 3.54 (2H, ABq, JAB = 14.6 Hz, Δδ = 0.06 ppm); 2.80-2.72 (3H, comp), 2.65-2.58 (2H, comp), 2.48-2.39 (2H, comp), 1.69 (1H, m), 1.49 (1H, ddd, J = 13.5, 10.5, 3.9 Hz), 1.27-1.18 (3H, comp), 1.09 (18H, d, J = 6.9 Hz), 1.06 (1H, m), 0.9 (6H, app d, J = 6.9 Hz); 13C NMR (CDCl3) δ 160.0 (C), 147.2 (C), 147.0 (C), 127.2 (C), 126.5 (C), 111.2 (CH), 109.3 (CH), 88.9 (CH2), 61.8 (CH2), 56.0 (CH2), 55.9 (CH3 x 2), 51.2 (CH2), 42.6 (CH), 39.8 (CH2), 28.8 (CH2), 25.4 (CH), 23.8 (CH3), 21.6 (CH3), 18.1 (CH3 x 6), 12.8 (CH x 3); IR (film) 2944, 2865, 1612 (weak), 1517, 1256, 1228, 1016, 753, 680 cm−1; HRMS (CI) m/z 474.3399 [C28H48NO3Si (M-1) requires 474.3403].
(±)-3-Isobutyl-9,10-dimethoxy-3,4,6,7-tetrahydro-1H-pyrido[2,1-a]isoquinolin-2(11bH)-one [(±)-tetrabenazine, TBZ, (±)-1]
A 25 mL round bottom flask was charged with TIPS enol ether 15 (240.4 mg, 0.505 mmol), Ru(bpy)3Cl2 (7 mg, 0.0093 mmol), and CH3CN/H2O (10:1, 4.4 mL). The flask was equipped with a magnetic stir bar and septa, which was pierced with an 18 gauge needle, and irradiated with the light produced from a 8.5 W blue LED strip for 16 h at 45 °C. CH2Cl2 (10 mL) and 2 M aqueous NaOH (5 mL) were added to the reaction mixture. The layers were separated and the aqueous phase was extracted with CH2Cl2 (4 x 10 mL). The combined organic extracts were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (alumina), eluting with EtOAc/hexanes (15:85) to give (±)-tetrabenazine (1) (89.2 mg, 56%) as a white crystalline solid (5:1 dr). mp 108–110 °C; Characterization is provided for the major diastereomer. 1H NMR (CDCl3) δ 6.62 (1H, s), 6.55 (1H, s), 3.86 (3H, s), 3.83 (3H, s), 3.51 (1H, br d, J = 10.5 Hz), 3.29 (1H, dd, J = 11.5, 6.4 Hz), 3.18-3.07 (2H, comp), 2.90 (1H, dd, J = 13.7, 3.0 Hz), 2.78-2.72 (2H, comp), 2.64-2.51 (2H, comp), 2.36 (1H, t, J = 11.5 Hz), 1.80 (1H, ddd, J = 14.0, 8.7, 5.5 Hz), 1.67 (1H, comp), 1.04 (1H, ddd, J = 13.7, 7.3, 5.9 Hz), 0.92 (3H, d, J = 5.0 Hz), 0.91 (3H, d, J = 5.0 Hz); 13C NMR (CDCl3) δ 210.2 (C), 147.7 (C), 147.4 (C), 128.4 (C), 126.0 (C), 111.3 (CH), 107.7 (CH), 62.4 (CH), 61.4 (CH2), 55.9 (CH3), 55.8 (CH3), 50.5 (CH2), 47.6 (CH2), 47.5 (CH), 35.0 (CH2), 29.3 (CH2), 25.3 (CH), 23.2 (CH3), 22.0 (CH3); IR (AT-IR) 2920, 2862, 1699, 1515, 1463, 1257, 882 cm−1; HRMS (CI) m/z 318.2073 [C19H28NO3 (M+1) requires 318.2069].
(±)-3-Isobutyl-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-2-ol [α-dihydrotetrabenazine, DTBZ, (±)-2]
A solution of (±)-1 (101.7 mg, 0.32 mmol) in EtOH (3 mL) was cooled to 0 °C and treated with NaBH4 (42 mg, 1.11 mmol). After 1 h, 2 M aqueous NaOH (15 mL) was added, and the resulting mixture was extracted with CH2Cl2 (4 x 10 mL). The combined organic extracts were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel), eluting with MeOH/CH2Cl2 (10:250) to give α-dihydrotetrabenazine (±)-2 (70.2 mg, 70%) as a white solid and 4.2 mg (4%) of the 2-epi-dihydrotetrabenazine. α-dihydrotetrabenazine (major diastereomer): 1H NMR (CDCl3) δ 6.68 (1H, s), 6.58 (1H, s), 5.30 (1H, s), 3.85-3.84 (6H, comp), 3.40 (1H, app td, J = 10.5, 4.6 Hz), 3.13 (1H, br d, J = 11.9 Hz), 3.11-2.98 (3H, comp), 2.64 (1H, br dd, J = 16.9, 4.1 Hz), 2.59 (1H, ddd, J = 12.4, 4.6, 2.3 Hz), 2.46 (1H, app td, J = 11.5, 4.1 Hz), 1.98 (1H, app t, J = 11.5 Hz), 1.77-1.65 (2H, comp), 1.58 (1H, ddd, J = 13.3, 10.0, 3.2 Hz), 1.49 (1H, app q, J = 11.5 Hz), 1.07(1H, ddd, J = 13.7, 10.0, 4.1 Hz), 0.94 (3H, d, J = 6.7 Hz), 0.92 (3H, d, J = 6.7 Hz); 13C NMR (CDCl3) δ 147.4 (C), 147.1 (C), 129.2 (C), 126.3 (C), 111.4 (CH), 107.8 (CH), 74.6 (CH), 60.9 (CH), 60.0 (CH2), 55.9 (CH3), 55.8 (CH3), 51.9 (CH2), 41.6 (CH), 40.5 (CH2), 39.6 (CH2), 29.1 (CH2), 25.3 (CH), 24.2 (CH3), 21.7 (CH3); IR (AT-IR) 3381, 1513, 1261, 1212, 1008, 767 cm−1; HRMS (CI) m/z 320.2219 [C19H30NO3 (M+1) requires 320.2226]. 2-epi-dihydrotetrabenazine (minor diastereomer): 1H NMR (CDCl3) δ 6.66 (1H, s), 6.58 (1H, s), 4.09 (1H, br d, J = 2.3 Hz), 3.844 (3H, s), 3.841 (3H, s), 3.54 (1H, br d, J = 9.2 Hz), 3.14 (1H, m), 3.00 (1H, m), 2.73-2.56 (3H, comp), 2.46 (1H, m), 2.44 (1H, app dt, J = 13.7, 2.8 Hz), 2.02 (1H, m), 1.76-1.64 (2H, comp), 1.31-1.24 (2H, comp), 1.20-1.13 (1H, m), 0.94 (3H, d, J = 7.0 Hz), 0.92 (3H, d, J = 7.0 Hz); 13C NMR (CDCl3) δ 147.4 (C), 147.1 (C), 126.7 (C), 111.4 (CH), 107.9 (CH), 67.9 (CH), 56.4 (CH), 56.2 (CH2), 55.9 (CH3), 55.8 (CH3), 52.4 (CH2), 39.0 (CH2), 38.8 (CH2), 37.6 (CH), 29.0 (CH2), 24.8 (CH), 22.9 (CH3 x 2). This data was consistent with previously reported data.18,26
Supplementary Material
Acknowledgments
This publication was made possible by the Arkansas INBRE program, supported by grant funding from the National Institutes of Health (NIH), National Institute of General Medical Sciences (NIGMS) (P20 GM103429)(formerly P20RR016460). L.R.O was supported in part by a grant from the Research Corporation for Science Advancement (ID: 21087). Hendrix College’s NMR facilities are supported by the National Science Foundation under grant No. 1040470.
Footnotes
General experimental procedures, 1H and 13C NMR spectra, and comparisons of the NMR data for (±)−1 and (±)−2 with literature data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pletscher A. Science. 1957;126:507–507. doi: 10.1126/science.126.3272.507. [DOI] [PubMed] [Google Scholar]
- 2.Brossi A, Chopard-dit-Jean LH, Schnider O. Helv Chim Acta. 1958;41:1793–1806. [Google Scholar]
- 3.Hayden MR, Leavitt BR, Yasothan U, Kirkpatrick P. Nat Rev Drug Discov. 2009;8:17–18. doi: 10.1038/nrd2784. [DOI] [PubMed] [Google Scholar]
- 4.Frank S, Jankovic J. Drugs. 2010;70:561–571. doi: 10.2165/11534430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Müller T. Expert Opin Invest Drugs. 2015;24:737–742. doi: 10.1517/13543784.2015.1029573. [DOI] [PubMed] [Google Scholar]
- 6.Zheng G, Dwoskin LP, Crooks PA. AAPS J. 2006;8:E682–E692. doi: 10.1208/aapsj080478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ugolev Y, Segal T, Yaffe D, Gros Y, Schuldiner S. J Biol Chem. 2013;288:32160–32171. doi: 10.1074/jbc.M113.502971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein AI, Stout KA, Miller GW. Neurochem Int. 2014;73:89–97. doi: 10.1016/j.neuint.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A, Lo ST, Öz OK, Sun X. Bioorg Med Chem Lett. 2014;24:5663–5665. doi: 10.1016/j.bmcl.2014.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin KJ, Weng YH, Hsieh CJ, Lin WY, Wey SP, Kung MP, Yen TC, Lu CS, Hsiao IT. PLoS ONE. 2013;8:e75952. doi: 10.1371/journal.pone.0075952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu L, Liu J, Kung HF. Bioorg Med Chem Lett. 2009;19:5026–5028. doi: 10.1016/j.bmcl.2009.07.048. [DOI] [PubMed] [Google Scholar]
- 12.Koeppe RA, Gilman S, Joshi A, Liu S, Little R, Junck L, Heumann M, Frey KA, Albin RL. J Nucl Med. 2005;46:936–944. [PubMed] [Google Scholar]
- 13.Zubieta JK, Taylor SF, Huguelet P, Koeppe RA, Kilbourn MR, Frey KA. Biol Psychiatry. 2001;49:110–116. doi: 10.1016/s0006-3223(00)00981-1. [DOI] [PubMed] [Google Scholar]
- 14.Kilbourn MR. Nuc Med Biol. 1997;24:615–619. doi: 10.1016/s0969-8051(97)00101-7. [DOI] [PubMed] [Google Scholar]
- 15.Kilbourn M, Lee L, Borght TV, Jewett D, Frey K. Eur J Pharmacol. 1995;278:249–252. doi: 10.1016/0014-2999(95)00162-e. [DOI] [PubMed] [Google Scholar]
- 16.Podurgiel SJ, Nunes EJ, Yohn SE, Barber J, Thompson A, Milligan M, Lee CA, López-Cruz L, Pardo M, Valverde O, Lendent C, Baqi Y, Müller CE, Correa M, Salamone JD. Neuroscience. 2013;250:507–519. doi: 10.1016/j.neuroscience.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Boldt KG, Biggers MS, Phifer SS, Brine GA, Rehder KS. Synth Commun. 2009;39:3574–3585. [Google Scholar]
- 18.Rishel MJ, Amarasinghe KKD, Dinn SR, Johnson BF. J Org Chem. 2009;74:4001–4004. doi: 10.1021/jo900480n. [DOI] [PubMed] [Google Scholar]
- 19.Paek SM, Kim NJ, Shin D, Jung JK, Jung JW, Chang DJ, Moon H, Suh YG. Chem—Eur J. 2010;16:4623–4628. doi: 10.1002/chem.200902591. [DOI] [PubMed] [Google Scholar]
- 20.Yu QS, Luo W, Deschamps J, Holloway HW, Kopajtic T, Katz JL, Brossi A, Greig NH. ACS Med Chem Lett. 2010;1:105–109. doi: 10.1021/ml1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Son YW, Kwon TH, Lee JK, Pae AN, Lee JY, Cho YS, Min SJ. Org Lett. 2011;13:6500–6503. doi: 10.1021/ol202792q. [DOI] [PubMed] [Google Scholar]
- 22.Johannes M, Altmann KH. Org Lett. 2012;14:3752–3755. doi: 10.1021/ol301612q. [DOI] [PubMed] [Google Scholar]
- 23.Reddy NSS, Reddy AS, Yadav JS, Reddy BVS. Tetrahedron Lett. 2012;53:6916–6918. [Google Scholar]
- 24.Lee LC, Vander Borght T, Sherman PS, Frey KA, Kilbourn MR. J Med Chem. 1996;39:191–196. doi: 10.1021/jm950117b. [DOI] [PubMed] [Google Scholar]
- 25.Boldt KG, Brine GA, Rehder K. Org Prep Proc Int. 2008;40:379–384. [Google Scholar]
- 26.Yao Z, Wei X, Wu X, Katz JL, Kopajtic T, Greig NH, Sun H. Eur J Med Chem. 2011;46:1841–1848. doi: 10.1016/j.ejmech.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng P, Lieberman BP, Choi SR, Plöessl K, Kung HF. Bioorg Med Chem Lett. 2011;21:3435–3438. doi: 10.1016/j.bmcl.2011.03.113. [DOI] [PubMed] [Google Scholar]
- 28.Burns NZ, Baran PS, Hoffmann RW. Angew Chem, Int Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 29.Bäckvall J-E, editor. Modern Oxidation Methods. 2. Wiley-VCH; Weinheim: 2011. [Google Scholar]
- 30.Boess E, Schmitz C, Klussmann M. J Am Chem Soc. 2012;134:5317–5325. doi: 10.1021/ja211697s. [DOI] [PubMed] [Google Scholar]
- 31.Sureshkumar D, Sud A, Klussmann M. Synlett. 2009;2009:1558–1561. [Google Scholar]
- 32.Shen Y, Tan Z, Chen D, Feng X, Li M, Guo CC, Zhu C. Tetrahedron. 2009;65:158–163. [Google Scholar]
- 33.Shen Y, Li M, Wang S, Zhan T, Tan Z, Guo C-C. Chem Commun. 2009:953–955. doi: 10.1039/b819657e. [DOI] [PubMed] [Google Scholar]
- 34.Zeng T, Song G, Moores A, Li CJ. Synlett. 2010;2010:2002–2008. [Google Scholar]
- 35.Ratnikov MO, Xu X, Doyle MP. J Am Chem Soc. 2013;135:9475–9479. doi: 10.1021/ja402479r. [DOI] [PubMed] [Google Scholar]
- 36.Huo C, Wu M, Jia X, Xie H, Yuan Y, Tang J. J Org Chem. 2014;79:9860–9864. doi: 10.1021/jo5017822. [DOI] [PubMed] [Google Scholar]
- 37.Dhineshkumar J, Lamani M, Alagiri K, Prabhu KR. Org Lett. 2013;15:1092–1095. doi: 10.1021/ol4001153. [DOI] [PubMed] [Google Scholar]
- 38.Nobuta T, Fujiya A, Yamaguchi T, Tada N, Miura T, Itoh A. RSC Adv. 2013;3:10189–10192. [Google Scholar]
- 39.Tanoue A, Yoo WJ, Kobayashi S. Org Lett. 2014;16:2346–2349. doi: 10.1021/ol500661t. [DOI] [PubMed] [Google Scholar]
- 40.Beatty JW, Stephenson CRJ. Acc Chem Res. 2015;48:1474–1484. doi: 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schultz DM, Yoon TP. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jahn E, Jahn U. Angew Chem, Int Ed. 2014;53:13326–13328. doi: 10.1002/anie.201408748. [DOI] [PubMed] [Google Scholar]
- 43.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi L, Xia W. Chem Soc Rev. 2012;41:7687–7697. doi: 10.1039/c2cs35203f. [DOI] [PubMed] [Google Scholar]
- 45.Xuan J, Xiao WJ. Angew Chem, Int Ed. 2012;51:6828–6838. doi: 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]
- 46.Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- 47.Condie AG, Gonzalez-Gomez JC, Stephenson CRJ. J Am Chem Soc. 2010;132:1464–1465. doi: 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]
- 48.Rueping M, Zhu S, Koenigs RM. Chem Commun. 2011;47:12709–12711. doi: 10.1039/c1cc15643h. [DOI] [PubMed] [Google Scholar]
- 49.Hari DP, Koenig B. Org Lett. 2011;13:3852–3855. doi: 10.1021/ol201376v. [DOI] [PubMed] [Google Scholar]
- 50.Xuan J, Feng ZJ, Duan SW, Xiao WJ. RSC Adv. 2012;2:4065–4068. [Google Scholar]
- 51.Rueping M, Koenigs RM, Poscharny K, Fabry DC, Leonori D, Vila C. Chem—Eur J. 2012;18:5170–5174. doi: 10.1002/chem.201200050. [DOI] [PubMed] [Google Scholar]
- 52.DiRocco DA, Rovis T. J Am Chem Soc. 2012;134:8094–8097. doi: 10.1021/ja3030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nie SZ, Sun X, Wei WT, Zhang XJ, Yan M, Xiao JL. Org Lett. 2013;15:2394–2397. doi: 10.1021/ol4008469. [DOI] [PubMed] [Google Scholar]
- 54.Mathis CL, Gist BM, Frederickson CK, Midkiff KM, Marvin CC. Tetrahedron Lett. 2013;54:2101–2104. [Google Scholar]
- 55.Miller LL, Stermitz FR, Becker JY, Ramachandran V. J Am Chem Soc. 1975;97:2922–2923. [Google Scholar]
- 56.Baslé O, Borduas N, Dubois P, Chapuzet JM, Chan TH, Lessard J, Li CJ. Chem—Eur J. 2010;16:8162–8166. doi: 10.1002/chem.201000240. [DOI] [PubMed] [Google Scholar]
- 57.Chow PK, To WP, Low KH, Che CM. Chem—Asian J. 2014;9:534–545. doi: 10.1002/asia.201301059. [DOI] [PubMed] [Google Scholar]
- 58.Jones KM, Klussmann M. Synlett. 2012;2012:159–162. [Google Scholar]
- 59.Freeman DB, Furst L, Condie AG, Stephenson CRJ. Org Lett. 2012;14:94–97. doi: 10.1021/ol202883v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao G, Yang C, Guo L, Sun H, Chen C, Xia W. Chem Commun. 2012;48:2337–2339. doi: 10.1039/c2cc17130a. [DOI] [PubMed] [Google Scholar]
- 61.Rueping M, Vila C, Koenigs RM, Poscharny K, Fabry DC. Chem Commun. 2011;47:2360–2362. doi: 10.1039/c0cc04539j. [DOI] [PubMed] [Google Scholar]
- 62.Pan Y, Kee CW, Chen L, Tan CH. Green Chem. 2011;13:2682–2685. [Google Scholar]
- 63.Kohls P, Jadhav D, Pandey G, Reiser O. Org Lett. 2012;14:672–675. doi: 10.1021/ol202857t. [DOI] [PubMed] [Google Scholar]
- 64.Perepichka I, Kundu S, Hearne Z, Li CJ. Org Biomol Chem. 2015;13:447–451. doi: 10.1039/c4ob02138j. [DOI] [PubMed] [Google Scholar]
- 65.Rusch F, Unkel LN, Alpers D, Hoffmann F, Brasholz M. Chem—Eur J. 2015;21:8336–8340. doi: 10.1002/chem.201500612. [DOI] [PubMed] [Google Scholar]
- 66.Andrews RS, Becker JJ, Gagné MR. Org Lett. 2011;13:2406–2409. doi: 10.1021/ol200644w. [DOI] [PubMed] [Google Scholar]
- 67.Bouzide A. Org Lett. 2002;4:1347–1350. doi: 10.1021/ol020032m. [DOI] [PubMed] [Google Scholar]
- 68.Baker BA, Bošković ŽV, Lipshutz BH. Org Lett. 2007;10:289–292. doi: 10.1021/ol702689v. [DOI] [PubMed] [Google Scholar]
- 69.Pelšs A, Kumpulainen ETT, Koskinen AMP. J Org Chem. 2009;74:7598–7601. doi: 10.1021/jo9017588. [DOI] [PubMed] [Google Scholar]
- 70.Ashby EC, Lin JJ. Tetrahedron Lett. 1975;16:4453–4456. [Google Scholar]
- 71.Ashby EC, Lin JJ, Kovar R. J Org Chem. 1976;41:1939–1943. [Google Scholar]
- 72.Leonard WR, Livinghouse T. J Org Chem. 1985;50:730–732. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.