

Abstract
Hepatitis C virus (HCV) poses a major health threat to the world. The recent development of direct-acting antivirals (DAAs) against HCV has markedly improved the response rate of HCV and reduced the side effects in comparison to the interferon-based therapy. Despite this therapeutic advance, there is still a need to develop new inhibitors that target different stages of the HCV life cycle because of various limitations of the current regimens. In this study, we performed a quantitative high-throughput screening of the Molecular Libraries Small Molecule Repository (MLSMR) of ~ 350,000 chemicals for novel HCV inhibitors using our previously developed cell-based HCV infection assay. Following confirmation and structural clustering analysis, we narrowed down to 158 compounds from the initial ~ 3,000 molecules that showed inhibitory activity for further structural and functional analyses. We were able to assign the majority of these compounds to specific stage(s) in the HCV life cycle. Three of them are direct inhibitors of NS3/4A protease. Most of the compounds appear to act on novel targets in HCV life cycle. Four compounds with novel structure and excellent drug-like properties, three targeting HCV entry and one targeting HCV assembly/secretion, were advanced for further development as lead hits. These compounds represent diverse chemotypes that are potential lead compounds for further optimization and may offer promising candidates for the development of novel therapeutics against HCV infection. In addition, they represent novel molecular probes to explore the complex interactions between HCV and the cells.
Keywords: antiviral, HCV inhibitors, high throughput screening, cell-based assay, viral life cycle
1. Introduction
Hepatitis C virus (HCV) infection affects more than 200 million people worldwide and poses a major health threat in the world (Liang et al., 2000). Persistent infection of HCV often leads to chronic liver diseases including cirrhosis with a risk of developing hepatocellular carcinoma. Since the introduction of interferon for clinical therapy of HCV in 1990, significant strides have been made in the treatment of HCV infection (Buti and Esteban, 2011). In recent years, development of direct-acting antivirals (DAAs) against HCV has yielded a multitude of new potent HCV inhibitors such as telaprevir, daclatasvir, simeprevir and sofosbuvir (Liang and Ghany, 2013). Combination of the new agents and the traditional HCV inhibitors has improved the cure rate from around 50% of the standard treatment (peginterferon and ribavirin) to about 90% in certain HCV genotypes and clinical conditions. By eliminating interferon, combination therapies with some of the new-generation DAAs have greatly reduced the side effects and improved efficacy (Liang and Ghany, 2014). Despite these encouraging progresses, various limitations still exist. Most DAAs in clinical use or clinical trials target enzymatic functions of viral-encoded proteins, such as viral protease and polymerase. These agents all inhibit the same stage of HCV life cycle and are associated with rapid emergence of drug-resistant viral mutations, as observed during monotherapy with these DAAs (Schiffer et al., 2011). To minimize the occurrence of drug resistance and achieve maximal efficacy, combination with one or more drugs are usually needed (Liang and Ghany, 2013). Furthermore, different genotypes of HCV may present with different profiles of sensitivity to the inhibitors. HCV infection associated with various clinical complications may also require adjustment of treatment regimen. Therefore, new HCV inhibitors that target different stages of the HCV life cycle, such as entry and assembly, may be needed to overcome these limitations. Targeting multiple key steps in the viral life cycle not only improves antiviral efficacy but also decreases the chance of developing drug resistance (Sarrazin and Zeuzem, 2010).
Various cell-based systems have been developed to screen anti-HCV molecules. The HCV replicon system (Kim et al., 2007; Lohmann et al., 1999), which is based on the subgenomic HCV RNA consisting of the nonstructural genes of the HCV, retains the replication capability in cell culture. On the other hand, HCV pseudoviral particles (HCVpp) (Bartosch and Cosset, 2009; Hsu et al., 2003), which contains the HCV envelop proteins in the particle, can mimic HCV entry process via interaction with the cell surface HCV receptors. Both systems have been used for HCV drug screening and mechanisms of action study of HCV inhibitors (Hao et al., 2007; Lemm et al., 2010; Lupberger et al., 2011). However, these systems only involve specific steps of viral life cycle and cannot target other HCV infection steps which span from viral entry, trafficking, replication, assembly to virion secretion (Liang and Ghany, 2013).
Previously, we and others developed a robust cell culture system for infectious HCV (HCVcc) (Wakita et al., 2005). Reporter genes inserted into certain locations of the HCV genome could yield viable HCV capable of infection and replication in the cell culture (Koutsoudakis et al., 2006). In recent years, several groups have applied either wild-type HCV or modified HCV with reporter genes to develop cell-based HCV infection systems for the screening of HCV inhibitors (Chockalingam et al., 2010; Gastaminza et al., 2010; Kim et al., 2007; Wichroski et al., 2012; Yu et al., 2012). Cell-based HCV infection systems have the advantage of targeting various stages of HCV life cycle, thus significantly improving the efficiency of the drug screening process.
Our group has developed a novel cell-based HCV infection system that is highly sensitive and suitable for quantitative high-throughput screening (qHTS) assay (Hu et al., 2014). The system comprises two parts: HCV-Cre virus and Huh7.5.1.LoxPGluc reporter cell line [stably transfected with Gaussia luciferase (Gluc)]. The expression of Gluc is silenced by LoxP sequences under normal conditions in cell culture. Upon infection of the LoxPGluc cells with the HCV-Cre, Cre recombinase specifically recognizes the LoxP sequence and removes the intervening stop sequence (Sauer and Henderson, 1988). Gluc is then expressed and secreted into the medium. The luciferase activity of the HCV-Cre infected cell reflects the level of HCV infection and replication (Hu et al., 2014; Sauer and Henderson, 1988). In the current study, we screened a small molecule library of about 350,000 chemicals using the qHTS platform of the HCV-Cre/LoxPGluc infection system. Our goal was to identify novel HCV inhibitors targeting various stages encompassing the entire HCV life cycle. Several promising compounds with novel properties were advanced for further development as lead hits.
2. Materials and methods
2.1. Cells and viruses
Human hepatoma cells Huh7.5.1, Huh7.5.1LoxPGluc reporter cells and other Huh7 derived cells were maintained in DMEM medium (Life Technologies, Carlsbad, CA) with 10% FBS and antibiotics in 5% CO2 at 37 °C. Wild-type HCVcc (HCV-WT, J6/JFH1 clone), HCV-WT-derived viruses including HCV-Cre and HCV-Luc, pseudo-typed HCV virus (HCVpp), HCV single cycle virus (HCVsc) and other control viruses VSV-Gpp were produced according to protocols described in the literatures (Chang et al., 1999; He et al., 1995; Hsu et al., 2003; Hu et al., 2014).
2.2. Compound library preparation and primary qHTS assay
Compounds from the Molecular Libraries Small Molecule Repository (MLSMR) was dissolved in DMSO at 10 mM concentration (2010-). The compounds were serially diluted in DMSO at a 1:5 ratio to give 12 stock concentrations ranging from 0.205 nM to 10 mM and formatted in 1536-well plates as compound source plates. The primary qHTS screen was performed as described previously using a fully automated robotic screening system (Inglese et al., 2006). Briefly, Huh7.5.1LoxPGluc cells were plated in white 1536-well plates in 3 μL of DMEM medium (750 cells/well) and cultured overnight. Then the library compounds from the source plates were added to the cell plates at 23 nL/well followed by addition of 2.5 μL/well HCV-Cre-containing medium (at ~ 0.05 m.o.i.). The primary screen was conducted in five serial plates, one DMSO control and four concentrations (0.34, 1.7, 8.5, 42 μM) of compounds. Cyclosporin A (Sigma-Aldrich, St. Louis, MO) was used as a positive control. The plates were harvested for luciferase assay 48 h after treatment.
2.3. qHTS data analysis
Primary screen data and curve fitting were analyzed with a customized software developed internally (Wang et al., 2010). The maximal response (100% activity) was set to the response to 10 μM cyclosporin A and the basal response (0% activity) to the DMSO control treatment. Efficacy of a compound refers to the highest response standardized to the maximal response to cyclosporin A (defined above) and is derived from the concentration-response curve (CRC) where the inhibition reaches the plateau of maximal response. The EC50 (compound concentration at 50% efficacy) and CC50 (compound concentration at 50% cytotoxicity) values of compounds were calculated from the CRC by nonlinear regression analysis using Prism software (GraphPad Software, San Diego, CA). The pattern of the CRC was categorized into 4 curve classes: Class 1 curves are well-fit (r2 ≥ 0.9) and have both upper and lower asymptotes with either complete response (efficacy > 80%, sub-class 1.1) or partial response (30–80%, subclass 1.2); class 2 curves are incomplete with only one asymptote and efficacy either > 80% (curve class 2.1) or < 80% (curve class 2.2); class 3 curves display activity only at the highest tested concentration and efficacy > 30%; class 4 curves are with low (efficacy < 30%) or no response and deemed as inactive (Inglese et al., 2006). Compounds in curve class 1.1, 1.2, 2.1, 2.2 with maximal response > 50% and EC50 < 10 μM were considered potential hits and then grouped in clusters based on their structure and cross-activity. Compounds with best efficacy and potency in each cluster were selected for further validation assay.
2.4. Confirmation assays
The selected potential hits from the primary screen were retested in the HCV-Cre/LoxPGluc infection assay platform using a full 12-concentration setting (starting from 42 μM and 1:5 serial dilutions thereafter) in triplicate plates. A parallel ATPlite cytotoxicity assay was carried out with ATPlite assay kit (PerkinElmer, Waltham, MA) to exclude the cytotoxic compounds from the primary hit pools. The cells were cultured for 48 hr following the compound treatment before being processed for each assay.
2.5. HCV core immunofluorescence staining assay
Validated compounds were further confirmed in 2-part HCV-WT infection assay with HCV core immunofluorescence staining as described previously (Li et al., 2009). Huh7.5.1 cells were plated in 384-well plates (3,000 cells/well) and infected with HCV-WT together with the selected compounds in 1:3 serial dilutions starting from 30 μM (part-one). After 48 h, the media from part-one were transferred onto a new replica plate with Huh 7.5.1 cells (part-two). After 48 h infection, the cells were processed for staining with anti-HCV core 6G7 monoclonal antibody and an anti-mouse Alexa Fluor 488 secondary antibody (Life Technologies). The cell nuclei were visualized with Hoechst dye (Life Technologies) staining. Cells were imaged with a fluorescent cell imager and analyzed with Metamorph Cell Scoring software (Molecular Devices, LLC, Sunnyvale, CA).
2.6. HCV entry and replication assays
HCV entry was tested with HCVpp-1a infection. VSV-Gpp infection was used as a control for HCV specificity. HCV replication assay was performed by transient transfection of HCV replicon genotype 2a containing a luciferase reporter. Bafilomycin and cyclosporine A were used as positive controls for HCVpp and replicon assays, respectively. The assays were performed as described previously (Hu et al., 2014).
2.7. HCV single-cycle infection assay
HCV single-cycle infection assay was performed with the infection of a single round defective HCV particle (HCVsc). HCVsc was generated by a replicon trans-packaging system as described previously (Masaki et al., 2010). The HCVsc can infect and replicate efficiently but cannot assemble new virions. Thus it can help distinguish whether a compound inhibitor is working at the stage of viral entry/replication or assembly/secretion. Cyclosporin A was used as a positive control. Luciferase signal was measured 48 h after the compound treatment.
2.8. HCV NS3/NS4A protease and NS5B polymerase assays
HCV NS3/4A protease activity was examined by using an in vitro FRET based assay kit (ProteinOne, Rockville, MD) in 96-well plate format according to manufacturer’s protocol. Briefly, FRET peptide substrate containing NS3-dependent cleavage site Asp/GluXaa4Cys/Thr-Ser/Ala was incubated in 100 μL assay buffer together with 10 μM of HCV inhibitors and 100 ng of recombinant HCV GT1b protease in triplicates at room temperature for 1 h. The fluorescence intensity was measured using a fluorescence plate reader (BMG Labtech, Ortenberg, Germany) with excitation at 485 nm and emission at 520 nm. Telaprevir (10 μM) was used as a positive control.
In vitro RNA-dependent RNA synthesis assay was performed to test the replication inhibitors on HCV NS5B polymerase activity. RNA synthesis by the 1b Con1 RdRp was performed as described in Yi et al. (Yi et al., 2012). Briefly, the 20 μL reaction mixture contains 20 mM sodium glutamate (pH 8.2), 12.5 mM dithiothreitol, 4 mM MgCl2, 1 mM MnCl2, 0.5% Triton X-100, 0.2 mM GTP, 0.1 mM ATP and UTP, 33 nM [α-32P]CTP (Perkin Elmer), 80 nM recombinant 1b/Con1 NS5B that lack the C-terminal 21-residues, 100 nM PE46, 50 nM LE19p and 10 μM of each compound. Benzodiathiadine (10 μM) was used as a positive control. The 3’sequence from the PE46b RNA will anneal intramolecularly to direct the HCV RdRp to synthesize a primer-extended product of 46-nt. The LE19P RNA will direct the HCV RdRp to synthesize a 19-nt de novo initiated RNA. The reaction mixture was incubated at 30°C for 1 h, and RNA products were separated by 7.5 M urea–20% polyacrylamide gels. The signals were quantified using PhosphorImager and ImageQuant software.
2.9. Molecular docking
The binding models of HCV NS3/4A protease were predicted using the AutoDock program (Vercauteren et al., 2015). Two crystal structures of NS3/4A (PDB code: 4A92 and 2GVF) were used with an ensemble-docking protocol (Hu et al., 2013). The three-dimensional structures were obtained from the Protein Data Bank (Berman et al., 2000; Wang et al., 2010). The active site of the protein was defined by a grid of 70 × 70 × 70 points with a grid spacing of 0.5 Å centered at the mass center of the corresponding ligand bound in the crystal structure. The Lamarckian Genetic Algorithm (LGA) (Morris et al., 1998) was applied with 100 runs and the maximum number of energy evaluations was set to 2 × 106. The interactions were further refined by MD stimulations using the AMBER12 package and the ff99SB force field.
2.10. Physicochemical properties
Microsomes prepared from rat liver tissues were used for the evaluation of the compound’s microsomal stability. The compounds were incubated with microsomes at 37°C in the presence of the co-factor, NADPH. The concentrations of compounds were measured by LC-MS/MS at 0, 5, 15, 30, and 45 min after the incubation. Half-life (t1/2) and intrinsic clearance (Clint) were calculated as described in the literature (Obach, 1999).
3. Results
3.1. Primary screen and hit selection
A flow chart outlining the primary screen and follow-up confirmation assays is summarized in Fig. 1. with the primary screen of ~ 350,000 compounds from the MLSMR library yielded ~ 3,000 hits (hit rate = 0.86%) based on the following criteria: robust CRC categories (1.1/1.2/2.1/2.2), maximal response > 50%, and potency (EC50) < 10 μM. To further triage the hits down to a manageable level for further study, we applied an approach combining structural clustering and cross-activity analysis to prioritize the primary hits. The cross-activity of the hits was inspected in seven other cell-based screens of the MLSMR library conducted for very different purposes. If a compound or a class of structurally similar compounds exerts any nonspecific effect resulting as a hit, either through cytotoxicity or other mechanisms, we would expect it being active in many of the disparate screens, thus potentially promiscuous. The 3,000 hits were first clustered based on their chemical structures (Fig. S1). Similar cross-activity pattern across the seven different assays can be seen in some clusters. To further quantify the cross-activity and cytotoxicity, promiscuity index was determined by the number of positive activities among the seven cell-based screen assays for each compound. Furthermore, clustering analysis was performed based on the CRCs of compounds in each assay and a “cytotoxicity score” was assigned to each cluster by calculating the mean of the promiscuity index of compounds in the cluster.
Figure 1.

Flowchart of the quantitative high throughput screening (qHTS) of the MLSMR library for HCV inhibitors.
The structural and cross-activity clustering enabled us to select the most promising hits with structural diversity and less cytotoxicity. While the compounds in the toxic clusters (score < 2.0) were flagged, representatives of compounds with the best efficacy and potency in each structural cluster were selected for further analyses. A few singletons with high potency (EC50 < 5 μM) were also included. In total, 650 hits (0.187%) from the primary screen were selected for follow-up study.
3.2. Hit confirmation
The 650 primary hits were retested in 12 concentrations in the HCV-Cre/LoxPGluc reporter platform. 615 (95%) of them were confirmed active, 90% of which were found in the robust curve class 1.1, 1.2, 2.1, and 2.2. Fig. 2 showed a plot of CRCs of the compounds tested, indicating a robust performance of the confirmation assay in validating the primary screen. In the parallel ATPlite cytotoxicity assay, 418 (64%) of the testing compounds were non-cytotoxic (curve class 4; CC50 > 100 μM), while 112 (17%) of the compounds showing weak cytotoxicity (curve class 1.1, 1.2, 2.1, and 2.2; CC50 > 20 μM). Only about 10% of compounds showed significant cytotoxicity (CC50 < 10 μM).
Figure 2.
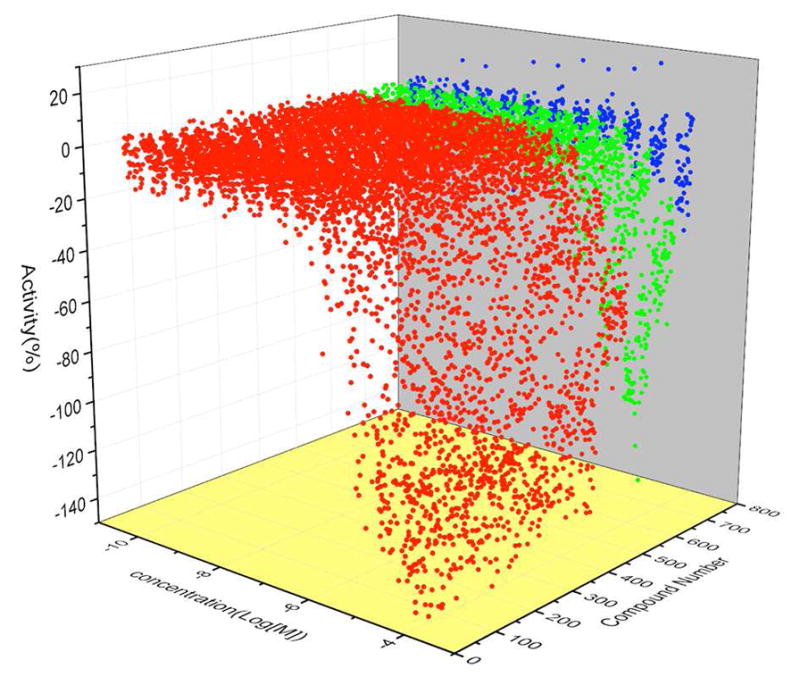
Waterfall plot of concentration response curves in the confirmation assay. Red: active compounds in curve class 1 and 2. Green: weakly active compounds in curve class 3. Blue: inactive compounds in curve class 4.
3.3. Secondary screen with HCV core staining assay
To eliminate the false-positives that may be resulted from the reporter gene, we applied an orthogonal assay to further validate the positive hits. A 2-part HCV core immunofluorescence staining assay in HCV-WT infected cells was used (Li et al., 2009). Compounds that were confirmed in the HCV-Cre/LoxPGluc reporter assay and showed non-cytototoxicity were selected. We prioritized a subset of the confirmed hits by considering structural diversity and drug-like properties. The Lipinski’s Rule of Five (MW < 500 Da, H-donor < 5, H-acceptor < 10, LogP < 5) and other properties were calculated and applied as a filter to further triage the confirmed hits (Bickerton et al., 2012). In total, 216 compounds were tested. Among them, 171 (79%) compounds scored positive, and 140 (65.8%) had robust CRCs. The remaining 45 (21%) showed little or no inhibition against HCV-WT and were thus excluded from follow-up analysis.
In the 2-part HCV infection assay, HCV core-positive cells decreased similarly in both parts if the inhibitor targets the early stage (entry or replication, such as MLS000714550) of the HCV life cycle. Late-stage inhibitors (targeting HCV assembly or secretion), such as MLS000763045, showed a more prominent inhibition in part-2 than in part-1 of the assay (Fig. 3). This assay, in combination with other HCV life cycle assays, is used to characterize the stage of HCV life cycle targeted by the confirmed hits.
Figure 3.

Hits validation with 2-part HCV infection and core immunofluorescence staining assay. Naïve Huh7.5.1 cells in 384-well plates were infected with wild-type HCVcc at 0.05 moi. Compounds in 1:3 serial dilutions starting at 30 μM were administered to the cells at the time of infection (part 1). After 48 h of culture, the media were transferred to a fresh plate of cells (part 2). Percentages of HCV core-positive cells were calculated and normalized against the DMSO control and plotted as CRCs. Two representatives of compounds targeting at early and late stages respectively are shown.
3.4. Target stages of selected hits in HCV life cycle
To investigate the mechanism of inhibition, we further tested the compounds with HCV life cycle assays, including HCV entry with HCVpp assay, replication with HCV replicon assay, and early-stage infection (HCV entry to replication) with HCVsc assay (Masaki et al., 2010). HCVpp assay is used to define inhibitors targeting the entry stage, although with some exceptions (Bartosch and Cosset, 2009; Sainz et al., 2012). HCV subgenomic replicon retains the property of HCV RNA replication and is used as an indicator of HCV replication (Lohmann et al., 1999). HCVsc is a replicon trans-packaging system and can infect and replicate but does not assemble new virions (Masaki et al., 2010). Thus these systems allow the distinction of viral entry/replication from assembly/secretion.
A total of 158 of the above 171 compounds validated from the secondary assay were examined in the three HCV life cycle assays (Fig. 1). The remaining compounds were not available for further testing. Each compound was assigned to a putative target stage of the HCV life cycle based on its effects on HCVsc, HCVpp infection and HCV replicon activity (Table S1). Activities in each assay with more than 50% decrease were regarded as significant inhibition. The criteria for categorizing the target stage of each compound are summarized in Table 1. Compounds targeting the late stages should have a preferential inhibition of HCV infection in part-two over part-one in the 2-part HCV infection assay (such as MLS000763045 in Fig. 3), but this trend was not always observed.
Table 1.
Criteria for the Designation of Target Stage Categories of Selected Compounds
| Life cycle stage | Criteria (% inhibition)*
|
Number of hits | ||
|---|---|---|---|---|
| HCVsc | HCVpp | HCV replicon | ||
| Entry | + | + | − | 39 |
| Replication | + | − | + | 14 |
| Entry and replication | + | + | + | 18 |
| Trafficking | + | − | − | 37 |
| Assembly/secretion | − | − | − | 47 |
+ : inhibition > 50%; − : inhibition < 50%.
Using the criteria set in Table 1, 108 hits among the 158 compounds were defined as early stage inhibitors (39 inhibit the entry stage, 14 the replication stage, 18 both entry and replication, 37 trafficking.) and 47 hits target the late stage (assembly or secretion) of the HCV life cycle (Table S1). Two compounds (MLS001217747-01, and MLS000040412-01) with a significant inhibition in HCVsc assay were categorized as entry inhibitors because they are structurally related to a group of entry inhibitors (MLS-000078670-01, MLS001139315-01, MLS001139388-01 and MLS000879011-01), although their inhibition in HCVpp assay was less than 50%. Additionally, 3 compounds could not be classified into a specific category based on the criteria set above (Table S1). Many compounds were defined as early stage inhibitors due to their significant inhibition of HCVsc activity had no effect on HCV entry or replication. These compounds are most likely involved in the trafficking post-HCV entry.
HCV life cycle is rather complex and involves many steps and factors. The above classification of targeting stage for each compound based on existing virologic assays is only a preliminary step in studying the modes of action of these inhibitors. Due to the diversity and complexity of the compound structures, the current HCV assays may not fully reflect the functional mechanisms for some of the compounds. For example, many of the compounds with a significant inhibition of HCVsc didn’t show very active inhibition in HCVpp or replicon assays as anticipated. Conversely, some compounds with very little effect on HCVsc assay turned out to have significant inhibition on HCVpp or replicon activity.
Fig. 4 shows the structures and activities of the representative hits targeting HCV entry (Fig. 4A)/trafficking (Fig. 4B) or assembly/secretion (Fig. 4C). The structures of these inhibitors are rather diverse and likely have novel mechanisms of action in inhibiting HCV. Most of the assembly/secretion inhibitors showed higher potency at part-2 of the HCV-WT infection assay, supporting that the compounds more likely act in the late stage. Interestingly, inhibitors D7 (MLS000552390-01) and D8 (MLS000763045-01) showed 10–50 fold of higher potency at part-2 when compared to that at part-1. The two compounds are structurally related with a sulfanyl-quinol-like scaffold.
Figure 4.

Molecular structure of representative compounds targeting early and late stages of HCV life cycle. A. Identified inhibitors targeting the entry stage of HCV infection; B. Identified inhibitors targeting the trafficking stage of HCV infection; C. Identified inhibitors targeting the assembly/secretion stage of HCV infection.
3.5. Mechanisms of action of replication inhibitors
To investigate the mechanisms of action of the replication stage inhibitors identified in our screen, we evaluated the activities of these inhibitors against HCV NS3/4A protease and NS5B polymerase using biochemical enzyme assays (Fatima et al., 2011; Yi et al., 2012). Of the 32 inhibitors targeting HCV replication, three compounds (MLS000730707, MLS001163541 and MLS002638531) showed activities against NS3/4A with 40–50% inhibition at 10 μM (Table S1); none of them showed inhibition in the NS5B assay (Table S1).
Most NS3/4A serine protease inhibitors known so far are peptidomimetics that either form covalent bond with the catalytic triad (S139/D81/H57), or non-covalent inhibitors, which are mostly macrocyclic compounds bound to the S2–S4 pocket (Arasappan et al., 2006). The identified inhibitor MLS000730707 is a chemotype of thiourea-based compounds that have recently reported to target the HCV NS3/4A protease (Yang et al., 2013). The other two compounds active in the protease assay are not structurally similar to the known protease inhibitors. Molecular modeling and Docking analysis of these two compounds showed that they appeared to fit well into the binding site of NS3/4A protease. As shown in Fig. 5, the carbonyl moiety of inhibitor MLS001163541 occupied the oxyanion hole by forming interactions with S139 and H57, while the trimethoxyphenyl group pointed into the S1 hydrophobic pocket, which formed a salt interaction with K136 and locked the C-clamp residue in place. Similar binding mode was observed with inhibitor MLS002638531. The catalytic S139 likely attack the electrophilic keto functionality in a reversible manner, and the dioxopiperidin group bound to the S1 pocket forming extensive hydrophobic interactions with I132, K136, F154, and A157. Moreover, the oxocyclohexyl ring formed interactions with R155, while the other phenyl ring reached the S1 pocket and formed interactions with H57 and Q41, which are key residues involved in substrate binding and catalytic process (Lee et al., 2009).
Figure 5.

Predicted binding model of inhibitors targeting HCV NS3/4A protease (PDB 4a92). (A) MLS001163541 and (B) MLS002638531. The protein is shown as ribbons and key residues are shown as sticks. Small molecule Inhibitor is shown in magenta (atoms N and O are shown in blue and red).
Inhibitors of NS5B polymerase consist of two main classes: nucleoside inhibitor (NI) and non-nucleoside inhibitor (NNI) (Sofia et al., 2012). The in vitro NS5B polymerase assay can only be used to test the activity of NNI because NI requires phosphorylation by cells to be active. However none of these 32 compounds targeting HCV replication stage in our screen have structures resembling nucleoside or nucleotide and in vitro NS5B polymerase assays were all negative for the 32 compounds. Thus it is unlikely that any of them targets NS5B directly. It is possible that some of the compounds may target other viral components, like NS2, helicase domain of NS3 or NS4B. Alternatively they may target the viral replicase complex (but not the individual subunits) or host factor that are involved in viral replication.
3.6. SARs and in vitro physicochemical study of the lead compounds
Based on potency, structural novelty and drug-like properties as well as our intent to focus on non-replication targets (entry, assembly and secretion), we selected 4 chemotypes as lead hits for additional studies (Table 2). The in vitro stability profiles of these compounds are shown in Table 2. The three entry-stage inhibitors have relatively good aqueous solubility and permeability, indicating that these are promising lead compounds amenable for drug development. The half-life (t1/2) of MLS000876570 and MLS000877128 in rat microsomes is greater than 30 min, while inhibitor MLS000049859 has a shorter t1/2 (7.9 min). However, the t1/2 of MLS000662842 in rat microsomes is only 2.3 min, suggesting stability may be an issue with this chemotype.
Table 2.
In Vitro Stability of Promising Lead Compounds for Probe Development
| Physicochemical properties | ||||||
|---|---|---|---|---|---|---|
| Chemotype | Sample ID | Life cycle stage | Structure | Microsomal t1/2 (m) | Permeability (1e-6cm/s) | Solubility (μg/ml) |
| 1 | MLS000876570-01 | Entry |
|
> 30.0 | < 30.9 | > 59.0 |
| 2 | MLS000877128-01 | Entry |
|
> 30.0 | 489.1 | 53.9 |
| 3 | MLS000049859-01 | Entry |
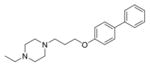
|
7.9 | >2183.0 | > 48.0 |
| 4 | MLS000662842-01 | Assembly/secretion |
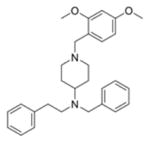
|
2.3 | N. D. | 49.3 |
Initial studies of the structurally-related analogs of the lead compounds from the existing MLSMR library indicated a clear structure-activity relationship (SAR) of the core structure and functionalities that are important to activity. A number of related compounds of inhibitors MLS000876570 and MLS000877128 in the primary screen showed HCV inhibition varied from low μM up to 100 μM of EC50 (Table 3). Interestingly, similar SARs can be seen among these two chemotypes of compounds. While the cores of dioxoquinazoline and oxazolpiperidine in the middle of molecule are important to its functional activity, the substitutions on two sides showed varied effects on its potency and efficacy. Both sides appear to be more effective when they possess aromatic and lipophilic group, and quite restricted with the length of substitutions. In addition, these small molecules typically adopt a linker molecule shape with a hydrophobic head and a varied length of lipophilic tail, suggesting that they likely have the same target and mechanism of action.
Table 3.
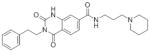
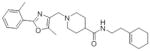
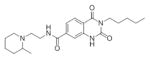
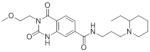
SAR Studies of the Entry Inhibitors MLS00876570 and MLS000877128
| Sample ID | Structure | EC50 (μM) | Max Resp | Sample ID | Structure | EC50 (μM) | Max Resp |
|---|---|---|---|---|---|---|---|
| MLS000876570-01* |
|
4.4668 | −76.038 | MLS000877128-01† |
|
1.2589 | −73.014 |
| MLS000948303-01 |
|
7.0795 | −55.607 | MLS000913279-01 |
|
1.4125 | −80.801 |
| MLS000086763-01 |
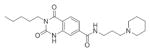
|
10 | −72.669 | MLS000095189-01 |
|
2.8184 | −67.008 |
| MLS000519917-01 |
|
3.9811 | −67.83 | MLS000095186-01 |
|
5.0119 | −42.656 |
| MLS000086784-01 |
|
6.3096 | −83.27 | MLS000095191-01 |
|
6.3096 | −73.5 |
| MLS000519916-01 |
|
8.9125 | −74.83 | MLS000117605-01 |
|
11.2202 | −64.827 |
| MLS000948325-01 |
|
22.3872 | −67.553 | MLS000095192-01 |
|
14.1254 | −75.426 |
| MLS000730063-01 |
|
31.6228 | −69.852 | MLS000878431-01 |
|
22.3872 | −38.647 |
| MLS000519908-01 |
|
> 100 | 12.531 | MLS000095190-01 |
|
> 100 | −23.229 |
| MLS000948314-01 |
|
> 100 | −1.231 | MLS000913230-01 |
|
> 100 | −27.788 |
Lead compound chemotype 1;
lead compound chemotype 2.
4. Discussion
Using a cell-based HCV infection qHTS platform, we identified a number of novel HCV inhibitors with diverse structure and functions. With the help of various HCV functional assays, we were able to classify most of these inhibitors into each targeting stage of the HCV life cycle, thus allowing us to have a preliminary understanding of the mechanism of these compounds and better prioritize lead compound selection for further drug development. Structure and functional analysis of these inhibitors also provided valuable information for the understanding of HCV life cycle and identification of potential new drug targets.
Of 158 tested hits in the HCV life cycle assays, 108 compounds were identified to affect the HCV entry and trafficking, which includes viral binding to cell surface receptors, entry and post-entry trafficking of the virus. Structures of these entry-stage inhibitors are rather diverse, including a number of interesting groups such as piperazine, piperidine, furan, and oxazole. A cluster of inhibitors tested in the HCV life cycle assay (MLS001139388-01, B6 as an example) were all found in the entry stage. These compounds are rather potent in the inhibition of HCV infection, with EC50 in a range of low micromole. SAR clearly indicated that the activity is dependent on the two side groups connecting to the tricyclic ring, where a polar and aromatic ground is favored. The oxo-pyrimidine tricyclic ring appeared to play a functional role in the inhibition of HCV. This chemotype compound has been reported as antagonists of GPR55 (PuchChem AID: 485287) and antagonist of Cannabinoid Receptor 1 (AID: 485285). Interestingly, a number of such oxoheterocyclic compounds, such as B7 and B8, were also found in this class, implying that they likely target the same pathway.
Another structural feature observed in these entry-stage inhibitors is that many compounds contain the piperazine or piperidine group. Such functionalities have been frequently observed with ligands of GPCRs screen. The quinazolindione derivatives B8 (MLS000876570, are potent 5-HT3A receptor antagonists (Lee et al., 2009). B5 (MLS000536284) showed activities against human M1 muscarinic receptor (AID: 624040) and D1 dopamine receptor (AID: 504660). B4 (MLS000877128) showed activities in activation of activator of the GIRK family of Potassium Channels and G-protein-linked glutamate receptors (AID 488969). H1 histamine receptor inhibitors of diverse chemical structures were revealed with anti-HCV activity in previous studies when screening with this assay system of NCATS Pharmaceutical Collection (He et al., 2015). These compounds seem to target the late entry stage of HCV infection. Therefore, GPCR may represent a potentially new pathway of HCV infection and another target for HCV drug development.
47 hits are inhibitors of HCV assembly/secretion. While these compounds are structurally diverse, common structural moiety are thiol- and thiourea. Analysis of the cross-activities of these compounds in PubChem and assayed in our in-house qHTS database indicated that most of them appeared to be involved in DNA repair activities. As shown in Fig. 4C, inhibitors D1 (MLS002302696-01) and D2 (MLS000592075-01) showed inhibitory activity of human tyrosyl-DNA phosphodiesterase 1 (AID 686978). D3 (MLS000121992-01) was an inhibitor of DNA polymerase ETA (AID 588591) and inhibited the assembly of the perinucleolar compartement (AID 2427). D4 (MLS000665896-01) and its derivatives are potent modulators of miRNAs (AID: 2289) and activators of 5′UTR stem-loop driven prion protein mRNA translation in H4 neuroglioblastoma cells (AID: 1814). It also targets posterior segregation family member (pos-1) binding to mex-3-RNA (PubChem AID: 1964). D5 (MLS000582548-01) has been reported as an inhibitor of respiratory syncytial virus (RSV) (AID: 2410) and HIV (AID:651610). It also showed inhibitory activities against human TDP1 and ELG1-dependent DNA repair. D6 is a piperazine-based compounds and showed activities against a broad range of targets including GPCRs, TDP1, ion channel, and choline transporter.
The HCV NS3/4A protease and NS5B polymerase are two crucial enzymatic targets involved in viral replication, and have been extensively explored in structure-based drug design. A large number of potent inhibitors and drug candidates have been developed (Yang et al., 2011). Majority of the compounds targeting HCV replication stage in this screen seems to be novel inhibitors since most of them showed little effects on the enzyme activities and had no structural similarity to the known replication inhibitors.
Although new generation of DAAs offers much improved profile of efficacy and side effect, rapid development of HCV resistant variants is a challenge (Schiffer et al., 2011). This issue is especially a concern for the difficult-to-cure patient groups and patients who have difficulty in therapy adherence. To suppress the emergence of drug resistance, combination of multiple antiviral agents with distinct mechanisms is usually needed (Liang and Ghany, 2014). One attractive approach is to combine with host targeting agents (HTAs) which usually have a higher genetic barrier for resistance than DAAs (Khattab, 2009; von Hahn et al., 2011). One recent preclinical in vivo study showed that addition of an entry inhibitor (the anti-scavenger receptor class B type I mAb1671) to an anti-HCV DAA regimen (monotherapy with the protease inhibitor ciluprevir) in human-liver mice infected with HCV of genotype 1b restricted the breakthrough of DAA-resistant viruses (Vercauteren et al., 2015). Our drug screen scheme targets all aspects of the HCV viral life cycle, including host antiviral factors. Actually, many of the compounds have unique structures that are potentially HTAs. Most entry inhibitors, in particular, probably target some of the cellular entry factors for the inhibition of HCV infection. Further understanding the mechanism of these compounds could greatly help design new combination strategy in HCV treatment.
The main goal of this study is to identify novel inhibitors of HCV as drug candidates for further development as well as molecular probes to study HCV infection. As most of the HCV drugs currently in clinical development and application are inhibitors of HCV RNA replication, the identified inhibitors targeting the other stages of HCV life cycle, such as the entry/trafficking and assembly/secretion stage, are more appealing for further development. We therefore prioritized 4 compounds that inhibit HCV entry or assembly/secretion as the lead hits for further study. Structural and functional characteristics of these small molecules may provide insight into the mechanism of viral infection and inhibition, and most importantly, offer promising candidates for the development of novel therapeutics against HCV infection.
In summary, we performed a large-scale qHTS of compounds for novel HCV inhibitors using a robust cell-based HCV infection assay. This qHTS of a large and diverse molecular library resulted in the identification and preliminary characterization of many novel HCV inhibitors. Inhibitors targeting all stages of HCV life cycle were demonstrated. Further characterization and SAR campaign of several lead compounds would likely generate novel classes of HCV antivirals that are suitable for clinical therapeutic development. The availability of new drugs with modes of action different from those of existing drugs would provide a desirable addition to the current armamentarium against HCV infection. In addition, these compounds may represent novel molecular probes to explore the complex interactions between HCV and the cells.
Supplementary Material
Figure S1. Structure clustering and promiscuity analysis of the primary screen hits. A) Structural clustering analysis; B) Promiscuity analysis using 7 cell-based assays (1-cmt, 2-ikapad, 3-miR21, 4-nucgrowth, 5-ror-vp16, 6-vsv-lassa, 7-plasmodium). The cross-activity score (0–7) is determined by the number of activity being inactive among the seven assays. Compounds with low scores are usually non-specific due to cytotoxicity. Compounds with score equal or less than 2 were flagged and excluded from the cherry-picked list for further analysis.
Table S1. Primary, secondary and tertiary assay results of selected 158 HCV inhibitors
158 inhibitors with diverse structure and functions were identified from HTS of a small molecule library.
We were able to assign majority of the compounds to a specific target stage(s) in the HCV life cycle.
Four compounds with novel structure and drug-like properties were selected as lead hits for further development.
Acknowledgments
We thank Baihua Zhang for her excellent technical work, Paul Shinn for his preparation of compounds, Sam Michael and Mike Balcom for operating robot-controlled compound administration and plate reading. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Center for Advancing Translational Sciences, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information (US); Bethesda (MD): [PubMed] [Google Scholar]
- 2.Arasappan A, Njoroge FG, Chen KX, Venkatraman S, Parekh TN, Gu H, Pichardo J, Butkiewicz N, Prongay A, Madison V, Girijavallabhan V. P2-P4 macrocyclic inhibitors of hepatitis C virus NS3-4A serine protease. Bioorganic & medicinal chemistry letters. 2006;16:3960–3965. doi: 10.1016/j.bmcl.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 3.Bartosch B, Cosset FL. Studying HCV cell entry with HCV pseudoparticles (HCVpp) Methods in molecular biology. 2009;510:279–293. doi: 10.1007/978-1-59745-394-3_21. [DOI] [PubMed] [Google Scholar]
- 4.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic acids research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4:90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buti M, Esteban R. 1990–2010: two decades of interferon-based therapy. Clinics in liver disease. 2011;15:473–482. doi: 10.1016/j.cld.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Chang LJ, Urlacher V, Iwakuma T, Cui Y, Zucali J. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 1999;6:715–728. doi: 10.1038/sj.gt.3300895. [DOI] [PubMed] [Google Scholar]
- 8.Chockalingam K, Simeon RL, Rice CM, Chen Z. A cell protection screen reveals potent inhibitors of multiple stages of the hepatitis C virus life cycle. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3764–3769. doi: 10.1073/pnas.0915117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fatima K, Tahir M, Qadri I. Development of robust in vitro serine protease assay based on recombinant Pakistani HCV NS3-4A protease. Virus Res. 2011;160:230–237. doi: 10.1016/j.virusres.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 10.Gastaminza P, Whitten-Bauer C, Chisari FV. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:291–296. doi: 10.1073/pnas.0912966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hao W, Herlihy KJ, Zhang NJ, Fuhrman SA, Doan C, Patick AK, Duggal R. Development of a novel dicistronic reporter-selectable hepatitis C virus replicon suitable for high-throughput inhibitor screening. Antimicrob Agents Chemother. 2007;51:95–102. doi: 10.1128/AAC.01008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He S, Lin B, Chu V, Hu Z, Hu X, Xiao J, Wang AQ, Schweitzer CJ, Li Q, Imamura M, Hiraga N, Southall N, Ferrer M, Zheng W, Chayama K, Marugan JJ, Liang TJ. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci Transl Med. 2015;7:282ra249. doi: 10.1126/scitranslmed.3010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A. 2003;100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu X, Compton JR, Abdulhameed MD, Marchand CL, Robertson KL, Leary DH, Jadhav A, Hershfield JR, Wallqvist A, Friedlander AM, Legler PM. 3-substituted indole inhibitors against Francisella tularensis FabI identified by structure-based virtual screening. Journal of medicinal chemistry. 2013;56:5275–5287. doi: 10.1021/jm4001242. [DOI] [PubMed] [Google Scholar]
- 16.Hu Z, Lan KH, He S, Swaroop M, Hu X, Southall N, Zheng W, Liang TJ. Novel Cell-Based Hepatitis C Virus Infection Assay for Quantitative High-Throughput Screening of Anti-Hepatitis C Virus Compounds. Antimicrob Agents Chemother. 2014;58:995–1004. doi: 10.1128/AAC.02094-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol. 2009;15:3472–3479. doi: 10.3748/wjg.15.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SS, Peng LF, Lin W, Choe WH, Sakamoto N, Kato N, Ikeda M, Schreiber SL, Chung RT. A cell-based, high-throughput screen for small molecule regulators of hepatitis C virus replication. Gastroenterology. 2007;132:311–320. doi: 10.1053/j.gastro.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 20.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. Journal of virology. 2006;80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee BH, Choi MJ, Jo MN, Seo HJ, Nah SY, Cho YS, Nam G, Pae AN, Rhim H, Choo H. Quinazolindione derivatives as potent 5-HT3A receptor antagonists. Bioorganic & medicinal chemistry. 2009;17:4793–4796. doi: 10.1016/j.bmc.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 22.Lemm JA, O’Boyle D, 2nd, Liu M, Nower PT, Colonno R, Deshpande MS, Snyder LB, Martin SW, St Laurent DR, Serrano-Wu MH, Romine JL, Meanwell NA, Gao M. Identification of hepatitis C virus NS5A inhibitors. J Virol. 2010;84:482–491. doi: 10.1128/JVI.01360-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med. 2013;368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang TJ, Ghany MG. Therapy of hepatitis C--back to the future. N Engl J Med. 2014;370:2043–2047. doi: 10.1056/NEJMe1403619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Annals of internal medicine. 2000;132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 27.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 28.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nature medicine. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masaki T, Suzuki R, Saeed M, Mori K, Matsuda M, Aizaki H, Ishii K, Maki N, Miyamura T, Matsuura Y, Wakita T, Suzuki T. Production of infectious hepatitis C virus by using RNA polymerase I-mediated transcription. Journal of virology. 2010;84:5824–5835. doi: 10.1128/JVI.02397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of computational chemistry. 1998;19:1639–1662. [Google Scholar]
- 31.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 32.Sainz B, Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarrazin C, Zeuzem S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology. 2010;138:447–462. doi: 10.1053/j.gastro.2009.11.055. [DOI] [PubMed] [Google Scholar]
- 34.Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schiffer JT, Scott J, Corey L. A siege of hepatitis: fighting a defiant virus. Nature medicine. 2011;17:253–254. doi: 10.1038/nm0311-253. [DOI] [PubMed] [Google Scholar]
- 36.Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. Journal of medicinal chemistry. 2012;55:2481–2531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 37.Vercauteren K, Brown RJ, Mesalam AA, Doerrbecker J, Bhuju S, Geffers R, Van Den Eede N, McClure CP, Troise F, Verhoye L, Baumert T, Farhoudi A, Cortese R, Ball JK, Leroux-Roels G, Pietschmann T, Nicosia A, Meuleman P. Targeting a host-cell entry factor barricades antiviral-resistant HCV variants from on-therapy breakthrough in human-liver mice. Gut. 2015 doi: 10.1136/gutjnl-2014-309045. [DOI] [PubMed] [Google Scholar]
- 38.von Hahn T, Ciesek S, Manns MP. Arrest all accessories--inhibition of hepatitis C virus by compounds that target host factors. Discov Med. 2011;12:237–244. [PubMed] [Google Scholar]
- 39.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nature medicine. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Jadhav A, Southal N, Huang R, Nguyen DT. A grid algorithm for high throughput fitting of dose-response curve data. Curr Chem Genomics. 2010;4:57–66. doi: 10.2174/1875397301004010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wichroski MJ, Fang J, Eggers BJ, Rose RE, Mazzucco CE, Pokornowski KA, Baldick CJ, Anthony MN, Dowling CJ, Barber LE, Leet JE, Beno BR, Gerritz SW, Agler ML, Cockett MI, Tenney DJ. High-throughput screening and rapid inhibitor triage using an infectious chimeric Hepatitis C virus. PloS one. 2012;7:e42609. doi: 10.1371/journal.pone.0042609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang PL, Gao M, Lin K, Liu Q, Villareal VA. Anti-HCV drugs in the pipeline. Curr Opin Virol. 2011;1:607–616. doi: 10.1016/j.coviro.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang W, Sun Y, Hou X, Zhao Y, Fabrycki J, Chen D, Wang X, Agarwal A, Phadke A, Deshpande M, Huang M. ACH-806, an NS4A antagonist, inhibits hepatitis C virus replication by altering the composition of viral replication complexes. Antimicrob Agents Chemother. 2013;57:3168–3177. doi: 10.1128/AAC.02630-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi G, Deval J, Fan B, Cai H, Soulard C, Ranjith-Kumar CT, Smith DB, Blatt L, Beigelman L, Kao CC. Biochemical study of the comparative inhibition of hepatitis C virus RNA polymerase by VX-222 and filibuvir. Antimicrob Agents Chemother. 2012;56:830–837. doi: 10.1128/AAC.05438-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu X, Sainz B, Jr, Petukhov PA, Uprichard SL. Identification of hepatitis C virus inhibitors targeting different aspects of infection using a cell-based assay. Antimicrob Agents Chemother. 2012;56:6109–6120. doi: 10.1128/AAC.01413-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Structure clustering and promiscuity analysis of the primary screen hits. A) Structural clustering analysis; B) Promiscuity analysis using 7 cell-based assays (1-cmt, 2-ikapad, 3-miR21, 4-nucgrowth, 5-ror-vp16, 6-vsv-lassa, 7-plasmodium). The cross-activity score (0–7) is determined by the number of activity being inactive among the seven assays. Compounds with low scores are usually non-specific due to cytotoxicity. Compounds with score equal or less than 2 were flagged and excluded from the cherry-picked list for further analysis.
Table S1. Primary, secondary and tertiary assay results of selected 158 HCV inhibitors