

Abstract
Reduced levels of adiponectin (APN) contribute to cardiovascular injury in the diabetic population. Recent studies demonstrate elevated circulating APN levels are associated with endothelial dysfunction during pre-diabetes, suggesting the development of APN resistance. However, mechanisms leading to, and the role of, vascular APN resistance in endothelial dysfunction remain unidentified. The current study determined whether diabetes cause endothelial APN resistance, and by what mechanisms. Under high glucose/high lipids (HG/HL), APN-stimulated nitric oxide production by HUVEC was decreased, phosphorylation of eNOS, AMPK, and Akt was attenuated (P<0.01), and APN’s anti-TNFα effect was blunted (P<0.01). APN receptor expression remained normal, whereas Cav1 expression was reduced in HG/HL cells (P<0.01). The AdipoR1/Cav1 signaling complex was dissociated in HG/HL cells. Knock-down of Cav1 inhibited APN’s anti-oxidative and anti-inflammatory actions. Conversely, preventing HG/HL-induced Cav1 downregulation by Cav1 overexpression preserved APN signaling in HG/HL cells. Knock-in of a wild type Cav1 in Cav1 knock-down cells restored caveolae structure and rescued APN signaling. In contrast, knock-in of a mutated Cav1 scaffolding domain restored caveolae structure, but failed to rescue APN signaling in Cav1 knock-down cells. Finally, AdipoR1/Cav1 interaction was significantly reduced in diabetic vascular tissue, and the vasorelaxative response to APN was impaired in diabetic animals. The current study demonstrates for the first time the interaction between AdipoR1 and Cav1 is critical for adiponectin-mediated vascular signaling. The AdipoR1/Cav1 interaction is adversely affected by HG/HL, due largely to reduced Cav1 expression, supporting a potential mechanism for the development of APN resistance, contributing to diabetic endothelial dysfunction.
Keywords: Caveolin, Adiponectin, Vascular Injury, Endothelial Function
Graphical abstract
Introduction
Cardiovascular disease (CVD) is the most significant cause of death in developed countries [18]. Despite extensive research worldwide in the past several decades, therapeutic interventions efficacious in reducing cardiovascular mortality remain limited. Type II diabetes is associated with atherosclerotic CVD [25]. Early interventions remain pivotal in preventing CVD. Endothelial dysfunction gives genesis to diabetic micro- and macro-vascular complications, the major causes of morbidity and mortality in diabetic patients [6]. Elucidating the specific mechanisms responsible for endothelial dysfunction in diabetes may yield viable targets for early therapeutic interventions.
Type II diabetes is characterized by a high glucose and high lipid (HG/HL) state [5]. HG/HL markedly increases oxidative/nitrative stress, which contributes centrally to diabetic endothelial dysfunction [12]. Additionally, chronic HG/HL induces a state of low grade inflammation in the vasculature, initiating inflammatory processes causing micro- and macro-vascular damage [1]. Concomitantly, systemic and vascular stress reduces endothelial nitric oxide (NO) bioactivity, and increases expression of cell surface adhesion molecules, ultimately augmenting atherosclerosis development. Constitutively expressed in endothelial cells, the intercellular adhesion molecule-1 (ICAM-1) is upregulated in human atherosclerotic lesions, which recruits leukocytes into the intima. As inflammatory cells migrate into the endothelium, proinflammatory cytokines (such as TNF-α) exacerbate atherosclerotic vascular disease [15,19,30].
Adiponectin (APN), a 30 kD protein of primarily adipose origin with multiple metabolic functions, protects the cardiovascular system against inflammation [2,11]. APN deficiency is associated with endothelial dysfunction and increased inflammatory responses in both diabetic rodent models and humans [16,22,24]. However, we recently demonstrated endothelial dysfunction occurs during early development of diabetes (pre-diabetes)[17]. Strikingly, plasma APN levels are significantly increased during this pre-diabetic stage. Moreover, there is no significant change in the expression levels of signaling molecules involved in APN vascular function, indicating the involvement of an unknown molecular mechanism. Interestingly, numerous recent epidemiological studies report elevated circulating APN levels in patients with heart failure, suggesting the onset of APN resistance with heart failure [3,8,14]. However, whether the metabolic derangements in pre-diabetes cause APN resistance (and are causatively related to subsequent endothelial dysfunction) has not been elucidated. Finally, we have recently demonstrated that AdipoR1 and Cav1 co-localize and co-precipitate in HUVECs, and the Cav1/AdipoR1 complex formation is critical for APN transmembrane signaling. However, whether this interaction is altered in diabetic condition, contributing to APN resistance and vascular injury, has not been previously investigated.
The objectives of the current study were 1) to determine whether HG/HL (mimicking the pre-diabetic stage) may cause APN resistance, and if so, by what molecular mechanisms, and 2) to assess potential therapeutic interventions capable of improving APN sensitivity and preserving endothelial function.
Materials and Methods
Materials
Human umbilical vein cells (HUVEC, 4–6 passages) and all cell culture reagents and medium were from Cell Applications (San Diego, CA). Cells were cultured in endothelial growth medium with 10% fetal bovine serum (Hyclone, Logan, CT), 2mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C and 5% CO2. Antibodies against APN receptor 1 (AdipoR1), APN receptor 2 (AdipoR2), endothelial NO synthase (eNOS), AMP activated protein kinase (AMPK), Akt, ICAM-1, GAPDH, eNOS phosphorylated at Ser1177, phosphorylated AMPK, and phosphorylated Akt were from Cell Signaling Technology (Beverly, MA). Antibody to Cav1 was from Abcam (Cambridge, MA). Antibody against gp91phox was from BioLegend Company (San Diego, CA). Antibody against Flag was from Sigma (St. Louis, MO). Antibody against AdipoR1 was from Bioss, Inc. (Woburn, MA) for Western analysis and IP.
HUVECs were randomized to receive one of the following treatments: Normal glucose/normal lipid culture for 72 hours (control, 5.5 mM D-glucose+19.5 mM L-glucose) followed by vehicle or APN (5μg/ml) treatment; or high glucose (HG, 25mM D-glucose)/high lipids (HL, 250 μM palmitates [23,28]) culture for 72 hours (HG/HL) followed by vehicle or APN treatment. The effect of HG/HL upon APN’s endothelial stimulatory effect was determined by production of NO, and phosphorylation of eNOS, AMPK and Akt. The effect of HG/HL upon APN’s anti-oxidative and anti-inflammatory actions was determined by superoxide production, peroxynitrite formation, and expression of gp91phox and ICAM-1.
Small Interfering RNA Transfection
RNA oligonucleotides complementary to Cav1 target sequences silenced respective gene expression. HUVECs were transfected with a siIMPORTER siRNA transfection kit (Qiagen Science Inc. Benelux, Venlo, Netherlands) per manufacturer’s protocol with siRNA duplexes against Cav1 (5′-UAUUAUGAGAUGGUAGGCAdTdT-3′) and universal control oligonucleotides (AllStars, Westerville, OH). Briefly, HUVECs were plated on six-well plates before transfection. After reaching 80% confluence, siRNA was applied to each well (final concentration 50 nM).
Plasmid Construction and Transfection
The cDNA encoding full-length (FL) human Cav1 and scaffolding domain mutated Cav1 (muCav1, in which 6 aromatic residues within the scaffolding domain responsible for Cav1 interaction with its partner proteins were converted to alanine [21]) were subcloned into pcDNA3.1 at Hind III and XbaI sites. Full length adiponectin receptor 1 (AdipoR1) protein was generated using wild type AdipoR1 cDNA (gift from Liu/Dong Labs, Department of Pharmacology, University of Texas Health Science Center, San Antonio, TX) as a template. Truncated protein plasmids were additionally generated: AdipoR1 (1-230), AdipoR1 (1-215), AdipoR1 (316-375), and AdipoR1 (336-375) were generated via PCR and cloned into pcDNA3.1. Cav1 full length (FL), Cav1 (1-101), Cav1 (1-81), Cav1 (1-61) were generated via PCR using wild-type Cav3/Cav1 cDNAs as a template, and cloned into p3XFLAG.
HUVECs in a 60-mm culture plate were transfected with 2μg plasmid DNA encoding muCav1 and Cav1 FL via LipofectAMINE (Qiagen) per manufacturer’s protocols. When cells were co-transfected with plasmid DNA, empty vector (no cDNA insert) served as control. Approximately 30 hours after transfection, culture medium was switched to 1% DMEM culture medium. Incubation commenced for 10 hours prior to any experiments.
HUVECs were grown to 90% confluence in 6-well dishes, and transfected by Lipofectamine 2000 Reagent. Specifically, DNA (2 μg of AdipoR1 expression vector, 2 μg of Cav1 expression vector, or empty vector: p3XFLAG, pcDNA3.1) was mixed with 10 μl transfection reagent (diluted in 100μl of serum-free medium) per manufacturer’s protocol. 293T cells were incubated with lipid-DNA complexes in serum-free DMEM for 8 hours. Following transfection, cells were twice washed with PBS, and cultured in 10% serum medium. Lysates were prepared for co-IP analysis.
Electron Microscopy
HUVECs were fixed overnight at 4°C in a solution of 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1M sodium cacodylate buffer, pH7.4. After subsequent buffer washes, the samples were post-fixed in 2.0% osmium tetroxide for 1 hour at room temperature, and rinsed in deionized H2O in preparation for block staining by 2% uranyl acetate. After dehydration by graded ethanol series, the tissue was infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Thin sections were stained with uranyl acetate and lead citrate, and evaluated by a JEOL 1010 electron microscope (fitted with a Hamamatsu digital camera running AMT Advantage image capture software).
Co-Immunoprecipitation and Immunoblotting
After treatment, HUVECs or 293T cells were lysed with cold lysis buffer, homogenized, and analyzed. Briefly, cell lysates were pre-cleared with corresponding nonimmune IgG incubated together with protein A plus-Sepharose for 30 minutes at 4°C. Cleared lysates were then incubated with 2 μg of anti-AdipoR1, anti-Cav1, anti-Flag, or anti-Myc antibodies. Cell lysates were then incubated with protein A plus-Sepharose overnight at 4°C. Non-immune rabbit IgG served as negative control. Protein A beads were then extensively washed with lysis buffer. Proteins were eluted from the beads, resolved by elusion buffer, and subjected to Western blot analysis.
Samples with 2XSDS sample buffer were heated and separated by electrophoresis. After transfer to PVDF membranes, proteins were immunoblotted with appropriate antibodies. Signals were quantified by densitometry. Results were expressed as ratios of phosphorylated to unphosphorylated protein, or normalized to GAPDH loading control.
Subcellular Fractionation by Sucrose Density Gradient Centrifugation: Whole hearts or caveolae-rich membrane fractions were isolated as previously described2. Briefly, 150-mm dishes of endothelial cells were grown to confluence, scraped into 2ml of MBS (MES-buffered saline [25mM MES (pH6.5), 150mM NaCl, 2mM EDTA, 250 mM NaCO3]) without detergent. Triton X-100 was replaced with sodium carbonate buffer. Samples were homogenized upon ice (three 3 second bursts at medium speed), sonicated (six 10-second pulses of 400 watts), and incubated on ice for 10 minutes with periodic tube inversion. The resulting cell lysates were mixed with equal amounts of 90% sucrose in MBS (end sucrose concentration 40%). Resulting lysates were transferred to a centrifuge tube, and overlaid with 4 ml each of 35% and 5% sucrose in MBS. The gradient was centrifuged at 39,000 rpm for 20 hours by a Beckman SW41Ti rotor at 4°C. Twelve 1-ml fractions were collected from the gradient bottom, and analyzed by Western blot analysis or immunoprecipitation. Fractions 4–6 were buoyant membrane fractions (BFs) enriched in caveolins and proteins associated with caveolins. Fractions 9–12 were nonbuyant fractions (non-BFs).
Confocal Microscopic Analysis
Endothelial cells were fixed with 4% paraformaldehyde/PBS for 15 minutes and rinsed in PBS. They were then treated with primary antibodies (rabbit anti-mouse Cav1 or goat anti-mouse APNR1 antibody at 1:200 dilution), then tetramethyl rhodamine (TRITC)-conjugated anti-mouse IgG and Cy5-conjugated anti-goat IgG (both at 1:200 dilution). After washing with PBS, coverslips were mounted using an anti-fade solution (KPL, Gaithersburg, MD). Negative control slides without primary antibodies were also stained and processed. Fluorescent images were acquired by Fluoview software (Olympus) on a FV1000 confocal microscope (Olympus, Tokyo, Japan).
Determination of NOx in Endothelial Cell Culture Medium
Endothelium-derived NO and its metabolic products (NO2 and NO3), collectively known as NOx, were determined using a chemiluminescent NO detector (Sierver 280i, Boulder, CO, USA) as described in our previous study [26]. Briefly, samples of 50 μl of culture medium were injected into a gas stripping apparatus containing 2 ml of a 1% solution of vanadium (III) chloride in hydrochloric acid at 90°C which was connected to an NO analyzer. NO was measured according to the instructions of the manufacturer. The sensitivity of the NO analyzer is < 10 pmol/ml, with a linearity of 4 orders of magnitude. Calibrations were made according to the manufacturer’s instructions with standard solutions of sodium nitrate and sodium nitrate (Sigma), respectively.
Animal model and Aortic Contractility and Relaxation Assay
All in vivo experiments were performed upon adult (8 weeks old) male C57BL/6 mice, in adherence with the National Institutes of Health Guidelines on the Use of Laboratory Animals, and were approved by the Thomas Jefferson University Committee on Animal Care. Experimental mice were randomized to receive a high fat diet (HFD) (60 kcal%) (Research Diets Inc. D12492i) or a 10 kcal% control normal diet (ND, D12450Bi) containing the same protein content as the HFD. 10 weeks after ND or HFD, mice were anesthetized with 2% isoflurane, and descending aortic segments were isolated. Aortic segments were maintained into ice-cold Krebs-Henseleit (K-H) buffer consisting of (in mM): NaCl (118), KCl (4.75), CaCl2.2H20 (2.54), KH2PO4 (1.19), MgSO4.7H20 (1.19), NaHCO3 (25), and glucose (10.0). Loose fat and connective tissue were removed, and 2–3 mm aorta rings were isometrically mounted upon a Muti-Wire Myograph System (DMT 620M, DMT-USA Inc, Ann Arbor, MI). Each aortic ring’s resting tension was set at 4 mN, and maintained throughout the experiment. During the one hour equilibration, the rings were exposed to K-H buffer in the tissue bath (replaced every 15 minutes). Phenylephrine (10−9 to 10−6 M) was applied to evaluate contraction response. Upon achieving stable contraction, globular APN (0.3, 1, 10, and 30 μM) was administered to determine APN-induced vasorelaxative response. After ring response to cumulative APN administration stabilized, bath washout occurred, and re-equilibration commenced. The procedure was then repeated with 1–100 μM dose increments of acidified NaNO2 (prepared by dissolving NaNO2 in 0.1 N HCl titrated to pH 2.0), an agent directly inducing vasorelaxation. Data was obtained and analyzed by a Powerlab system (ADInstruments, Colorado Springs, CO).
Statistical analyses
Quantitative results are expressed as mean±SEM. Comparisons between two groups were analyzed by t test, comparisons among more than two groups were made by one-way analysis of variance, and comparisons between each group were made by post hoc analysis via Tukey test. P values less than 0.05 were considered statistically significant. All statistical analyses were performed via GraphPad Prism6.
Results
HG/HL treatment decreased APN signaling response in endothelial cells
Initiation and progression of endothelial dysfunction in type II diabetes is characterized by decreased bioavailability of endothelium-derived NO. Therefore, we determined whether HG/HL treatment altered APN signaling response in endothelial cells. In HUVECs cultured with control medium (Control), APN treatment increased NO production, enhanced eNOS phosphorylation at Ser 1177, increased AMPK phosphorylation, and enhanced Akt phosphorylation (Figure 1). Importantly, exposing HUVECs to HG/HL for 72 hours significantly blunted or virtually abolished APN signaling. Specifically, gAPN-mediated NO production and phosphorylation of eNOS, AMPK, Akt and VASP were all markedly blunted compared to APN response in HUVECs cultured with normal medium (decreased to 43.4%, 22.3%, 36.2%, and 29.1% of control cell response, respectively). These results demonstrate impaired endothelial NO production in response to APN in a condition mimicking diabetic pathology, initiating endothelial dysfunction.
Figure 1.

APN significantly increased NO production (chemiluminescent NO detector, A) and stimulated phosphorylation of eNOS (B), AMPK (C), Akt (D) and VASP (E) in control cells cultured with normal glucose/normal lipid (Control). After 72 hours of HUVEC exposure to HG/HL, APN biologic function was determined. NO production, eNOS, AMPK, Akt and VASP phosphorylation were significantly attenuated. N=6–8 dishes/group from at least 4 different cell batches. *P<0.05, **P<0.01 vs. respective vehicle control; ##P<0.01 vs. control group treated with APN.
HG/HL inhibits APN’s anti-inflammatory effects
It is well known APN and TNFα exert reciprocal effects at multiple levels. Moreover, diabetes itself exacerbates inflammation, oxidative/nitrative stress, and increases TNFα production. However, whether the anti-TNFα effect of APN is impaired under diabetic conditions has not been previously investigated. HUVECs were cultured with normal culture medium or HG/HL medium in the presence or absence of TNFα. The effect of APN upon nitrotyrosine content and adhesion molecule expression was subsequently determined. As we and others have reported, TNF-α markedly increased nitrotyrosine production and ICAM-1 expression (Figure 2). After HG/HL administration, TNF-α further augmented nitrotyrosine production and ICAM-1 expression (1.22 and 1.27-fold, respectively). More importantly, whereas APN significantly decreased nitrotyrosine production and ICAM-1 expression in control HUVECs, HG/HL treatment reduced APN’s inhibitive effect upon nitrotyrosine formation (50.2% inhibition), and virtually abolished its effect upon ICAM-1 expression (Figure 2A–2B). These results suggest HG/HL decreases APN’s suppressive effect upon TNF-α induced inflammation and oxidative/nitrative stress.
Figure 2.

APN attenuated TNF-α induced nitrotyrosine formation (A) and ICAM-1 expression (B) in control cells cultured with normal glucose/normal lipid (Control). After 72 hours of HUVEC exposure to HG/HL, APN failed to reduce TNF-α mediated nitrotyrosine production (A) and ICAM-1 expression (B). No significant alteration was observed in expression levels of AdipoR1, AdipoR2 and APPL1 (C). N=6–8 dishes/group from at least 4 different cell batches.**P<0.01 vs. TNFα+Vehicle.
Expression of APN signaling molecules are unaltered 72 hours after HG/HL culture, but interaction between AdipoR1 and Cav1 is significantly impaired
The results presented in Figures 1 and 2 demonstrate HG/HL impairs APN function, contributing to diabetic endothelial dysfunction and inflammatory response. To determine the molecular mechanisms leading to impaired APN function (APN resistance), we determined the expression levels of molecules known to be involved in APN signaling. Somewhat to our surprise, no significant changes were observed in the expression levels of AdipoR1, AdipoR2, APPL1, AMPK, or eNOS after 72 hours of HG/HL culture (Figure 2C), indicating mechanisms other than altered APN signaling molecule expression are responsible for impaired APN signaling in HG/HL-cultured HUVEC. We recently reported AdipoR1 directly interacts with Cav3 in adult cardiomyocytes, facilitating APN cardiac signaling [27]. However, whether AdipoR1 may form a signaling complex with endothelial-expressed Cav1 to facilitate endothelial APN signaling (and more importantly, whether this interaction is altered by HG/HL) has not been previously investigated. To evaluate these possibilities, several experiments were performed.
Confocal image overlay reveals AdipoR1 and Cav1 reside in the same cell membrane region (Figure 3A). To further establish the relationship between AdipoR1 and Cav1, HUVEC lysates were immunoprecipitated with an antibody against Cav1 or nonimmune IgG. AdipoR1/Cav1 proteins were detected in precipitates via Western blot. As shown in Figure 3B, AdipoR1 was detected in lysates immunoprecipitated with an antibody against Cav1, but was not detected in lysates immunoprecipitated with a non-immune IgG. When analyzed in reverse (immunoprecipitating the lysates with an antibody against AdipoR1 first), Cav1 protein was detected by Western blot (Figure 3B). To confirm the caveolar distribution of the AdipoR1/Cav1 complex, a caveolae-rich detergent-resistant membrane fraction was prepared from HUVECs. As illustrated in Figure 3C, AdipoR1 and Cav1 proteins were associated with the detergent-resistant membrane fractions (fractions 5–7).
Figure 3.

Adiponectin receptor 1 (AdipoR1) co-localizes (A) and interacts with Cav1 (B). AdipoR1 was mainly distributed in the caveolae-rich fraction of detergent-resistant fractions (5–6), but seldom in detergent-soluble fractions (C). AdipoR1/Cav1 interaction was detected in HUVECs co-transfected with Cav1/AdipoR1 plasmid including full length AdipoR1 (FL), truncated peptide1-230, truncated peptide316-375, but not in cell lysates expressing truncated AdipoR1 peptide1-215 and truncated AdipoR1337-375 (D). Conversely, Cav1/AdipoR1 interaction was identified in HUVECs co-transfected with full length AdipoR1 and full length Cav1(FL), truncated Cav1 peptides61-101, truncated Cav11-101, but not in truncated Cav11-81 (E). N=4–5 dishes/group from at least 4 different cell batches. Abbreviations: Ir, irrelevant IgG; FL, full length; IP, immunoprecipitation; IB, immunoblot.
It has been demonstrated previously that membrane proteins interact with the caveolin scaffolding domain via a specific caveolin-binding motif (ϕXϕXXXXϕ or ϕXXXXϕXXϕ, where ϕ is an aromatic residue)20, 21. Sequence analysis revealed 2 potential caveolin-binding motifs in AdipoR1 (220FVPWLYYSF228 and 325FFPGKFDIW333). To determine whether AdipoR1 binds Cav1 via these specific motifs, vectors expressing Myc-tag full-length AdipoR1 or truncated AdipoR1 were co-transfected with Flag-tag full length Cav1 into HUVECs. A co-immunoprecipitation assay demonstrated Cav1 protein was detected in cell lysates expressing full length AdipoR1, truncated AdipoR11-230, or truncated AdipoR1316-375, but not in cell lysates expressing truncated AdipoR11-215 or truncated AdipoR1337-375 (Figure 3D). These results demonstrate AdipoR1 interacts with Cav1 via the specific Cav1 binding motif. To determine whether the Cav1 scaffolding domain (residues 81-101) is required for AdipoR1 binding, we co-transfected Flag tag labeled truncated Cav1 protein and Myc tag labeled full length AdipoR1 into 293T cells (Figure 3E). AdipoR1 protein was detected in cell lysates with full length Cav1 or truncated Cav161-101 and Cav11-101, but not in cell lysates with truncated Cav11-81, signifying AdipoR1 specifically binds to the Cav1 scaffolding domain.
Cav1 signaling is required for APN’s anti-inflammatory and anti nitrative/oxidative effect in HUVECs
To test whether Cav1/AdipoR1 interaction is altered in the diabetic condition, the effect of HG/HL upon Cav1/AdipoR1 complex formation was determined. A co-immunoprecipitation assay demonstrated markedly reduced interaction between Cav1 and AdipoR1 after 72 hours of HG/HL treatment. Detection of Cav-1/AdipoR1 interaction in reverse sequence confirmed a similar result (Figure 4B). To determine the mechanism underlying impaired Cav1/AdipoR1 interaction after HG/HL culture, the expression levels of Cav1 and AdipoR1 was determined. Interestingly, although AdipoR1 expression level remains unaltered, Cav1 expression decreased 63.5% after 72 hours HG/HL treatment (Figures 4C and 4D respectively).
Figure 4.

AdipoR1 interacts with Cav1 to form a protein complex, an interaction markedly reduced when HUVECs were treated with HG/HL for 72 hours (A). Cav1 interacted with AdipoR1, an interaction decreased after 72 hours HG/HL treatment (B). AdipoR1 expression was unchanged after 72 hours HG/HL treatment of HUVECs (C). Cav1 expression was markedly decreased after 72 hours HG/HL treatment of HUVECs (D). N=6–8 dishes/group from at least 4 different cell batches. Abbreviations: Con, control. **P<0.01 vs. vehicle control.
To investigate whether reduced Cav1 expression and impaired Cav1/AdipoR1 interaction contributes to the loss of APN-induced anti-inflammatory and anti-nitrative/oxidative stress when exposed to HG/HL, loss- and gain-of function approaches were employed by utilizing Cav1 siRNA or Cav1 plasmid. Cav1 knockdown (Figure 5C, Cav1) abolished the attenuating effect of APN upon TNFα-induced nitrotyrosine formation (Figure 5A), superoxide production (Figure 5B), gp91phox expression (Figure 5C/D), and ICAM-1 expression (Figure 5C/E). Conversely, when HUVECs overexpressing Cav1 were exposed to HG/HL for 72 hours, APN markedly stimulated eNOS and AMPK phosphorylation (Figure 6A/B). Moreover, APN’s anti-TNFα effects (as determined by ICAM-1 expression and nitrotyrosine formation) were fully recovered (Figure 6C/D). These data suggest Cav1 is critical for APN-induced anti-nitrative/oxidative stress and anti-inflammatory response in endothelial cells.
Figure 5.

Cav1 knockdown abolished APN’s anti-nitrative stress (determined by nitrotyrosine content) (A) and anti-oxidative stress (determined by superoxide generation) (B), and markedly blunted APN’s anti-NADPH oxidase (gp91phox) expression (C= representive blots; D=bar graph) and ICAM-1 expression (C= representive blots; E=bar graph) in HUVECs pretreated with gAPN 2 hours, and subjected to TNF-α (10 hours). N=6–8 dishes/group from at least 4 different cell batches. *P<0.05, **P<0.01 vs. vehicle control.
Figure 6.

Cav1 overexpression (Cav-1OE) preserved APN-induced eNOS and AMPK phosphorylation (A, B) in HUVECs cultured in HG/HL for 72 hours. Cav-1OE restored APN’s anti-nitrative and anti-inflammatory effect (C/D) following TNFα treatment in HUVECs cultured with HG/HL medium. N=6–8 dishes/group from at least 4 different cell batches. #P<0.05, ##P<0.01 vs. APN+TNFα in control group (normal culture medium); $P<0.05, $$P<0.01 vs. HUVECs transfected with empty plasmid (Empty).
Cav1 scaffolding domain facilitates adiponectin signaling transduction
The above results concerning the interaction between AdipoR1 and Cav1 suggest Cav1 plays an important role in downstream APN signaling transduction. To further distinguish adiponectin signaling transduction dependency upon caveolar structure or caveolin scaffolding-mediated protein-protein interaction, additional experiments were performed. First, HUVEC endogenous Cav-1 expression was knocked down via Cav-1 siRNA. 72 hours after Cav1 siRNA, a wild-type or mutated Cav-1 was knocked-in. 72 hours after Cav1 knock-in, caveolae structure was determined via electron microscopy. APN signaling was determined by AMPK and eNOS phosphorylation. Cav1 knockdown significantly inhibited caveolae formation, which was restored by knock-in of either wild-type or mutated Cav1 (Figure 7A). However, attenuated AMPK and eNOS phosphorylation in response to APN was only rescued by knock-in of wild type Cav1, not mutated Cav1 (Figure 7B–D). These data importantly indicate the protein-protein interaction mediated by Cav1 is more essential than cell membrane caveolar structure in mediating adiponectin signaling transduction.
Figure 7.

Knock-in of a wild type Cav1 (Cav1-OE) in Cav1 knock-down cells restored caveolae structure (A) and rescued APN signaling as determined by AMPK and eNOS phosphorylation (B, C, D). In contrast, knock-in of a mutated Cav1 scaffolding domain (Mu Cav1-OE) restored caveolae structure (A), but failed to rescue APN signaling in Cav1 knock-down cells (B, C, D). Arrows in panel A indicate caveolae structures in the cell surface. N=6–8 dishes/group from at least 4 different cell batches. **P<0.01 vs. respective control.
AdipoR1/Cav1 interaction is reduced and vasorelaxative response to APN is virtually abolished in the pre-diabetic vessel
The in vitro experimental results presented above demonstrate AdipoR1/Cav1 interaction is critical in APN signaling, a function impaired by HG/HL incubation. To clarify the in vivo pathologic relevance of this in vitro finding, a high-fat-diet induced diabetic animal model was utilized. In aortic samples from ND mice, AdipoR1/Cav1 complex was clearly detected (Figure 8A, ND). This interaction was significantly diminished in aortic samples from HFD mice (Figure 8A, HFD). Moreover, Cav1 expression is significantly reduced 10 weeks after HFD (Figure 8B). More importantly, in aortic rings freshly isolated from ND mice, APN induces concentration-dependent vasorelaxation (Figure 8C/D, ND). At the maximal APN concentration studied (30 μM), APN induced 51.2±6.9% vasorelaxation. However, APN-induced vasorelaxation was virtually abolished in aortic segments isolated from 10 week-HFD mice (Figure 8C, HFD). The vasorelaxative response to acidified NaNO2 is normal in all aortic segments studied, indicating preserved smooth muscle function in all tested vascular segments.
Figure 8.

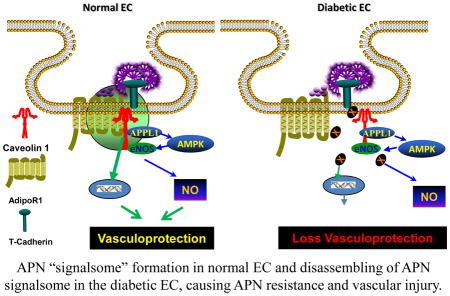
High-fat-diet (10 weeks) induced diabetes impaired AdipoR1/Cav1 integration decreased Cav1 expression, and and blunted APN-induced vasorelaxation. (A) Aortic samples from ND (left two lanes) and HFD (right two lanes) were immunoprecipitated by antibody against AdipoR1, followed by immunoblotting with antibodies against Cav1 or immunoprecipitated by antibody against Cav1, followed by immunoblotting with antibodies against AdipoR1; (B) Cav1 expression was determined by Western blot analysis; (C) Original recording of APN-induced vasorelaxation in aortic segments isolated from ND and HFD mice. (D) Dose-response curves of APN-(left) and acidified NaNO2-induced vasorelaxation in aortic segments isolated from ND and HFD mice (n=10–12 aortic segments/group from at least 4 animals/group). *P<0.05, **P<0.01 vs. ND group. Abbreviations: Ir, irrelevant IgG. (E) Graphic illustration of AdipoR1/Cav1 signalsome in APN transmembrane signaling under normal condition (left) and its impairment under diabetic condition (right).
Discussion
This study investigated the mechanisms of endothelial dysfunction associated with systemic adiponectin resistance, a phenomenon garnering interest given the current obesity epidemic and prevalent associated vascular disease and endothelial dysfunction.
Our study demonstrates high glucose/high lipid impedes adiponectin receptor signaling transduction, compromising APN’s downstream vascular protective effects. The interaction between AdipoR1 and Cav1 is requisite for adiponectin-mediated vasculoprotection. HG/HL conditions interfered with AdipoR1/Cav1 interaction to such extent that exogenous APN supplementation could not restore vascular function to control standards. This finding is consistent with a recent clinical observation [13] and experimental study [29] that elevated plasma adiponectin fails to provide protection in diabetic patients and animals. We demonstrate Cav1 overexpression may restore the anti-inflammatory, anti-oxidative, anti-nitrative effects of APN in an endothelial environment subjected to HG/HL conditions. The vascular protective effects of adiponectin are not solely reliant upon its circulating concentration, but its intact signaling machinery. It is worth noting that Cav1 siRNA alone moderately increased nitrotyrosine content and superoxide generation (Figure 5B/C). Although we do not have detailed molecular mechanism yet, our results suggest that physiological level of Cav1 is important in maintaining anti-oxidative signaling under basal condition.
Significant clinical data supports diabetes as a disease of chronic inflammation and nitrative/oxidative stress. It is accepted that prolonged exposure of the vascular endothelium to elevated levels of circulating inflammatory mediators perturbs endothelial cell homeostasis, provoking endothelial dysfunction [20]. Nitrotyrosine and ICAM-1 are indices of nitrative/oxidative stress and inflammation, processes implicated in accelerated atherogenesis and plaque formation [4]. Both are markedly upregulated in the diabetic vascular endothelium [7,10]. Our study demonstrated hyperglycemia/hyperlipidemia altered the interaction between caveolin-1 and adiponectin receptor 1, deleteriously affecting the anti-inflammatory and vaso-relaxative effects of adiponectin, evidenced by increased nitrotyrosine and ICAM-1. Such results prove for the first time that adiponectin resistance during diabetes has intricate relationship with caveolin family derangement in the setting of unchanged adiponectin receptor function.
We demonstrate the anti-nitrative/oxidative/inflammatory effects of adiponectin are reduced in a pre-diabetic environment, contributing to adiponectin resistance. To further elucidate the responsible molecular mechanisms, caveolin-1 expression was determined during HG/HL pretreatment. Cav1 expression reached nadir after 72 hours HG/HL exposure. Interestingly, AdipoR1 levels remained unchanged throughout HG/HL treatment, evidence that loss of adiponectin’s anti-inflammatory properties cannot be attributed to receptor expression alteration. Caveolins are integral membrane proteins regulating intracellular signal transduction via caveolin scaffolding domains [9]. We previously demonstrated the imperative role Cav3 plays in adiponectin signal regulation in cardiomyocytes [27]. In the current study, by employing an isolated fresh vessel ring experiment, we demonstrated adiponectin enhanced the vasorelaxative effect of acetylcholine, which is lost during HG/HL conditions. In summation, we provide evidence Cav1 facilitates regulation of APN signaling at the plasma membrane of endothelial cells, and beyond.
The present study clarifies two novel aspects of the relationship between caveolin-1 and adiponectin receptor 1 in endothelial cells. Our experiments utilized truncated protein fragments to reveal the Cav1 scaffolding domain can interact with AdipoR1’s caveolin-binding motif; we further demonstrated the interaction was abrogated by mutated Cav1. The necessity of intact Cav1-AdipoR1 interaction for advantageous APN downstream signaling is exhibited many times over in this study; the Cav1-AdipoR1 relationship maintains APN’s attenuation of both TNFα-induced ICAM-1 expression and HG/HL-exacerbated nitrotyrosine production. The second novel finding concerns the facilitation of adiponectin signaling transduction by the Cav1 scaffolding domain, not caveolae itself. The caveolae structure does not modulate adiponectin signaling transduction- proper Cav1 protein expression establishes the foundation for efficient and complete adiponectin signaling pathway transduction.
In summation, Cav-1/AdipoR1 interaction is impaired by in vitro HG/HL exposure, or in vivo HFD. Dissociation of Cav1/AdipoR1 impairs APN signaling, contributing to APN resistance and vascular injury (Figure 8E). Bolstering Cav1 signaling may be a novel approach reducing vascular injury in diabetic patients.
highlights.
The AdipoR1/Cav1 complex is critical for adiponectin signaling
Diabetes downregulated Cav1 expression and separated AdipoR1/Cav1 complex
Diabetes attenuates adiponectin’s nitric oxide stimulatory and anti-oxidant actions
Adiponectin resistance contributes to diabetic endothelial dysfunction
Acknowledgments
We thank Ray Meade and Biao Zuo (of the Electron Microscopy Resource Laboratory of the University of Pennsylvania) for their gracious provision of technical support.
We also thank Dr. Lily Dong (Department of Pharmacology, University of Texas Health Science Center, San Antonio, TX) for her kind gift of wild type AdipoR1 cDNA.
Funding Sources
This work was supported by the following grants: American Diabetes Association 1-14-BS-218, Natural Science Foundation of China 81170199, 31322026 (Y. Wang), 81270185, 81470020 (J. Zhao), National Institutes of Health HL-96686, HL-123404, and American Diabetes Association 1-15-BS-122 (X.L. Ma).
NONSTANDARD ABBREVIATIONS
- AdipoR1
Adiponectin receptor 1
- AdipoR2
Adiponectin receptor 2
- AMPK
AMP activated protein kinase
- APN
Adiponectin
- BFs
Buoyant membrane fractions
- Cav1
Caveolin-1
- CVD
Cardiovascular disease
- eNOS
Endothelial nitric oxide synthase
- HG/HL
High glucose and high lipid
- HUVEC
Human umbilical vein cells
- ICAM-1
Intercellular adhesion molecule-1
- NO
Nitric oxide
Footnotes
Disclosures
The authors declare no competing interests, financial or otherwise.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aljada A. Endothelium, inflammation, and diabetes. Metab Syndr Relat Disord. 2003;1:3–21. doi: 10.1089/154041903321648225. [DOI] [PubMed] [Google Scholar]
- 2.Antoniades C, Antonopoulos AS, Tousoulis D, Stefanadis C. Adiponectin: from obesity to cardiovascular disease. Obes Rev. 2009;10:269–279. doi: 10.1111/j.1467-789X.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- 3.Antonopoulos AS, Margaritis M, Coutinho P, Digby J, Patel R, Psarros C, Ntusi N, Karamitsos TD, Lee R, De SR, Petrou M, Sayeed R. Demosthenous, M., Bakogiannis, C., Wordsworth, P. B., Tousoulis, D., Neubauer, S., Channon, K. M., Antoniades, C. Reciprocal effects of systemic inflammation and brain natriuretic peptide on adiponectin biosynthesis in adipose tissue of patients with ischemic heart disease. Arterioscler. Thromb Vasc Biol. 2014;34:2151–2159. doi: 10.1161/ATVBAHA.114.303828. [DOI] [PubMed] [Google Scholar]
- 4.Bachschmid M, Thurau S, Zou MH, Ullrich V. Endothelial cell activation by endotoxin involves superoxide/NO-mediated nitration of prostacyclin synthase and thromboxane receptor stimulation. FASEB J. 2003;17:914–916. doi: 10.1096/fj.02-0530fje. [DOI] [PubMed] [Google Scholar]
- 5.Betteridge DJ. Lipid control in patients with diabetes mellitus. Nat Rev Cardiol. 2011;8:278–290. doi: 10.1038/nrcardio.2011.23. [DOI] [PubMed] [Google Scholar]
- 6.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 7.Cassuto J, Dou H, Czikora I, Szabo A, Patel VS, Kamath V, Belin de Chantemele E, Feher A, Romero MJ, Bagi Z. Peroxynitrite Disrupts Endothelial Caveolae Leading to eNOS Uncoupling and Diminished Flow-Mediated Dilation in Coronary Arterioles of Diabetic Patients. Diabetes. 2014;63:1381–1393. doi: 10.2337/db13-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celik T, Yaman H. Elevated adiponectin levels in patients with chronic heart failure: an independent predictor of mortality or a marker of cardiac cachexia? Int J Cardiol. 2010;144:319–320. doi: 10.1016/j.ijcard.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Remessy AB, Tawfik HE, Matragoon S, Pillai B, Caldwell RB, Caldwell RW. Peroxynitrite mediates diabetes-induced endothelial dysfunction: possible role of Rho kinase activation. Exp Diabetes Res. 2010;2010:247861. doi: 10.1155/2010/247861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med. 2009;6:27–35. doi: 10.1038/ncpcardio1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haidara MA, Yassin HZ, Rateb M, Ammar H, Zorkani MA. Role of oxidative stress in development of cardiovascular complications in diabetes mellitus. Curr Vasc Pharmacol. 2006;4:215–227. doi: 10.2174/157016106777698469. [DOI] [PubMed] [Google Scholar]
- 13.Hung WC, Wang CP, Lu LF, Yu TH, Chiu CA, Chung FM, Chen HJ, Houng JY, Shin SJ, Lee YJ. Circulating adiponectin level is associated with major adverse cardiovascular events in type 2 diabetic patients with coronary artery disease. Endocr J. 2010;57:793–802. doi: 10.1507/endocrj.k10e-020. [DOI] [PubMed] [Google Scholar]
- 14.Laoutaris ID, Vasiliadis IK, Dritsas A, Mavrogeni S, Kallistratos MS, Manginas A, Chaidaroglou A, Degiannis D, Panagiotakos DB, Cokkinos DV. High plasma adiponectin is related to low functional capacity in patients with chronic heart failure. Int J Cardiol. 2010;144:230–231. doi: 10.1016/j.ijcard.2008.12.126. [DOI] [PubMed] [Google Scholar]
- 15.Lee S, Zhang H, Chen J, Dellsperger KC, Hill MA, Zhang C. Adiponectin abates diabetes-induced endothelial dysfunction by suppressing oxidative stress, adhesion molecules, and inflammation in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2012;303:H106–H115. doi: 10.1152/ajpheart.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, Wang WQ, Zhang H, Yang X, Fan Q, Christopher TA, Lopez BL, Tao L, Goldstein BJ, Gao F, Ma XL. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. Am J Physiol Endocrinol Metab. 2007;293:E1703–E1708. doi: 10.1152/ajpendo.00462.2007. [DOI] [PubMed] [Google Scholar]
- 17.Li R, Xu M, Wang X, Wang Y, Lau WB, Yuan Y, Yi W, Wei X, Lopez BL, Christopher TA, Wang XM, Ma XL. Reduced vascular responsiveness to adiponectin in hyperlipidemic rats-mechanisms and significance. J Mol Cell Cardiol. 2010;49:508–515. doi: 10.1016/j.yjmcc.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 19.Munzel D, Lehle K, Haubner F, Schmid C, Birnbaum DE, Preuner JG. Impact of diabetic serum on endothelial cells: an in-vitro-analysis of endothelial dysfunction in diabetes mellitus type 2. Biochem Biophys Res Commun. 2007;362:238–244. doi: 10.1016/j.bbrc.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 20.Rask-Madsen C, King GL. Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- 21.Razani B, Lisanti MP. Two distinct caveolin-1 domains mediate the functional interaction of caveolin-1 with protein kinase A. Am J Physiol Cell Physiol. 2001;281:C1241–C1250. doi: 10.1152/ajpcell.2001.281.4.C1241. [DOI] [PubMed] [Google Scholar]
- 22.Shimabukuro M, Higa N, Asahi T, Oshiro Y, Takasu N, Tagawa T, Ueda S, Shimomura I, Funahashi T, Matsuzawa Y. Hypoadiponectinemia Is Closely Linked to Endothelial Dysfunction in Man. J Clin Endocrinol Metab. 2003;88:3236–3240. doi: 10.1210/jc.2002-021883. [DOI] [PubMed] [Google Scholar]
- 23.Staiger H, Staiger K, Stefan N, Wahl HG, Machicao F, Kellerer M, Haring HU. Palmitate-induced interleukin-6 expression in human coronary artery endothelial cells. Diabetes. 2004;53:3209–3216. doi: 10.2337/diabetes.53.12.3209. [DOI] [PubMed] [Google Scholar]
- 24.Su H, Lau WB, Ma XL. Hypoadiponectinaemia in diabetes mellitus type 2: molecular mechanisms and clinical significance. Clin Exp Pharmacol Physiol. 2011;38:897–904. doi: 10.1111/j.1440-1681.2011.05606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sumbayev VV. Activities of apoptotic signal 1-regulating protein kinase and poly-(ADP-ribose) polymerase and internucleosomal DNA fragmentation in rat liver during oxidative stress induced by cobalt chloride. Bulletin Of Experimental Biology And Medicine. 2001;131:119–120. doi: 10.1023/a:1017571323899. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ, Lopez BL, Christopher TA, Tian R, Koch W, Ma XL. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation. 2009;119:835–844. doi: 10.1161/CIRCULATIONAHA.108.815043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Wang X, Jasmin JF, Lau WB, Li R, Yuan Y, Yi W, Chuprun K, Lisanti MP, Koch WJ, Gao E, Ma XL. Essential role of caveolin-3 in adiponectin signalsome formation and adiponectin cardioprotection. Arterioscler Thromb Vasc Biol. 2012;32:934–942. doi: 10.1161/ATVBAHA.111.242164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–9788. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 29.Yi W, Sun Y, Gao E, Wei X, Lau WB, Zheng Q, Wang Y, Yuan Y, Wang X, Tao L, Li R, Koch W, Ma XL. Reduced Cardioprotective Action of Adiponectin in High-Fat Diet-Induced Type II Diabetic Mice and Its Underlying Mechanisms. Antioxid Redox Signal. 2011;15:1779–1788. doi: 10.1089/ars.2010.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang P, Wang Y, Fan Y, Tang Z, Wang N. Overexpression of adiponectin receptors potentiates the antiinflammatory action of subeffective dose of globular adiponectin in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:67–74. doi: 10.1161/ATVBAHA.108.178061. [DOI] [PubMed] [Google Scholar]