

Abstract
In recent years, mGlu4 has received great research attention because of the potential benefits of mGlu4 activation in treating numerous brain disorders, such as Parkinson’s disease (PD). A specific mGlu4 PET radioligand could be an important tool in understanding the role of mGlu4 in both healthy and disease conditions, and also for the development of new drugs. In this study, we synthesized four new N-(methylthiophenyl)picolinamide derivatives 11–14. Of these ligands, 11 and 14 showed high in vitro binding affinity for mGlu4 with IC50 values of 3.4 nM and 3.1 nM, respectively, and suitable physicochemical parameters. Compound 11 also showed enhanced metabolic stability and good selectivity to other mGluRs. [11C]11 and [11C]14 were radiolabeled using the [11C]methylation of the thiophenol precursors 20a and 20c with [11C]CH3I in 19.0% and 34.8% radiochemical yields (RCY), and their specific activities at the end of synthesis (EOS) were 496 ± 138 GBq/μmol (n=6) and 463 ± 263 GBq/μmol (n=4), respectively. The PET studies showed that [11C]11 accumulated fast into the brain and had higher uptake, slower washout and 25% better contrast than [11C]2, indicating improved imaging characteristics as PET radiotracer for mGlu4 compared to [11C]2. Therefore, [11C]11 will be a useful radioligand to investigate mGlu4 in different biological applications.
Keywords: PET, metabotropic glutamate receptor subtype 4 (mGlu4), positive allosteric modulator (PAM)
Graphical Abstract
As the most abundant excitatory neurotransmitter in the central nervous system (CNS) of vertebrates, L-glutamate mediates more than 50% of all synapses.1, 2 Metabotropic glutamate receptors (mGluRs) and ionotropic glutamate receptors (iGluRs) are two major classes of glutamate receptors. The mGluRs belong to the class C G protein-coupled receptors (GPCR) superfamily, which have a distinct large extracellular N-terminus. The mGluRs can be further divided into three subgroups including eight known receptor subtypes (group I: mGlu1 and mGlu5, group II: mGlu2 and mGlu3, and group III: mGlu4, mGlu6, mGlu7 and mGlu8) based on their structural similarity, ligand specificity, and preferred coupling mechanisms.3 The mGluRs have distinctive biodistribution in CNS depending on subtypes and subgroups and are involved in glutamate signaling in almost every excitatory synapse in CNS.4
In recent years, mGlu4 has received a lot of research attention because of the potential benefits of mGlu4 activation in treating several brain disorders, such as Parkinson’s disease (PD).5–7 As a group III mGluR, mGlu4 interacts with the Gαi/o subunit of G-protein which negatively couples with adenylate cyclase to inhibit cAMP dependent signal pathways.8, 9 The mGlu4 is expressed at multiple synapses throughout the basal ganglia, mainly localized presynaptically and expressed in the striatum, hippocampus, thalamus, and cerebellum.3, 10, 11 Its activation reduces neurotransmitter release, a mechanism implicated in the pathophysiology of PD. The activation of mGlu4 can be accomplished by two different mechanisms: orthosteric agonists (competing with L-glutamate at the extracellular N-terminal’s Venus Flytrap Domain) or noncompetitive positive allosteric modulators (PAMs). Previous orthosteric ligands of mGlu4 lack clear subtype selectivity and blood-brain barrier (BBB) penetration. However, noteworthy examples of selective and brain penetrant orthosteric agonists such as LSP4-2022 exist.12, 13
Much recent effort has been focused on the development of allosteric modulators, which target the seven-transmembrane spanning domain. In particular, the allosteric modulation of mGlu4 has prompted intense interest after (−)-PHCCC (1), a partially selective mGlu4 PAM, was discovered and demonstrated activity in models of neuroprotection and PD.14 There has been substantial progress in identifying PAMs for mGlu4, and hundreds of mGlu4 PAMs have been reported and/or patented since 2009.5, 15, 16 Fig. 1 shows representative mGlu4 PAMs.5, 15, 17–20 Subsequent results with mGlu4 PAMs have further validated the antiparkinsonian activity in animal models of PD.11, 19, 21–24 This approach has opened a new avenue for developing nondopaminergic treatments for PD and for identifying novel disease modifying therapeutics.
Figure 1.

Representative mGlu4 PAMs.
As a noninvasive medical and molecular imaging technique and a powerful tool in neurological research, positron emission tomography (PET) offers a possibility to visualize and analyze the target receptor expression under physiological and pathophysiological conditions. PET has mostly been used to detect disease-related biochemical changes before the disease-associated anatomical changes can be found using standard medical imaging modalities. Moreover, PET radiotracers serve as invaluable biomarkers during the development of potential therapeutic drugs. Thus, extensive research effort has been directed towards the development of PET radioligands suitable for probing mGlu1 and mGlu5, in which many PET radiotracers have been reported and several of which, such as [18F]FIMX25 for mGlu1, and [18F]FPEB26,27, [18F]SP20328 and [11C]ABP68829 for mGlu5, have been advanced for human clinical trials.
Although many PET probes have been developed for mGlu1 and mGlu5, mGlu4 still lacks useful PET radioligands for clinical study. A specific mGlu4 PET radioligand could be an important tool for understanding the role of mGlu4 in healthy and disease conditions, and also for the development of new drugs targeting this receptor. Recently, we reported a carbon-11 labeled PET radioligand [11C]230 and a fluorine-18 labeled PET radioligand [18F]831 (Fig. 2). These compounds exhibited some favorable features as PET radioligands such as fast uptake into the brain and specific accumulation in mGlu4-rich regions of the brain. However, in comparison to one of the best mGlu5 PET radiotracers [18F]FPEB26, 27, these compounds showed decreased retention time in the brain, which may affect the quality of imaging. The results indicate that the affinity and metabolic stability of this class of radiotracers need further optimization.
Figure 2.

The PET radiotracers for mGlu4.
Thus, we have carried out the structure-affinity relationship (SAR) study of a series of new N-phenylpicolinamide derivatives.32 It was then discovered that the 3-methylthio group was superior to the 3-methoxy group for mGlu4 affinity by comparing compounds 9 (IC50=13.7 nM) and 10 (IC50=4.9 nM), showing a 2.8 fold enhancement in affinity. Metabolic stability was considered one of the major issues for ML128 (2), in which the 3-methoxy group was recognized as the metabolic soft group. As the N-methylthiophenyl derivatives may have a different metabolic profile from the corresponding N-methoxyphenyl analogs, it is interesting to understand its effect on the metabolic stability of the picolinamide derivatives. Based on these reasons, we extended our SAR study to four new N-(methylthiophenyl)picolinamide derivatives 11–14 (Fig. 3).
Figure 3.

The N-phenylpicoliamide derivatives for SAR study.
Compounds 11 and 12 have a Cl- or F-substitution at the 4-phenyl position, respectively, in which the same substitutions have enhanced the affinity for N-(3-methoxyphenyl)picolinamide and they may have a similar effect for N-(3-(methylthio)phenyl)picolinamide. The 4-phenyl position of the N-phenylpicolinamide was tolerated with a relatively large substitution as demonstrated in compounds 6 and 7, so we synthesized compounds 13 and 14, which are the regioisomers of 10 and 11, respectively. Furthermore, these compounds can be easily radiolabeled by carbon-11 on the thiomethyl group.
Since poor BBB permeability and high nonspecific binding (NSB) are among the frequent causes for failure in CNS PET radioligand development, it is necessary to consider some important physicochemical parameters such as MW, cLogP, tPSA, and HBD at the design stage. It has been proposed that more desirable ranges for CNS drugs are tPSA < 90; HBD < 3; 2 < cLogP < 5; and MW < 450.33 As shown in Fig. 3, these compounds possess favorable physicochemical parameters, making them good candidates for CNS ligand development.
The syntheses of compounds 11–14 are illustrated in Scheme 1. Compounds 11 and 12 were synthesized in six steps (Scheme 1a). 5-Amino-2-halo-thiophenol (17a and 17b) were prepared by the chlorosulfonylation of 4-halo-nitrobenzene (15a and 15b) followed by tin(II) chloride mediated reduction in acidic media. To prevent nucleophilic attack on the thiol group during the amide coupling reaction, 17a and 17b were oxidized to form a disulfide bond. The resulting amino-dimer 18a and 18b were coupled with picolinic acid or its acid chloride under the corresponding amide coupling conditions to give 19a and 19b. The disulfide bond in 19a and 19b was reduced using either sodium borohydride or tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) to give the corresponding thiophenol derivatives, 20a and 20b, respectively. 20a and 20b were methylated using iodomethane (CH3I) in presence of potassium carbonate (K2CO3) or diisopropylethylamine (DIPEA) to give 11 and 12, respectively. Compound 13 was synthesized by an amide coupling reaction between acid chloride of picolinic acid and aniline 21 in 60% yield (Scheme 1b). Compound 14 was synthesized from 22 in four steps. 22 was derivatized with ammonium thiocyanate to give 23 in 65% yield. Picolinic acid was converted to acid chloride and then coupled with 23 to form amide 24 in 88% yield. The nitrile group in 24 was removed by sodium borohydride to give 20c in 75% yield. The methylation of 20c was carried out using CH3I and K2CO3 to give 14 in 28% yield (Scheme 1c).
Scheme 1.

Syntheses of 11–14 and thiophenol precursors, 20a and 20c.
Compounds, 2 and 9–14, were evaluated with competitive binding studies using mGlu4 transfected CHO cells by increasing the concentration of test materials from 0.01 nM to 10 μM in the presence of 2 nM of [3H]2.31 As shown in Table 1, the IC50 values of 11 and 14 are 3.4 and 3.1 nM, respectively, in which the affinity is enhanced by 1.5–1.6 fold compared to 2 (IC50 = 5.1 nM). Compound 10 (IC50 = 4.9 nM) has a similar affinity to 2, while compounds 12 and 13 show reduced affinity. Compounds 10–14 were further tested for their metabolic stability in microsome from rat liver extract. Compound 2 was also examined at the same time as a reference. As Table 1 shows, the metabolic stabilities are in the following order: 11>2>10>12>13>14. Although the half-life difference between compounds 2 and 11 is not significant, we anticipate that the difference of the half-life in the brain between these two compounds could be more significant. It is interesting to note that as the regioisomers of 10 and 11, compounds 13 and 14 display a substantial decreased trend in metabolic stability. The results show that compound 11 has the most promising enhanced affinity and/or increased in vitro microsomal stability compared to other compounds (significance p<0.05, t-test) in this series.
Table 1.
In vitro properties.
| Affinitya | Stabilityb | ||||
|---|---|---|---|---|---|
| IC50 (nM) | Log(IC50 ± SEM) | T1/2 (min) | κc (min−1) | SEM(κ) | |
| 2 (ML 128) | 5.1 | −8.29 ± 0.09e | 5.99 | 0.116 | 0.007 |
| 9 | 13.7 | −7.86 ± 0.10e | n.d.d | n.d. | n.d. |
| 10 | 4.9 | −8.31 ± 0.07e | 5.83 | 0.119 | 0.009 |
| 11 | 3.4 | −8.46 ± 0.07 | 6.54 | 0.106 | 0.005 |
| 12 | 7.8 | −8.11 ± 0.10 | 5.73 | 0.121 | 0.008 |
| 13 | 13.3 | −7.88 ± 0.09 | 4.14 | 0.168 | 0.013 |
| 14 | 3.1 | −8.50 ± 0.09 | 3.86 | 0.179 | 0.016 |
Binding inhibition assay
Microsomal stability assay (rat)
The decay constant that is slope of log concentration vs time profile (t1/2=ln2/κ).
Not determined
Ref. 32
The selectivity of 11 to other mGluRs including group I (mGlu1 and mGlu5), II (mGlu2) and III (mGlu6 and mGlu8) were checked (Supplementary material). The assays were carried out at eight different concentrations between 0.1 nM and 30 μM of 11. The results indicated that compound 11 did not have agonist and antagonist activities (>30 μM) toward mGlu1, mGlu5, mGlu2 and mGlu6, but displayed a weak agonist activity to mGlu8 (EC50 = 14 μM).34, 35 The selectivity data supports compound 11 as the most promising imaging agent candidate.
Since compounds 11 and 14 displayed the most promising affinity, they were selected for in vivo evaluation as potential mGlu4 radiotracers and were compared to [11C]2 to understand how their structure, affinity, and metabolic stability affect PET imaging.
Carbon-11 labeling was performed by methylation of the thiophenol precursors (20a and 20c) using [11C]CH3I and K2CO3 in acetone at 50 °C for 3 min (Scheme 2). All radiosyntheses took 35–40 min from the end of bombardment (EOB) to the end of synthesis (EOS). The carbon-11 labeled compounds [11C]11 and [11C]14 were obtained in 19.0 ± 7.7% (n=7) and 34.8 ± 8.0% (n=4) RCYs from [11C]CO2, respectively. Their specific activities were 496 ± 138 GBq/μmol (n=6) and 463 ± 263 GBq/μmol (n=4) at EOS, respectively.
Scheme 2.

Radiosyntheses of [11C]11 and [11C]14.
Fig. 4a indicates that the difference in retention times between the precursors (20a and 20c) and the corresponding radiolabeled products ([11C]11 and [11C]14) was big enough to purify the radiotracers as a baseline separation. The HPLC profile of [11C]14 was similar to that of [11C]11. The identities of [11C]11 and [11C]14 were confirmed by coinjection with the corresponding unlabled compound on a radio-HPLC. As shown in Fig. 4b, the retention time of [11C]11 was the same as that of coinjected nonradioactive standard 11. The QC analysis of [11C]14 was also conducted in the same way. The radiochemical purities of [11C]11 and [11C]14 determined by radio-TLC were 98.0 ± 0.5% (n = 7), and 97.5 ± 1.0% (n = 4), respectively. Similar results were also observed in the radio-HPLC analyses.
Figure 4.

The UV and radioactivity profiles (a) the reaction mixture was purified by a semi-preparative HPLC and (b) the QC analysis of the purified [11C]11 was carried out by an analytical HPLC. (a) Gemini-NX C18 semi-preparative column (Phenomenex, 250 mm × 10 mm, 5 μm), ammonium formate solution (0.1 M) : acetonitrile = 40:60, 4 mL/min (b) Alltima C18 analytical column (150 mm × 4.6 mm, 5 μm), 0.1% TFA solution : acetonitrile = 35:65, 1 mL/min.
We determined the LogD7.4 values for [11C]11 and [11C]14 as 3.15 ± 0.04 (n = 4) and 3.00 ± 0.05 (n = 4) using octanol and phosphate buffered saline (PBS, pH7.4), which lie in the range normally considered favorable for a PET radioligand. The calculated cLogP values of 11 and 14 are both 3.63 (Fig. 3), which is higher than the experimental values.
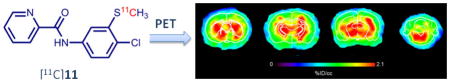
PET imaging studies were conducted using male Sprague Dawley rats. The in vivo uptake and kinetics were examined using small-animal PET. The PET images (Fig. 5) indicated that [11C]11 and [11C]14 crossed the BBB quickly and were mainly accumulated in the thalamus, hippocampus, cerebellum, and striatum, which were reported as the mGlu4-rich regions of the rat brain.10, 36–38 We also acquired PET images of [11C]2 in the same time frames for comparison. To confirm the corresponding brain regions, anatomical templates of rat brains were used for the images with [11C]11 (Fig. 5). The results suggested that [11C]11 gave the best contrast among the three radiotracers and displayed better images in the hippocampus, striatum, thalamus and cerebellum. Using the mGlu4-negligible region in the brain as a reference, we calculated the uptake ratio (contrast) of radioactivity between the mGlu4-rich and the reference regions (areas immediately outside the cortical areas) from the reconstructed PET images. The maximum ratio was 1.67 ± 0.08 (n = 30; 10 images in 3 baseline studies) for [11C]2 and 2.10 ± 0.25 (n = 30) for [11C]11. The results show that the signal to noise ratio of [11C]11 was enhanced up to 25% compared to [11C]2.
Figure 5.

Color-coded PET images of [11C]11, [11C]14, and [11C]2 at different brain levels (striatum, thalamus, hippocampus, and cerebellum). The images represent distribution of radioligand from 5 to 10 min after administration. All the images were normalized to the highest voxel value (%ID/cm3) of the whole data set. These images demonstrate improved imaging characteristics of [11C]11 compared to [11C]14 and [11C]2 based on the high accumulation and slow washout. The thickness of each slice was 0.625 mm.
The regional time-activity curves (TACs) of PET with [11C]2, [11C]11, and [11C]14 in the hippocampus, striatum, thalamus and cerebellum of rat brains are given in Fig. 6. The %ID/cc values at each time point obtained from the different PET images were averaged for each radiotracer and converted into TACs. As shown in the TACs, the radioactivity of [11C]2, [11C]11, and [11C]14 quickly increased in the brain within 2 min after a tail vein injection and then washed out within 20 min. The maximum radioactivity uptake depends on the region in the brain. The highest uptake for [11C]11 was seen in the thalamus, followed by the striatum, hippocampus, and cerebellum. The results indicate that [11C]11 has better brain uptake than [11C]2 and [11C]14 and is an improved PET radioligand for mGlu4.
Figure 6.

Brain regional TACs of PET with [11C]2, [11C]11, and [11C]14 in (a) hippocampus, (b) striatum, (c) thalamus and (d) cerebellum of rat brain. The %ID/cc values at each time point were averaged from different studies for constructing TACs.
A blocking experiment for [11C]11 was carried out 2 h after the baseline scans to compare blocking images with baseline images. An mGlu4 PAM (3·HCl) was used as an mGlu4 blocking agent. The blocking agent was administered intravenously 1 min before [11C]11 injection.
The mGlu4 blocking effects with compound 3 were 19–24% for first 5 min in selected regions of interest (ROIs). (Fig. 7) The incomplete blocking effect by compound 3 might be attributed to its relatively weak affinity and fast kinetics.17,32
Figure 7.

Blocking studies of [11C]11 with an mGlu4 PAM (3·HCl, 10 mg/kg, iv) during first 5 min after injection of [11C]11. Each rat was scanned for baseline and individually normalized (whole brain was set as 1.0). The blocking study was done with same instrumentation and settings. The blocking results were normalized to corresponding baseline results. Integrals were calculated over 0–5 min time periods. (n=4 blotted with S.D and significances were calculated using t-test, ** = p<0.01)
In this study, we synthesized four new N-(methylthiophenyl)picolinamide derivatives 11–14. Of these ligands, 11 and 14 showed high in vitro binding affinity for mGlu4 together with suitable physicochemical parameters. Compound 11 also showed enhanced metabolic stability and good selectivity to other mGluRs. [11C]11 and [11C]14 were radiolabeled via the [11C]methylation of the thiophenol precursors, 20a and 20c, with [11C]CH3I in reliable RCYs and specific activities. The PET studies showed that [11C]11 accumulated fast into the brain and had higher uptake, slower washout and 25% better contrast than [11C]2, indicating improved imaging characteristics as a PET radiotracer for mGlu4 compared to [11C]2. [11C]11 will be a useful radioligand to investigate mGlu4 in different biological applications.
Supplementary Material
Acknowledgments
Funding Sources
NIBIB-R01EB012864 and NIMH-R01MH91684 to A-L Brownell.
The authors would like to thank the technical support of the PET/MRI and radiochemistry facility at the Athinoula A. Martinos Center for Biomedical Imaging at Massachusetts General Hospital for the operation of cyclotron and synthesis modules. The authors are also grateful to Drs. Tanabe and Nakanishi’s laboratory (Osaka Bioscience Institute, Osaka, Japan) for providing the vector of mGlu4 from the rat as a gift. The following supporting instrument grants (1S10RR029495-01, 1S10RR026666-01, and 1S10RR023452-01) are also appreciated. The authors appreciate the generous help of the NIMH PDSP program (Contract # HHSN-271-2008-00025-C) led by Dr. Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and the Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. Finally, the authors would like to express their appreciation of the financial support for PP from The Orion Farmos Research Foundation, Saastamoinen Foundation, Sigrid Juselius Foundation, Osk Huttunen Foundation and Kuopio University Foundation.
Footnotes
Notes
The authors declare no competing financial interest.
Supplementary material (experimental procedures, spectroscopic characterization, HPLC data, in vitro selectivity data, and TACs for mGlu4 blocking studies) associated with this article can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Conn PJ. Ann N Y Acad Sci. 2003;1003:12. doi: 10.1196/annals.1300.002. [DOI] [PubMed] [Google Scholar]
- 2.Watkins JC, Jane DE. Br J Pharmacol. 2006;147:S100. doi: 10.1038/sj.bjp.0706444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conn PJ, Pin JP. Annu Rev Pharmacol Toxicol. 1997;37:205. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 4.Riedel G, Platt B, Micheau J. Behav Brain Res. 2003;140:1. doi: 10.1016/s0166-4328(02)00272-3. [DOI] [PubMed] [Google Scholar]
- 5.Robichaud AJ, Enger DW, Lindsley CW, Hopkins CR. ACS Chem Neurosci. 2011;17:433. doi: 10.1021/cn200043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amalric M, Lopez S, Goudet C, Fisone G, Battaglia G, Nicoletti F, Pin JP, Acher FC. Neuropharmacol. 2013;66:53. doi: 10.1016/j.neuropharm.2012.05.026. [DOI] [PubMed] [Google Scholar]
- 7.Huang X, Dale E, Brodbeck RM, Doller D. Curr Top Med Chem. 2014;14:1755. doi: 10.2174/1568026614666140902143830. [DOI] [PubMed] [Google Scholar]
- 8.Fettagutti F, Balani-Guerra B, Corsi M, Nakanishi S, Corti C. Eur J Neurosci. 1999;11:2073. doi: 10.1046/j.1460-9568.1999.00626.x. [DOI] [PubMed] [Google Scholar]
- 9.Niswender CM, Conn PJ. Annu Rev Pharmacol Toxicol. 2010;50:295. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corti C, Aldegheri L, Somogyi P, Ferraguti F. Neuroscience. 2002;110:403. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- 11.Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. J Pharmcol Exp Ther. 2005;313:1296. doi: 10.1124/jpet.104.080481. [DOI] [PubMed] [Google Scholar]
- 12.Goudet C, Vilar B, Courtiol T, Deltheil T, Bessiron T, Brabet I, Oueslati N, Rigault D, Bertrand HO, McLean H, Daniel H, Amalric M, Acher F, Pin JP. FASEB J. 2012;26:1682. doi: 10.1096/fj.11-195941. [DOI] [PubMed] [Google Scholar]
- 13.Cajina M, Nattini M, Song D, Smagin G, Jørgensen EB, Chandrasena G, Bundgaard C, Toft DB, Huang X, Acher F, Doller D. ACS Med Chem Lett. 2014;5:119. doi: 10.1021/ml400338f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marino MJ, Valenti O, Conn PJ. Drug & Aging. 2003;20:377. doi: 10.2165/00002512-200320050-00006. [DOI] [PubMed] [Google Scholar]
- 15.Lindsley CW, Hopkins CR. Expert Opin Ther Pat. 2012;22:461. doi: 10.1517/13543776.2012.679437. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Brownell A-L. In: Neuroimaging - clinical applications. Bright P, editor. Intech; Rijeka, Croatia: 2012. p. 499. [Google Scholar]
- 17.Engers DW, Niswender CM, Weaver CD, Jadhav S, Menon UN, Zamorano R, Conn PJ, Lindsley CW, Hopkins CR. J Med Chem. 2009;52:4115. doi: 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalinichev M, Le Paul E, Boléa C, Girard F, Campo B, Fonsi M, Royer-Urios I, Browne SE, Uslaner JM, Davis MJ, Raber J, Duvoisin R, Bate ST, Reynolds IJ, Poli S, Celanire S. J Pharmacol Exp Ther. 2014;350:495. doi: 10.1124/jpet.114.214437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Poul E, Bolea C, Girard F, Poli S, Charvin D, Campo B, Bortoli J, Bessif A, Luo B, Koser AJ, Hodge LM, Smith KM, DiLella AG, Liverton N, Hess F, Browne SE, Reymonds IJ. J Parmacol Exp Ther. 2012;343:167. doi: 10.1124/jpet.112.196063. [DOI] [PubMed] [Google Scholar]
- 20.Hong SP, Liu KG, Ma G, Sabio M, Uberti MA, Bacolod MD, Peterson J, Zou ZZ, Robichaud AJ, Doller D. J Med Chem. 2011;54:5070. doi: 10.1021/jm200290z. [DOI] [PubMed] [Google Scholar]
- 21.Jones CK, Engers DW, Thompson AD, Field JR, Blobaum AL, Lindsley SR, Zhou Y, Gogliotti RD, Jadhav S, Zamorano R, Bogenpohl J, Smith Y, Morrison R, Daniels JS, Weaver CD, Conn PJ, Lindsley CW, Niswender CM, Hopkins CR. J Med Chem. 2011;54:7639. doi: 10.1021/jm200956q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valenti O, Marino MJ, Wittmann M, Lis E, DiLella AG, Kinney GG, Conn PJ. J Neurosci. 2003;23:7218. doi: 10.1523/JNEUROSCI.23-18-07218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennouar K-E, Uberti MA, Melon C, Bacolod MD, Jimenez HN, Cajina M, Kerkerian-Le Goff L, Doller D, Gubellini P. Neuropharmacol. 2013;66:158. doi: 10.1016/j.neuropharm.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 24.Iderberg H, Maslava N, Thompson AD, Bubser M, Niswender CM, Hopkins CR, Lindsley CW, Conn PJ, Jones CK, Cenci MA. Neuropharmacol. 2015;95:121. doi: 10.1016/j.neuropharm.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu R, Zanotti-Fregonara P, Zoghbi SS, Gladding RL, Woock AE, Innis RB, Pike VW. J Med Chem. 2013;56:9146. doi: 10.1021/jm4012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel S, Hamill T, Connolly B, Jagoda E, Li W, Gibson R. Nucl Med Biol. 2007;34:1009. doi: 10.1016/j.nucmedbio.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Tueckmantel W, Zhu A, Pellegrino D, Brownell AL. Synapse. 2007;61:951. doi: 10.1002/syn.20445. [DOI] [PubMed] [Google Scholar]
- 28.Brown A, Kimura Y, Zoghbi SS, Siméon FG, Liow JS, Kreisl WC, Taku A, Fujita M, Pike VW, Innis RB. J Nucl Med. 2008;49:2042. doi: 10.2967/jnumed.108.056291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ametamey SM, Treyer V, Streffer J, Wyss MT, Schmidt M, Blagoev M, Hintermann S, Auberson Y, Gasparini F, Fischer UC, Buck A. J Nucl Med. 2007;48:247. [PubMed] [Google Scholar]
- 30.Kil KE, Zhang Z, Jokivarsi K, Gong C, Choi JK, Kura S, Brownell AL. Bioorg Med Chem. 2013;21:5955. doi: 10.1016/j.bmc.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kil K-E, Poutiainen P, Zhang Z, Zhu A, Choi J-K, Jokivarsi K, Brownell A-L. J Med Chem. 2014;57:9130. doi: 10.1021/jm501245b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Kil KE, Poutiainen P, Choi JK, Kang HJ, Huang XP, Roth BL, Brownell AL. Bioorg Med Chem Lett. 2015;25:3956. doi: 10.1016/j.bmcl.2015.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hitchcock SA. Curr Opin Chem Biol. 2008;12:318. doi: 10.1016/j.cbpa.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 34.Roth BL. National Institute of Mental Health Psychoactive Drug Screening Program Assay Protocol Book. http://pdsp.med.unc.edu/PDSP%20Protocols%20II%202013-03-28.pdf.
- 35.Shi Q, Savage JE, Hufeisen SJ, Rauser L, Grajkowska E, Ernsberger P, Wroblewski JT, Nadeau JH, Roth BL. J Pharmacol Exp Ther. 2003;305:131. doi: 10.1124/jpet.102.047092. [DOI] [PubMed] [Google Scholar]
- 36.Bradley SR, Standaert DG, Rhodes KJ, Rees HD, Testa CM, Levey AI, Conn PJ. J Comp Neurol. 1999;407:33. [PubMed] [Google Scholar]
- 37.Kinoshita A, Ohishi H, Nomura S, Shigemoto R, Nakanishi S, Mizuno N. Neurosci Lett. 1996;207:199. doi: 10.1016/0304-3940(96)12519-2. [DOI] [PubMed] [Google Scholar]
- 38.Lavreysen H, Dautzenberg FM. Curr Med Chem. 2008;15:671. doi: 10.2174/092986708783885246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.