

Abstract
In this review, we summarize some of the recent insight into pharmacological targeting of hypoxia in disease models. Studies from cultured cell systems, animal models as well as translation to human patients have revealed that post-translational modifications of individual proteins within NF-κB and hypoxia-inducible factor (HIF) pathways serve as ideal targets for analysis in disease models. Studies defining differences and similarities between these responses have taught us a number of important lessons about the complexity of the inflammatory response. A clearer definition of these pathways has provided new insight into disease pathogenesis and importantly, the potential for new therapeutic targets.
Introduction
Ongoing inflammatory responses are characterized by dramatic shifts in tissue metabolism. These changes include lactate accumulation with resultant metabolic acidosis and diminished availability of oxygen (hypoxia) 1, 2. Such shifts in tissue metabolism result, at least in part, from profound recruitment of inflammatory cell types, particularly myeloid cells such as neutrophils (PMN) and monocytes. The vast majority of inflammatory cells are recruited to, as opposed to being resident at, inflammatory lesions 3. As such, it is important to understand the interactions between localized metabolic changes (e.g. hypoxia) as they relate to recruitment signals and molecular mechanisms utilized by myeloid cells during inflammation.
It was recently shown that in acute inflammatory disease, infiltrating myeloid cells (esp. neutrophils) “mold” the tissue microenvironment in ways that significantly promote the stabilization of hypoxia-inducible factor (HIF) and HIF-dependent transcriptional responses4. Microarray analysis of epithelial cells following PMN transmigration revealed the induction of a prominent cohort of HIF target genes. Utilizing HIF reporter mice, Gp91phox−/− mice (lack a respiratory burst) and PMN depletion strategies in intestinal inflammation models, these studies revealed that transmigrating neutrophils rapidly deplete the microenvironment of molecular oxygen in an NADPH-oxidase-dependent manner and “imprint” a molecular fingerprint that reflects PMN-mediated induction of HIF target genes onto the surrounding tissue. Importantly, these studies implicated a significant contribution of HIF to inflammatory resolution. For example, Gp91phox−/− mice developed more severe inflammation with exaggerated PMN infiltration, diminished tissue hypoxia and increased microbial invasion. Here we summarize how these recent findings might be integrated to target hypoxia in inflammation.
Functional HIF targets in mucosal inflammation
In the mucosa, HIF triggers the expression of genes that enable intestinal epithelial cells to function as an effective barrier 5–8. Originally shown by microarray analysis of hypoxic intestinal epithelial cells 7, these studies have been validated in animal models of intestinal inflammation 9–14 and in inflamed human intestinal tissues 15–17. The functional proteins encoded by HIF-dependent mRNAs localize primarily to the most luminal aspect of polarized epithelia. Molecular studies of these hypoxia-elicited pathway(s) have shown a dependence on HIF-mediated transcriptional responses. The HIF-regulated pathways tend to influence overall tissue integrity, ranging from increased mucin production, 18 including molecules that modify mucins, such as, intestinal trefoil factor5, to xenobiotic clearance by P-glycoprotein,6 to nucleotide metabolism (by ecto-5′-nucleotidase and CD73)7, 8 and nucleotide signaling through the adenosine A2B receptor8.
As an extension of the original studies identifying HIF induction within the intestinal mucosa, Karhausen, et al. generated mice lacking expression of intestinal epithelial Hif1a (causing constitutive repression of Hif1a) or constitutive expression of HIF-1 in intestinal epithelia (via targeting of the von Hippel-Lindau gene)11. Loss of epithelial HIF-1α resulted in a more severe colitic phenotype than wild-type animals, with increased weight loss, decreased colon length and increased intestinal permeability, whereas constitutively active intestinal epithelial HIF was protective for each of these parameters. These findings may be somewhat model-dependent, since epithelial HIF-based signaling has also been shown to promote inflammation in other studies14, 19. However, the findings confirmed that intestinal epithelial cells can adapt to hypoxia and that HIF contributes to such adaptation.
HIF hydroxylation as a pharmacological target in hypoxia
It is now appreciated that the oxygenation profile of given tissues may provide important insight into disease pathogenesis. Breathable air at sea level contains a partial O2 pressure (pO2) of ~145 mmHg (approximately 21% O2). Measurements of the healthy lung alveolus have revealed a pO2 of 100–110 mmHg 20. By contrast, the most luminal aspect of the healthy colon exits at a pO2 of less than 20 mmHg 21, 22. Such differences reflect a combination of O2 sources, local metabolism and the anatomy of blood flow. It is thought that the steep gradient between the highly metabolic serosa and the anaerobic lumen of the gut primes the intestinal epithelium for rapid responses to changes in tissue oxygenation1, 2. In particular, inflammatory processes can rapidly increase the demand for oxygen in inflamed tissue, thereby leading to profound hypoxia 23, so called “inflammatory hypoxia” 24. Adaptation to hypoxia is, at least in part, mediated by HIF 25, 26.
HIF-1α was the original isoform purified by oligonucleotide binding to the 3′ region of the EPO gene 27. HIF-2α was subsequently identified by homology searches and as a binding partner for the heterodimeric partner HIF-1β 28. It was originally thought that the HIF-2α isoform was expressed only in endothelial cells (hence the name endothelial PAS protein or EPAS) 29. HIF-3α is a more distantly related isoform and when spliced appropriately, can encode a protein that antagonizes HRE-dependent gene induction 30. It is more recently appreciated that many cell types express both HIF-1 and HIF-2 and murine knockout studies suggest that these proteins have non-redundant roles 30. Some have suggested that distinct transcriptional responses mediated by HIF-1 and HIF-2 may be integrated in ways that support particular adaptations to hypoxia. For example, the transcriptional responses which coordinate the glycolytic pathways includes more than 11 target genes and appear to be more selective for the HIF-1α than for the HIF-2α isoform 30. Conversely, studies addressing the selectivity of the two isoforms for erythropoietin induction has suggested a more important role for the HIF-2α isoform 30. Currently, this specificity is not well understood. Some have suggested that binding of HIF-1α or HIF-2α to other transcriptional co-factors at the site of DNA binding could determine such specificity, but this is not conclusive.
Many cell types, including intestinal epithelial cells (IEC) 31, express both HIF1α and HIF2α and murine genetic studies suggest that these proteins have non-redundant roles 30. Some have suggested that distinct transcriptional responses mediated by HIF1α and HIF2α may be integrated in ways that support particular adaptations to hypoxia. For example, the transcriptional responses that coordinate the glycolytic pathways include more than 11 target genes and seem to be more selective for the HIF-1α than for the HIF-2α isoform 30. Likewise, studies addressing the selectivity of the two isoforms of HIFα suggest greater selectivity of HIF-2 for both erythropoietin production 30 and for intestinal iron transport 32.
The stability of the HIFα subunit is post-translationally regulated by three proly-hydroxylases (PHD1-3, Figure 1) and one aspariginyl-hydroxylase (Factor Inhibiting HIF, FIH), all of which are present in intestinal epithelial cells 9, 13, 33, 34. Under normoxic conditions, these enzymes hydroxylate HIF-α at specific prolines (PHDs) and/or at a specific asparaginyl (FIH) residue 25, 33. This hydroxylation leads to interaction with the von Hippel-Lindau protein, poly-ubiquitination of HIF-α subunit and subsequent proteasomal degradation (Figure 2)35. Several studies have shown that HIF triggers the transcription of a number of genes that enable IEC to function as an effective barrier. Guided initially by microarray analysis of hypoxic IEC 7, these studies have been validated in animal models of intestinal inflammation 9–14 and in human intestinal inflammation tissues 15–17. Interestingly, the functional proteins encoded by a number of uniquely hypoxia-inducible genes in intestinal epithelia localize primarily to the most luminal aspect of polarized epithelia, providing significant support for the hypothesis that hypoxia supports a barrier-protective phenotype. Molecular studies of these hypoxia-elicited pathway(s) have shown a dependence on HIF-mediated transcriptional responses. Notably, epithelial barrier protective pathways driven by HIF tend not to be the classical regulators of barrier function, such as the tight junction proteins occludin or claudins. Rather, the HIF-regulated molecules include molecules which support overall tissue integrity and include increased mucin production, 18 molecules that modify mucin (e.g. intestinal trefoil factor)5, promote xenobiotic clearance via P-glycoprotein 6, enhance nucleotide metabolism (by ecto-5′-nucleotidase and CD73)7, 8 and drive nucleotide signaling (e.g. adenosine A2B receptor) 8.
Figure 1. Functional features of hypoxia-inducible factor (HIF) and mechanism of HIF stabilization.
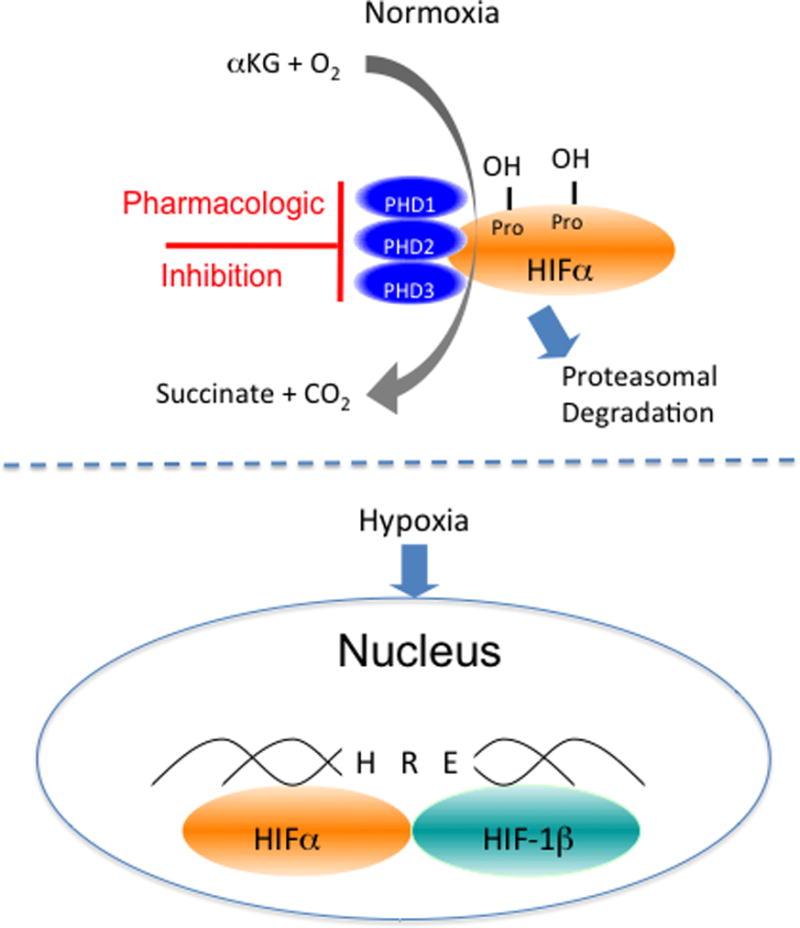
Depiction here is the biochemical pathyway of HIF hydroxylation by the combination of alpha-ketoglutarate (αKG), molecular oxygen (O2) and the prolyl-hydroxylase (PHD) enzymes in normoxia. When O2 becomes limiting (hypoxia), the alpha subunit is stabilized, binds to the HIF-1 beta subunit within the nucleus where it becomes transcriptionally active upon binding to the hypoxia-response element (HRE) consensus sequence on DNA.
Figure 2. Components of NF-κB and HIF E3 ligases.

Shown on the right is the HIF complex: The HIFα subunit in its hydroxylated form is degraded by the proteasome after ubiquitination via the Cul-2-Nedd8-pVHL complex. Pharmacological inhibition of Cul-2 neddylation using MLN4924 stabilizes cellular HIFα levels, leading to increased transcription of HIF target genes. Shown on the right is NF-κB: Activating stimuli facilitate the phosphorylation of IκB, leading to the recognition of p-IκB by the Cul-1-Nedd8-βTRCP complex, culminating in its polyubiquitination and proteasomal degradation. Pharmacological inhibition of Nedd8 conjugation by MLN4924 through inhibition of the Nedd8-activating enzyme (NAE, not shown here), prevents the activation of Cul-1, preventing the liberation of NF-κB from IκB.
As an extension of the original studies identifying HIF stabilization within the intestinal mucosa, transgenic mice expressing either mutant Hif1a (causing constitutive repression of HIF-1α) or mutant von Hippel-Lindau (causing constitutive overexpression of HIF) were targeted to the IEC 11. Loss of epithelial HIF-1α resulted in a more severe colitic phenotype than wild-type animals, including increased epithelial permeability, enhanced loss of bodyweight, and decreased colon length. Constitutively active intestinal epithelial HIF (mutant Vhl) was protective for each of these individual parameters. These findings may be somewhat model-dependent, since epithelial HIF-based signaling has also been shown to promote inflammation in another study 14. Nonetheless, these findings have revealed that IEC can adapt to even severe hypoxia and that HIF contributes in fundamental ways to this adaptation.
The identification of HIF-selective PHDs has provided unique opportunities for the development of PHD-based therapies 36, 37. While there is wide interest in developing HIF inhibitors as potential cancer therapies, opportunities also exist to selectively stabilize HIF in an attempt to promote inflammatory resolution 38. For example, 2-OG analogues stabilize HIF-α 36 and effectively promote the resolution of colitis in mouse models 9. Interestlingly, the protection afforded by PHD inhibitors (e.g. decreased tissue inflammatory cytokines, increased barrier function, decreased epithelial apoptosis) may involve both HIF and NF-κB activities. For example, in a genetic screen of PHD isoform deficient animals, Tambuwala et al. revealed that Phd1−/− mice were less susceptible to the development of DSS colitis, likely through decreased epithelial cell apoptosis 39, which was originally shown to be NF-κB-dependent 9. It has also been shown that hydroxylase inhibiton inhibits TNF-α induced barrier breakdown. Hindryckx and colleagues demonstrated, that DMOG repressed FADD (Fas-associated death domain protein), a linkage protein for the TNFα-receptor-1. This inhibition reduced TNF-α induced apoptosis and restored, or prevented loss of, epithelial barrier function. This response was HIF1-α mediated, and not dependent on abrogation of the NFκB pathway, since siRNA inhibition of HIF1-α diminished the protective function of DMOG despite a fully functional NFκB pathway40. To date, selective inhibitors for particular PHD isoforms have not become available.
There are likely a number of indications where uncontrolled stimulation of erythropoiesis (e.g. with HIF-2 stabilizer) is unwarranted. Some recent work has identified PHD inhibitors with relative selectivity for HIF-1 versus HIF-2. AKB-4924, a relatively HIF-1-selective PHD inhibitor, has been explored in mucosal infection and inflammation models 41, 42. The basis for HIF-1 selectivity over HIF-2 is not currently known. AKB-4924 has been shown to enhance phagocyte antibacterial function against of variety of pathogens and holds promise in enhancing overall innate immune response to microbial threats 41, 42. Use of AKB-4924 in models of murine colitis augmented epithelial barrier function and led to an approximately 50-fold reduction in serum endotoxin during colitis. AKB-4924 also decreased cytokines involved in pyrogenesis and hypothermia, significantly reducing serum levels of pro-inflammatory cytokines, while increasing anti-inflammatory IL-10. Interestingly, AKB-4924 offered no protection against colitis in epithelial-specific HIF-1α deficient mice, strongly implicating epithelial HIF-1α as the tissue target for AKB-4924-mediated protection in colitis. Such findings may provide the basis for a therapeutic use of PHD inhibitors in inflammatory and infectious disease.
Targeting protein neddylation in inflammation
There is much recent interest in targeting protein neddylation, i.e. the reversible conjugation of a NEDD8 (Neural precursor cell expressed, developmentally down-regulated 8) 43 during inflammation 44. Neddylation and deneddylation responses are higshly conserved between cell types 45 and species 46–49. Activating the inactive Nedd8-precursor through cleavage a carboxy-terminal glycine residue by Deneddylase-1 (DEN1, also called SENP8) enables Nedd8 to be conjugated to the E1 UBA3-APPBP1 heterodimer 50–53. Subsequently Nedd8 is conjugated to its specific E2 Ubc12 (ubiquitin conjugating enzyme) 54 and afterwards linked to the E3 complex 55, 56. Neddylation constitutes a central role in the post-translational modification of Cullin-RING-ligases 57 involved in the ubiquitin pathway. Cullins act as scaffolding proteins and are essential for the assembly of the ubiquitin E3 ligase complex conjugating ubiquitin to target proteins and thus marking them for proteasomal degradation 58.
New insights into potential roles for Cullin-deneddylation in inflammation have come of interest in recent years. Original work by Collier-Hyams et al. alluded to above have demonstrated that commensal bacteria-associated attenuation of NFκB is Cullin-de-neddylation-dependent 59 (see Figure 2). Furthermore, Kumar et al. were able to demonstrate that commensal bacteria can influence the neddylation status of Cullin-1 (Cul1) through generation of reactive oxygen species (ROS) and resulted in a transient and reversible deneddylation of Cul1 and subsequent decrease of NFκB pathway end products60. Interestingly, they were able to show, that different commensal bacterial strains differ in the amount of ROS they generate. Since there is an altered microbiota in patients with IBD compared to healthy subjects, and commensal bacterial strains also differ in their primary location in the gut, there might be different amounts of ROS in different parts of the intestine altering the inflammatory response in IBD60.
Adenosine receptor signaling has also been linked to neddylation. While signal transduction through the various adenosine receptors is well characterized, less is known about post-receptor events 61. One particularly intriguing mechanism suggests that adenosine inhibits NF-κB through actions on proteasomal degradation of IkB proteins 62. These findings were based on studies addressing adenosine signaling mechanisms which revealed that adenosine and adenosine analogs display a dose-dependent deneddylation of Cul-1 with rank order of receptor potencies A2BAR >A1AR>>A2AAR = A3AR 62. Our current understanding is that deneddylation reactions on Cullin targets via CSN-associated proteolysis is increasingly implicated as a central point for Cullin-mediated E3 ubiquitylation 57. Notably, other pathways for deneddylation have been reported. For example, the identification of the Nedd8-specific proteases NEDP1 and DEN1 have provided new insight into this emerging field. NEDP1/DEN1 appear to contain isopeptidase activity capable of directly deneddylating Cullin targets 63, 64. How adenosine influences DEN1 activity is not currently known.
Neddylation of other Cullin proteins (i.e. Cul-2) have also been implicated in mucosal inflammation, particularly related to HIF (see Figure 2). The proteasomal degradation of α-subunit of HIF provides a particularly intriguing example of post-translational modification. The E3 SCF ubiquitin ligase specific to HIFα-family members are comprised of Elongin B/C, RBX, CUL2, and the F-box domain of pVHL, and are responsible for the polyubiquitination of HIFα 45. Regulation of the E3 SCF is maintained by the covalent modification of NEDD8. The functional E3-SCF requires the COP9 signalosome (CSN) to bind Nedd8 to Cul2, which can be de-neddylated by DEN11/SENP8 65, 66. Work by MacManus et al 67, for example, showed that the HIF target gene adrenomedullin (ADM) functions as an endogenously generated vascular mediator that serves as a mucosal protective factor through fine-tuning of HIF. The underlying mechanism involved ADM-mediated deneddylation of Cul2, resulting in less pVHL activity and subsequent fine-tuning of HIF expression. Exogenous administration of ADM in a DSS colitis model resulted in decreased tissue and serum levels of pro-inflammatory cytokines, identifying the Cul2 pathway as another potential therapeutic target for IBD67. Likewise, Ehrentraut et al demonstrated that pharmacological targeting of neddylation with the AMP analog MLN4924 significantly abrogated NF-κB responses, induced HIF-1α promoter activity and reduced secretion of TNF-α-elicited pro-inflammatory cytokines in vitro. MLN4924 stabilized HIF and abrogated pro-inflammatory responses in vivo68. More recently, Curtis et al utilized loss and gain of function analysis to reveal that MLN4924-potently induces HIF in vitro (IC50 = 4.7nM) and that in vivo administration of MLN4924 abrogates disease severity in mucosal inflammation models69.
Conclusion
Numerous studies have implicated a prominent role for hypoxia in the inflammatory response. In this review, we have outlined the evidence for protein post-translational modifications, focused on hydroxylation and neddylation), as potential targets for the development of therapeutics. Animal models, particularly conditional deletion mutants, have been revealing and demonstrated an almost uniformly beneficial influence of HIF stabilization on mucosal inflammatory disease endpoints. The intense interest in development of HIF stabilizing agents (e.g. PHD inhibitors) have been insightful and show promise for near future clinical development. Ongoing studies to define differences and similarities between the various targets will undoubtedly teach us important lessons about the complexities and pathogenesis of inflammatory disease, and likely provide novel targets as templates for the development of therapies.
Acknowledgments
This work was supported by National Institutes of Health Grants DK50189, DK104713, and DK095491 as well as VA Merit Award I01BX002182.
Footnotes
Disclosure statement: The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as influencing the objectivity of this review.
References
- 1.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9:609–17. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med. 2008;85:1295–1300. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 3.Lewis JS, Lee JA, Underwood JC, et al. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999;66:889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- 4.Campbell EL, Bruyninckx WJ, Kelly CJ, et al. Transmigrating Neutrophils Shape the Mucosal Microenvironment through Localized Oxygen Depletion to Influence Resolution of Inflammation. Immunity. 2014 doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furuta GT, Turner JR, Taylor CT, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Ex Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comerford KM, Wallace TJ, Karhausen J, et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–94. [PubMed] [Google Scholar]
- 7.Synnestvedt K, Furuta GT, Comerford KM, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 (HIF-1) mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eltzschig HK, Ibla JC, Furuta GT, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Ex Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cummins EP, Seeballuck F, Keely SJ, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–65. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Han IO, Kim HS, Kim HC, et al. Synergistic expression of inducible nitric oxide synthase by phorbol ester and interferon-gamma is mediated through NF-kappaB and ERK in microglial cells. J Neurosci Res. 2003;73:659–69. doi: 10.1002/jnr.10706. [DOI] [PubMed] [Google Scholar]
- 11.Karhausen JO, Furuta GT, Tomaszewski JE, et al. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morote-Garcia JC, Rosenberger P, Nivillac NM, et al. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–18. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 13.Robinson A, Keely S, Karhausen J, et al. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–55. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah YM, Ito S, Morimura K, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134:2036–48. 2048 e1–3. doi: 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giatromanolaki A, Sivridis E, Maltezos E, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56:209–13. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mariani F, Sena P, Marzona L, et al. Cyclooxygenase-2 and Hypoxia-Inducible Factor-1alpha protein expression is related to inflammation, and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett. 2009;279:221–9. doi: 10.1016/j.canlet.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Matthijsen RA, Derikx JP, Kuipers D, et al. Enterocyte shedding and epithelial lining repair following ischemia of the human small intestine attenuate inflammation. PLoS One. 2009;4:e7045. doi: 10.1371/journal.pone.0007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louis NA, Hamilton KE, Canny G, et al. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem. 2006;99:1616–27. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 19.Xue X, Ramakrishnan S, Anderson E, et al. Endothelial PAS Domain Protein 1 Activates the Inflammatory Response in the Intestinal Epithelium to Promote Colitis in Mice. Gastroenterology. 2013;145:831–841. doi: 10.1053/j.gastro.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaible B, Schaffer K, Taylor CT. Hypoxia, innate immunity and infection in the lung. Respir Physiol Neurobiol. 2010;174:235–43. doi: 10.1016/j.resp.2010.08.006. Epub 2010 Aug 13. [DOI] [PubMed] [Google Scholar]
- 21.Albenberg L, Esipova TV, Judge CP, et al. Correlation Between Intraluminal Oxygen Gradient and Radial Partitioning of Intestinal Microbiota. Gastroenterology. 2014;18:1055–1063. doi: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karhausen J, Furuta GT, Tomaszewski JE, et al. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatoum OA, Binion DG, Gutterman DD. Paradox of simultaneous intestinal ischaemia and hyperaemia in inflammatory bowel disease. Eur J Clin Invest. 2005;35:599–609. doi: 10.1111/j.1365-2362.2005.01567.x. [DOI] [PubMed] [Google Scholar]
- 24.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–65. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2:336–61. doi: 10.1002/wsbm.69. [DOI] [PubMed] [Google Scholar]
- 26.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–54. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang GL, Jiang BH, Rue EA, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular oxygen tension. Proc Natl Acad Sci. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ema M, Taya S, Yokotani N, et al. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:4273–8. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu YZ, Moran SM, Hogenesch JB, et al. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998;7:205–13. [PMC free article] [PubMed] [Google Scholar]
- 30.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest. 2007;117:862–5. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mastrogiannaki M, Matak P, Keith B, et al. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119:1159–1166. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mastrogiannaki M, Matak P, Keith B, et al. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119:1159–66. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 34.Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365:537–47. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 36.Mole DR, Schlemminger I, McNeill LA, et al. 2-oxoglutarate analogue inhibitors of HIF prolyl hydroxylase. Bioorg Med Chem Lett. 2003;13:2677–80. doi: 10.1016/s0960-894x(03)00539-0. [DOI] [PubMed] [Google Scholar]
- 37.Masson N, Ratcliffe PJ. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O(2) levels. J Cell Sci. 2003;116:3041–9. doi: 10.1242/jcs.00655. [DOI] [PubMed] [Google Scholar]
- 38.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–7. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tambuwala MM, Cummins EP, Lenihan CR, et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology. 2010;139:2093–101. doi: 10.1053/j.gastro.2010.06.068. [DOI] [PubMed] [Google Scholar]
- 40.Hindryckx P, De Vos M, Jacques P, et al. Hydroxylase inhibition abrogates TNF-alpha-induced intestinal epithelial damage by hypoxia-inducible factor-1-dependent repression of FADD. J Immunol. 2010;185:6306–16. doi: 10.4049/jimmunol.1002541. [DOI] [PubMed] [Google Scholar]
- 41.Keely S, Campbell EL, Baird AW, et al. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol. 2014;7:114–23. doi: 10.1038/mi.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okumura CY, Hollands A, Tran DN, et al. A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J Mol Med. 2012;28:1079–1089. doi: 10.1007/s00109-012-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–61. doi: 10.1016/0006-291x(92)91747-e. [DOI] [PubMed] [Google Scholar]
- 44.Kamitani T, Kito K, Nguyen HP, et al. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. 1997;272:28557–62. doi: 10.1074/jbc.272.45.28557. [DOI] [PubMed] [Google Scholar]
- 45.Mikus P, Zundel W. COPing with hypoxia. Semin Cell Dev Biol. 2005;16:462–73. doi: 10.1016/j.semcdb.2005.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones D, Crowe E, Stevens TA, et al. Functional and phylogenetic analysis of the ubiquitylation system in Caenorhabditis elegans: ubiquitin-conjugating enzymes, ubiquitin-activating enzymes, and ubiquitin-like proteins. Genome Biol. 2002;3:RESEARCH0002. doi: 10.1186/gb-2001-3-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osaka F, Saeki M, Katayama S, et al. Covalent modifier NEDD8 is essential for SCF ubiquitin-ligase in fission yeast. EMBO J. 2000;19:3475–84. doi: 10.1093/emboj/19.13.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tateishi K, Omata M, Tanaka K, et al. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J Cell Biol. 2001;155:571–9. doi: 10.1083/jcb.200104035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ou CY, Lin YF, Chen YJ, et al. Distinct protein degradation mechanisms mediated by Cul1 and Cul3 controlling Ci stability in Drosophila eye development. Genes Dev. 2002;16:2403–14. doi: 10.1101/gad.1011402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wada H, Kito K, Caskey LS, et al. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem Biophys Res Commun. 1998;251:688–92. doi: 10.1006/bbrc.1998.9532. [DOI] [PubMed] [Google Scholar]
- 51.Huang DT, Miller DW, Mathew R, et al. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927–35. doi: 10.1038/nsmb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mendoza HM, Shen LN, Botting C, et al. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–43. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- 53.Wu K, Yamoah K, Dolios G, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–91. doi: 10.1074/jbc.M302888200. [DOI] [PubMed] [Google Scholar]
- 54.Liakopoulos D, Doenges G, Matuschewski K, et al. A novel protein modification pathway related to the ubiquitin system. EMBO J. 1998;17:2208–14. doi: 10.1093/emboj/17.8.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hori T, Osaka F, Chiba T, et al. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene. 1999;18:6829–34. doi: 10.1038/sj.onc.1203093. [DOI] [PubMed] [Google Scholar]
- 56.Jones J, Wu K, Yang Y, et al. A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. J Proteome Res. 2008;7:1274–87. doi: 10.1021/pr700749v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parry G, Estelle M. Regulation of cullin-based ubiquitin ligases by the Nedd8/RUB ubiquitin-like proteins. Semin Cell Dev Biol. 2004;15:221–9. doi: 10.1016/j.semcdb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 59.Collier-Hyams LS, Sloane V, Batten BC, et al. Cutting edge: bacterial modulation of epithelial signaling via changes in neddylation of cullin-1. J Immunol. 2005;175:4194–8. doi: 10.4049/jimmunol.175.7.4194. [DOI] [PubMed] [Google Scholar]
- 60.Kumar A, Wu H, Collier-Hyams LS, et al. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. Embo J. 2007;26:4457–66. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eltzschig HK, Rivera-Nieves J, Colgan SP. Targeting the A2B adenosine receptor during gastrointestinal ischemia and inflammation. Expert Opin Ther Targets. 2009;13:1267–77. doi: 10.1517/14728220903241666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khoury J, Ibla JC, Neish AS, et al. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest. 2007;117:703–11. doi: 10.1172/JCI30049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mendoza HM, Shen LN, Botting C, et al. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–43. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- 64.Wu K, Yamoah K, Dolios G, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–91. doi: 10.1074/jbc.M302888200. [DOI] [PubMed] [Google Scholar]
- 65.Chiba T, Tanaka K. Cullin-based ubiquitin ligase and its control by NEDD8-conjugating system. Curr Protein Pept Sci. 2004;5:177–84. doi: 10.2174/1389203043379783. [DOI] [PubMed] [Google Scholar]
- 66.Wei N, Deng XW. The COP9 signalosome. Annu Rev Cell Dev Biol. 2003;19:261–86. doi: 10.1146/annurev.cellbio.19.111301.112449. [DOI] [PubMed] [Google Scholar]
- 67.MacManus CF, Campbell EL, Keely S, et al. Anti-inflammatory actions of adrenomedullin through fine tuning of HIF stabilization. FASEB J. 2011;25:1856–64. doi: 10.1096/fj.10-170316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ehrentraut SF, Kominsky DJ, Glover LE, et al. Central role for endothelial human deneddylase-1/SENP8 in fine-tuning the vascular inflammatory response. J Immunol. 2013;190:392–400. doi: 10.4049/jimmunol.1202041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Curtis VF, Ehrentraut SF, Campbell EL, et al. Stabilization of HIF through inhibition of Cullin-2 neddylation is protective in mucosal inflammatory responses. FASEB J. 2015;29:208–215. doi: 10.1096/fj.14-259663. [DOI] [PMC free article] [PubMed] [Google Scholar]