

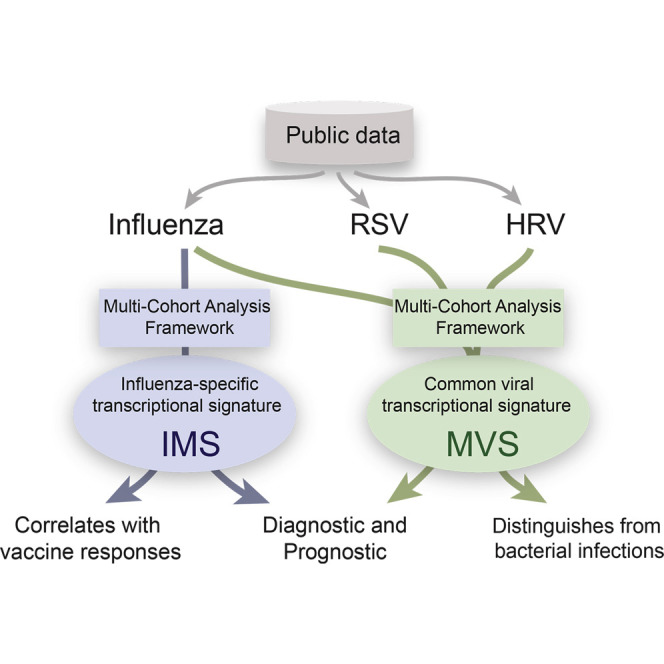
Abstract
Respiratory viral infections are a significant burden to healthcare worldwide. Many whole genome expression profiles have identified different respiratory viral infection signatures, but these have not translated to clinical practice. Here, we performed two integrated, multi-cohort analyses of publicly available transcriptional data of viral infections. First, we identified a common host signature across different respiratory viral infections that could distinguish (1) individuals with viral infections from healthy controls and from those with bacterial infections, and (2) symptomatic from asymptomatic subjects prior to symptom onset in challenge studies. Second, we identified an influenza-specific host response signature that (1) could distinguish influenza-infected samples from those with bacterial and other respiratory viral infections, (2) was a diagnostic and prognostic marker in influenza-pneumonia patients and influenza challenge studies, and (3) was predictive of response to influenza vaccine. Our results have applications in the diagnosis, prognosis, and identification of drug targets in viral infections.
Graphical Abstract
Highlights
-
•
MVS is a common transcriptional host response to respiratory viral infection
-
•
MVS could be used in clinics as a diagnostic and/or prognostic biomarker
-
•
IMS distinguishes influenza from other viral and bacterial infections
-
•
IMS correlates with infection symptomatology and vaccine response
Clinically relevant respiratory viral signatures have not been defined. Khatri and colleagues identified host transcriptional responses common to multiple respiratory viruses (MVS) or specific to influenza (IMS) by leveraging heterogeneity present in public datasets. Both signatures distinguish viral from bacterial infections and IMS also distinguishes influenza from other viral infections.
Introduction
Respiratory viruses such as influenza and SARS pose a major threat to global health, yet antiviral drugs have been difficult to develop. In addition, treating potential pandemic viral strains is problematic because of the many unknowns about the pathogenesis of infection. Current anti-viral drugs, which target a pathogen’s enzymatic functions and provide a “one-bug-one-drug” approach, use resources inefficiently and are often limited by the emergence of viral resistance (Locarnini and Warner, 2007, Richman et al., 2004). The drug-development process requires the ability to identify specific host factors that are necessary for viral growth and virulence that could also be potential drug targets. In light of the large unmet need for novel antiviral strategies, an efficient solution would be to repurpose currently approved drugs as broad-spectrum, host-centered antivirals that could impair viral transmission and prevent clinical pathology by identifying host factors that are targeted by existing drugs and are required for viral growth.
The prevailing approach for studying gene expression profiles is limited in its ability to identify these would-be targets for broad-spectrum antiviral therapeutics. Many gene expression microarray studies have proposed distinct gene signatures to discriminate different viral infections (Zaas et al., 2009) or influenza from bacterial infections (Parnell et al., 2012, Parnell et al., 2011, Ramilo et al., 2007). However, these experiments aim to reduce the effect of various biological and technical confounding factors as much as possible by focusing on only one viral infection in one tissue and using one type of microarray. This standard, single-cohort approach increases the risk of confounding factors on gene expression profiles from the specific tissue, technologies, demographics, and inclusion criteria of the respective studies or by other unknown biological and technical factors (Parnell et al., 2011, Parnell et al., 2012, Ramilo et al., 2007), all of which can mask the broad pathways used by multiple viruses to establish infection.
We have developed an integrated, multi-cohort analysis framework that leverages the heterogeneity present in public data repositories (e.g., GEO and ArrayExpress), which in turn increases sample size and allows for the identification and validation of robust and reproducible signatures of a disease phenotype. We have demonstrated the utility of this framework in identifying novel drug targets, diagnostic biomarkers, and repurposing FDA-approved drugs (Chen et al., 2014, Khatri et al., 2013, Li et al., 2015, Mazur et al., 2014, Sweeney et al., 2015).
We applied our method for two different hypotheses. First, to obtain a common transcriptional signature across all respiratory viral infections, we applied our method to three gene expression datasets of 205 human blood samples from three viral infections (influenza, human rhinovirus [HRV], and respiratory syncytial virus [RSV]), measured on two different microarray platforms in three countries to identify a robust 396-gene meta-virus signature (MVS) of respiratory viral infections. We tested this signature against 14 independent cohorts composed of 1,087 blood samples to show that it was not confounded by sample tissue, treatment, viral strain, or microarray technology.
We performed a separate multi-cohort analysis of influenza infection studies to illustrate that there were virus-specific signatures encompassing smaller subsets of genes. We applied our method to five influenza gene expression datasets consisting of 292 samples and identified an 11-gene influenza meta-signature (IMS). Using 11 additional independent cohorts, we showed that this influenza-specific signature was able to discriminate (1) symptomatic from asymptomatic subjects, (2) influenza infection from other respiratory viral infections, and (3) patients with mixed influenza and/or bacterial pneumonia from those with bacterial pneumonia alone. Finally, we bridge the gap between influenza infection and vaccination by demonstrating that the influenza infection signature is also increased significantly in influenza vaccine responders compared to non-responders.
These two multi-cohort analyses showed that (1) there was a conserved host response to respiratory viral infections and (2) there were virus-specific responses that could distinguish different virus types. Both have significant potential for use in the diagnosis and treatment of viral infections.
Results
Integrated, Multi-cohort Analysis of Viral Infections Identifies Broad Anti-virus Responses
We downloaded 18 microarray gene expression datasets from the NCBI GEO (Barrett et al., 2005) database comprising 2,939 samples obtained from whole blood, PBMCs, or primary epithelial cells (Table S1; Bermejo-Martin et al., 2010, Franco et al., 2013, Herberg et al., 2013, Hu et al., 2013, Ioannidis et al., 2012, Li et al., 2011, Loveday et al., 2012, Mejias et al., 2013, Parnell et al., 2011, Parnell et al., 2012, Ramilo et al., 2007, Reghunathan et al., 2005, Shapira et al., 2009, Sutejo et al., 2012, Tsang et al., 2014, Woods et al., 2013, Zaas et al., 2009). These datasets included healthy controls; individuals with various viral infections, bacterial infections, or non-infectious systematic inflammatory response syndrome (SIRS); individuals that were vaccinated for influenza; and in vitro transfection experiments expressing different viral antigens.
We used five cohorts from three datasets, composed of 205 samples for studying three respiratory viral infections to identify a potential common viral response (Table S1; Herberg et al., 2013, Hu et al., 2013, Ioannidis et al., 2012). We refer to each study (unique GSE ID) in GEO as a dataset and a set of samples for each comparison within a dataset as a cohort. Unlike a single-cohort experiment, where the goal is to control as many confounding factors as possible, we included broad biological and technical heterogeneity, such as treatment protocols and demographics, observed in the population by choosing discovery cohorts that were collected at different centers (with different treatment protocols and demographics) and that profiled more than one viral infection (influenza, RSV, HRV) across different age groups (infant and pediatric). We incorporated technical heterogeneity in our samples by choosing datasets that were profiled using microarrays from two different manufacturers and represented different technological confounding factors (e.g., length of oligonucleotide probes, sample preparation protocols). In order to avoid the potential influence of a single cohort on the results due to unequal sample sizes or other unknown confounding factors among cohorts, we performed a “leave-one-cohort-out” analysis. We hypothesized that the resulting set of genes that were significantly differentially expressed, irrespective of the set of cohorts analyzed, would constitute a robust signature of respiratory viral infection.
We identified 396 differentially expressed genes (161 over- and 235 underexpressed, p < 3 × 10−5, FDR < 1%; Figure 1 A and Table S2) during respiratory viral infection, many of which have been previously identified as differentially expressed after viral infection such as OASL, TYK2, toll-like receptors (TLRs), and interferon induced transmembrane proteins (IFITMs) (Parnell et al., 2011, Parnell et al., 2012, Ramilo et al., 2007, Woods et al., 2013, Zaas et al., 2009). We refer to the 396-gene set as a meta-virus signature (MVS). KEGG pathway analysis using iPathwayGuide (Draghici et al., 2007, Khatri et al., 2008, Tarca et al., 2009) of the MVS identified 18 significant pathways, including pathways for viral infections such as Epstein-Barr virus, influenza A, herpes simplex, and measles (Table S3). Other significant and relevant KEGG pathways included cell cycle, NF-κB signaling, toll-like receptor signaling, lysosome, and sphingolipid metabolism.
Figure 1.

Discovery and Validation of Meta Virus Signature
Effect size heatmaps of 396-gene MVS in 5 discovery (A) and 10 validation (B) cohorts. Each column is a gene and row is a cohort. The first row in both heatmaps displays summary effect size for each gene in discovery or validation cohorts. Genes are sorted in decreasing order of their summary effect size in discovery cohorts for both heatmaps.
Next, we analyzed the expression of these 396 MVS genes in 10 additional independent cohorts consisting of 329 PBMC or whole blood samples (226 viral infection samples, 103 controls). 136 out of 161 MVS overexpressed genes (84.5%), and 139 out of 235 underexpressed MVS genes (59.14%) were statistically significant (p < 0.05) in the validation cohorts (Figure 1B). These data indicated that the MVS defined broad immunological responses in the host to respiratory viral infection.
The MVS Is Specific to Viral Infection
Differential diagnosis of bacterial versus viral infection is confounded by similar clinical symptoms and underlying conditions such as immunosuppression and extrapulmonary complications (Babcock et al., 2008, Ison and Lee, 2010, Parnell et al., 2011, Parnell et al., 2012, Ramilo et al., 2007). Therefore, we explored whether the MVS could distinguish viral infections from bacterial infections.
First, we examined an independent cohort of children under 3 years of age (GSE: GSE40396) (Hu et al., 2013). For each sample, we defined an MVS score as the difference between the geometric mean of the 161 overexpressed genes and 235 underexpressed genes in the MVS. As expected, the MVS scores in virus-infected children were significantly higher than those in virus-negative controls (p = 6.88 × 10−7; receiver operating characteristic [ROC] area under the curve [AUC] = 1). The MVS scores were significantly higher in afebrile RSV-infected children than in those with febrile bacterial infections (p = 6.21 × 10−4) and distinguished both groups with high accuracy (ROC AUC = 0.98) (Figures 2 A and S1A). These results suggest that the MVS score is not confounded by the febrile status of children.
Figure 2.

MVS Scores in Four Independent Cohorts of Blood Samples
(A) Comparison of MVS scores in virus-negative, afebrile controls, and patients infected with bacteria or HRV.
(B) Comparison of MVS scores in healthy controls and patients infected with SARS coronavirus. Error bars indicate mean ± SE for a given group of samples. Width of a violin plot indicates density of samples, where each dot represents a sample.
(C and D) MVS scores in symptomatic and asymptomatic subjects inoculated with influenza (H3N2 in C or H1N1 in D). Smoothed lines indicate loess curves for symptomatic and asymptomatic subjects. Gray bars indicate 95% confidence intervals for each group.
See also Figure S1.
The MVS scores were also higher in another independent cohort (GSE: GSE1739) (Reghunathan et al., 2005), comprised of patients infected with severe acute respiratory syndrome (SARS) coronavirus (Figures 2B and S1B). Interestingly, in GSE40396, the MVS scores were also higher for children with other viral infections (adenovirus, HHV6, and enterovirus) compared to virus-negative controls (p = 1.02 × 10−8) and those with bacterial infections (p = 0.012), although none of these infections were used to define the MVS (Figure S1C). The MVS scores also distinguished samples with these viral infections from those with bacterial infections and samples from healthy controls with relatively high accuracy (Figure S1D). This indicates that the MVS might be more broadly applicable than respiratory viruses.
Next, we used an influenza challenge study (GSE: GSE52428), which inoculated healthy adults with H3N2 or H1N1 to evaluate changes in the MVS scores over the course of infection (Woods et al., 2013). The MVS scores remained unchanged over time in asymptomatic subjects that were not shedding any virus in both groups. However, the MVS scores increased significantly for virus-shedding symptomatic subjects over 24–72 hr and began to decline toward asymptomatic baseline levels as symptoms resolved (Figures 2C, 2D, S1E, and S1G). Specifically, the MVS scores for six of the nine H3N2 symptomatic volunteers (67%) were higher than those of the H3N2 asymptomatic volunteers at 36 hr after inoculation (Figure 2C). Similarly, the MVS scores for 9 of the 12 H1N1 symptomatic volunteers (75%) were higher than those of the H1N1 asymptomatic volunteers at 53 hr after inoculation (Figure 2D). The median onset time of symptoms for H3N2- and H1N1-inoculated volunteers was 49.3 hr (range 24–84 hr) and 61.3 hr (range 24–108 hr), respectively. Hence, the increase in the MVS scores preceded respiratory infection symptom onset in both strains and distinguished H3N2 and H1N1 symptomatic from asymptomatic volunteers with high specificity and sensitivity with ROC AUC for H3N2 and H1N1 of 0.94 at 36 hr (p = 0.009) and 0.84 at 53 hr (p = 0.008), respectively (Figures S1F and S1H).
Three H1N1-inoculated subjects (one asymptomatic and two symptomatic) showed MVS score profiles that were the opposite of their respective group (Figure S1G). Further examination of these individuals revealed that the asymptomatic subject, who followed a trajectory similar to the symptomatic group, was shedding the virus. The original study referred to this subject as an “asymptomatic shedder” (Woods et al., 2013). Similarly, one of the symptomatic subjects, who followed a trajectory similar to asymptomatic group, was not shedding any virus, and was therefore referred to as a “symptomatic non-shedder” in the original study. These results provide strong evidence of the accuracy of MVS score in correctly identifying infected individuals independent of their symptoms.
Collectively, our results showed that the MVS was a common transcriptional signature of a respiratory viral infection, independent of the subjects’ symptoms. The MVS was also able to identify symptomatic subjects prior to symptom onset in an influenza challenge study. Further, higher MVS scores in other viruses (adenovirus, enterovirus, and HHV6) in addition to influenza, RSV, and HRV suggested that the MVS might be more broadly applicable than respiratory viruses. However, a larger systematic analysis of diverse viruses would be necessary to identify such a core signature. Our results also suggest that the MVS might be able to distinguish other viral infections prior to symptom onset similar to influenza, though additional challenge studies using other viruses are needed for validation.
Identification of an Influenza-Specific Response Signature
A number of studies have previously reported virus- and strain-specific signatures (Hu et al., 2013, Zaas et al., 2009). Therefore, we hypothesized that despite a common transcriptional response to most respiratory viral infections, there might be a virus-specific transcriptional response. We applied our method to several influenza infection studies to see whether we could identify an influenza meta-signature (IMS).
As before, we chose expression profiles from 292 blood samples in five cohorts from three countries and profiled using two types of microarrays to represent biological and technical heterogeneity. In the discovery cohorts, we used samples from healthy individuals, patients with bacterial infection, and day 0 (pre-inoculation) individuals as controls. We used samples with influenza infection and individuals after inoculation as cases.
We identified 127 genes (FDR < 0.5%) as significantly overexpressed (Table S4 and Supplemental Experimental Procedures). Although our very stringent criterion might have left out some genes with varying expression in influenza, it allowed for the identification of a reproducible transcriptional profile that was found in all five influenza discovery cohorts despite the presence of significant heterogeneity.
These 127 genes include RIG-1-like receptor (RLR) molecules (DDX60, DHX58, IFIH1), transcription factors known to be overexpressed during influenza infection (IRF7, STAT1), interferon-alpha inducible genes (IFI44, IFI44L, IFI6), transport molecules (RAB8A), and antiviral molecules such as myxovirus resistance gene (MX1), 2′-5′-oligoadenylate synthetases (OAS1, OAS2, OAS3), guanylate-binding protein 1 (GBP1), and RSAD2. Many of these genes have been shown to be overexpressed after influenza infection (Ramilo et al., 2007), confirming the validity of our results. Network analysis of these 127 genes by Ingenuity Pathway Analysis (IPA) confirmed that 71 of the 127 genes are part of a network involved in innate virus sensing and initiation of antiviral response pathways (Figure S2). Thus, our analysis was able to capture the known biology of the response to influenza by defining a gene subset of the larger, common MVS.
We then applied a leave-one-cohort-out strategy to avoid the undue influence of a single cohort on the results. We identified 16 overexpressed genes: CD38, HERC5, HERC6, IFI44L, IFI6, IFIH1, IFIT1, LGALS3BP, LY6E, MX1, OAS1, OAS2, PARP12, RTP4, XAF1, and ZBP1, using a very stringent FDR < 0.01%. Of these 16 genes, 5 (IFI44L, IFIT1, OAS1, OAS2, and XAF1) had significant heterogeneity in effect size (p < 0.025) and were removed from further analysis. The remaining 11 genes were homogeneously overexpressed in patients infected with influenza across all discovery cohorts (Figures 3 A–3K; Table S5). Network analysis by IPA showed that 10 of these 11 genes were interconnected, with IRF7 and STAT1 forming a central axis of transcriptional regulation (Figure 3L). We will now refer to these 11 genes as the IMS.
Figure 3.

11-Gene Influenza Meta Signature
(A–K) 11 genes were significantly overexpressed during influenza infection in all discovery cohorts analyzed. The x axes represent standardized mean difference between influenza and control samples, computed as Hedges’ g, in log2 scale. The size of the blue rectangles is inversely proportional to the SEM in the study. Whiskers represent the 95% confidence interval. The orange diamonds represent overall, combined mean difference for a given gene. Width of the diamonds represents the 95% confidence interval of overall mean difference.
(L) Network analysis by Ingenuity Pathway Analysis showed that 10 out of the 11 genes are part of a single regulatory network. Blue nodes in the network represent IMS genes, and red nodes represent genes that were significantly overexpressed during influenza infection but are not included in the IMS.
The IMS Discriminates Influenza Infection from Bacterial Infections with High Specificity and Sensitivity
We asked whether the IMS could distinguish between influenza and bacterial infections, similar to the MVS. Therefore, we defined the geometric mean of the 11 IMS genes as the IMS score of a sample. In GSE: GSE40012 (Parnell et al., 2012), the IMS scores of influenza pneumonia patients were significantly higher than healthy controls, bacterial pneumonia patients, and non-infectious SIRS (p = 1 × 10−11; Figure 4 A). Importantly, there was no significant difference between the IMS scores of influenza pneumonia patients and mixed pneumonia patients (individuals with both influenza and bacterial infections). These data suggested that even with concurring bacterial infection, in the same patient the IMS score could still identify influenza infection. We also showed that using only the samples obtained within 24 hr of hospitalization, the IMS score identified influenza pneumonia patients with high specificity and sensitivity (AUC = 0.92; Figure 4B). These results validate the utility of the IMS as an index of influenza infection that can distinguish it from bacterial infection in clinical settings.
Figure 4.

The IMS Discriminates Influenza Infection from Bacterial Infections with High Specificity and Sensitivity in Multiple Cohorts
ROCs are for distinguishing patients with influenza infection from all other samples in a given cohort.
(A and B) For GSE40012, only samples on the first day of admission are used for violin plot (A) and ROC (B).
(C–H) Violin plots (C, E, G) and ROCs (D, F, H) for samples profiled with three microarray platforms in GSE6269 are displayed individually.
Error bars indicate mean ± SE for a given group of samples. Width of a violin plot indicates density of samples, where each dot represents a sample. See also Figure S3.
Next, we compared the IMS scores from PBMCs of pediatric patients (age ≤ 18 years) with influenza or bacterial infection (Staphylococcus aureus, Streptococcus pneumoniae, and Escherichia coli) in an independent cohort (GSE: GSE6269) (Ramilo et al., 2007). Despite the presence of biological (age, pathogens, antibiotics) and technological (three microarray platforms) confounding factors, the IMS scores of the patients with influenza infection were significantly higher than those of healthy controls and patients with bacterial infections (p ≤ 2.83 × 10−3; Figures 4C, 4E, and 4G), with high specificity and sensitivity in distinguishing influenza patients from those with a variety of bacterial infections (AUC range 0.86–0.97; Figures 4D, 4F, and 4H). The lack of statistical significance in Figure 4G is an artifact of a very small sample size (n = 3) in the S. pneumoniae group with an outlier.
Collectively, these results provide strong evidence that the IMS (1) is able to specifically distinguish influenza infection from bacterial infections and (2) is not confounded by age or the array technology used for profiling.
The IMS Can Distinguish Influenza from Other Respiratory Viruses
The IMS scores of patients with viral infections were not different from those of virus-negative controls and patients with bacterial infections (Figure S3A) or SARS coronavirus (Figure S3B). In fact, the IMS scores were significantly lower in virus-infected patients than virus-negative controls (p = 0.003; Figure S3A). In contrast, the MVS scores were significantly higher in both of these cohorts. These results suggested that the IMS might be specific to influenza, because none of the patients in these cohorts were infected with influenza.
Therefore, we compared the IMS scores of influenza-infected patients with those of patients with other respiratory viral infections (RSV and HRV) using three datasets (GSE: GSE34205, GSE42026, and GSE38900). These datasets profiled whole blood samples from children (age < 17 years). We note that influenza-infected samples and healthy control samples from GSE34205 and GSE42026 were used as discovery cohorts for identification of IMS (Table S1). The IMS scores were significantly higher in influenza samples compared to RSV samples in GSE34205 and GSE38900 (p < 0.0002) and marginally significant in GSE42026 (p = 0.061; Figures 5 A–5C). Influenza samples also had an IMS score that was significantly higher than that for HRV samples in GSE38900 (p = 2.33 × 10−5; Figure 5C). Notably, in these three datasets, the MVS scores of subjects with any viral infection were significantly higher than the healthy controls (p < 1 × 10−6) but were similar when compared to each other (p > 0.05; Figures 5D–5F), with the exception of HRV and RSV in GSE38900 (p = 0.002; Figure 5F). These data confirmed that the genes in the IMS were able to accurately discriminate influenza from other viral infections.
Figure 5.

The IMS Scores Distinguish Influenza-Infected Patients from Healthy Controls and Patients with Other Respiratory Viral Infections but the MVS Scores Do Not
(A–C) In GSE34205 (A) and GSE42026 (B), samples from influenza patients and healthy controls were used for discovery of the IMS, but samples from RSV-infected patients were not used. None of the samples from GSE38900 (C) were used for discovery of the IMS.
(D–F) All samples from GSE34205 (D) and GSE42026 (E) were used for identification of the MVS. None of the samples from GSE38900 (F) were used for identification of MVS.
Error bars indicate mean ± SE for a given group of samples. Width of a violin plot indicates density of samples, where each dot represents a sample. See also Figure S3.
The IMS Score Distinguishes Symptomatic and Asymptomatic Individuals with High Specificity and Sensitivity
Similar to the MVS, we further explored the change in the IMS scores after inoculation in an influenza challenge study (GSE: GSE52428) and two influenza and bacterial pneumonia cohorts (GSE: GSE20346 and GSE40012). Both pneumonia cohorts profiled longitudinal samples that were obtained from patients within 24 hr of their admission to an intensive care unit. The challenge study allowed for analysis of the IMS scores during the initial acute phase of infection, whereas the pneumonia cohorts allowed for analysis of the IMS scores in patients with an established infection as they progress through recovery and resolution of symptoms.
Similar to the MVS scores, the IMS scores were unchanged for asymptomatic volunteers inoculated with H3N2 or H1N1 but increased significantly for symptomatic patients (Figures 6A and 6B). Furthermore, an increase in the average IMS score of symptomatic volunteers also preceded symptom onset in both groups. For example, the IMS scores for all H3N2 symptomatic volunteers were higher than for asymptomatic volunteers at 69.5 hr after inoculation (AUC = 1; Figure S4A), suggesting that the IMS score correctly identified symptomatic volunteers prior to symptom onset. For H1N1 volunteers, who showed symptoms later than the H3N2 group (Woods et al., 2013), the IMS score also began to increase at a later time point as compared to the H3N2 group, achieving maximum discrimination between symptomatic and asymptomatic volunteers at 60 hr (AUC = 0.9; Figure S4B), which was also prior to symptom onset in all H1N1 volunteers. Thus, the kinetics of IMS gene expression might provide prognostic capability in the clinic for tracking potential morbidity after a known exposure by identifying susceptible individuals prior to symptom onset.
Figure 6.

IMS Score Is a Prognostic Marker of Influenza Infection
(A and B) IMS scores in symptomatic and asymptomatic subjects inoculated with influenza A (H3N2 in A; H1N1 in B). Smoothed lines indicate loess curves for symptomatic and asymptomatic subjects. Gray bars indicate 95% confidence interval.
(C and D) Comparison of MVS and IMS scores in the same subjects inoculated with H3N2 (C) or H1N1 (D).
(E and F) Change in IMS scores in healthy controls and patients with non-infectious systemic inflammatory response, influenza pneumonia, bacterial pneumonia, or both (influenza and bacterial pneumonia) during their stay in the hospital.
See also Figure S4.
However, MVS and IMS behaved differently in the GSE52428 challenge study, where MVS achieved higher discriminatory power than IMS at the same time point after inoculation (Figures 6C and 6D). For instance, 36 hr after inoculation, ROC AUCs were 0.94 and 0.65 in the H3N2 cohort for MVS and IMS, respectively. Similarly for H1N1, 45.5 hr after inoculations, ROC AUCs were 0.81 and 0.64 for MVS and IMS, respectively. Furthermore, IMS scores of symptomatic volunteers remained higher than those of asymptomatic volunteers at the end of the time course (Figures 6A and 6B) and is in contrast to MVS scores that began to decrease toward asymptomatic baseline levels at the same time point (Figures 2C and 2D). These results suggested that the MVS, a common transcriptional response to viral infection, was turned on earlier than the influenza-specific response and then also returned to baseline earlier than the influenza-specific response.
In the pneumonia cohorts, our analysis showed that the IMS score remained unchanged from baseline in healthy controls and patients with bacterial pneumonia or SIRS (Figures 6E and 6F). In contrast, the IMS scores of the influenza pneumonia patients were significantly higher than those of bacterial pneumonia (p = 3.35 × 10−13 in GSE40012 and p = 0.0017 in GSE20346) at the time of admission and progressively decreased over time in the influenza pneumonia patients (p = 1.3 × 10−5 in GSE40012 and p = 4.23 × 10−5 in GSE20346) (Figures 6E and 6F). The mean IMS score in mixed pneumonia patients, defined as those with both bacterial and influenza infections, were also significantly higher at admission, decreased over time, and were indistinguishable from those of influenza pneumonia patients, suggesting that the IMS score is not confounded by co-infection with bacterial pathogens.
IMS Score Is Significantly Higher in Vaccine Responders than Non-responders
The ultimate goal of influenza vaccination is to induce the same immune response as infection to induce a memory response, but without the corresponding pathology. Despite a large number of influenza infection (Mejias et al., 2013, Parnell et al., 2012, Ramilo et al., 2007) and vaccination (Franco et al., 2013, Furman et al., 2013, Nakaya et al., 2011, Tsang et al., 2014) studies, no common transcriptional signature between influenza and vaccination has been proposed. Therefore, we explored the change in the IMS scores after influenza vaccination in three independent cohorts of 310 individuals.
Tsang et al. (2014) (GSE: GSE47353) divided 63 vaccinated (Fluvirin, Novartis) healthy individuals into three groups (low, moderate, and high responders) that were defined as the lowest 20th percentile, 21st to 80th percentile, and above 80th percentile of microneutralization titers, respectively. The IMS scores increased significantly for the high (paired t test p = 0.008) and moderate (paired t test p = 6.36 × 10−6) responders, but not for low responders (paired t test p > 0.05) on day 1 after vaccination compared to day 0 (Figure 7 A).
Figure 7.

IMS Score Increases Significantly in Vaccine Responders
(A) Change in IMS scores on day 1 after vaccination for high (p = 0.008), moderate (p = 6.36 × 10−6), and low (p > 0.05) responders defined based on microneutralization titers.
(B and C) Change in IMS scores for vaccine responders and non-responders, defined based on HAI titers, in a female cohort after influenza vaccination (H1N1 in B; H3N2 in C).
(D and E) Change in IMS scores for vaccine responders and non-responders, defined based on HAI titers, in a male cohort after influenza vaccination (H1N1 in D; H3N2 in E).
See also Figure S5.
Unlike in Tsang et al. (2014), typically vaccine responders and non-responders are defined based on 4-fold change in hemagglutination-inhibition (HAI) assay titers 28 days after vaccination. Therefore, we used this definition to categorize responders and non-responders in two independent vaccination cohorts (GSE: GSE48018 and GSE48023 from Franco et al., 2013) and explored whether the IMS scores showed difference between vaccine responders and non-responders prior to the 28-day mark. In a cohort of 128 females (GSE48023), the IMS scores increased significantly on day 3 after vaccination in responders for H1N1 (paired t test p = 0.002) and H3N2 (paired t test p = 0.015), but not in non-responders (paired t test p > 0.2) (Figures 7B and 7C). Similarly, in a cohort of 119 males (GSE48018), the IMS scores increased significantly in responders for H1N1 and H3N2 (paired t test p < 2.2 × 10−16) on day 1 after vaccination for both H1N1 and H3N2 (Figures 7D and 7E). However, the IMS scores also increased significantly in non-responders (paired t test p < 0.01) for H1N1 and H3N2 in the male cohort, although the increase in the non-responders after vaccination was lower than the responders (Figures 7D and 7E). When we used 4-fold increase in the microneutralization titers on 28 days after vaccination to define responders, the IMS scores showed similar results for both cohorts (Figure S5). These results show that the IMS score increases significantly in vaccine responders and could potentially serve as a marker for successful vaccination.
The IMS Score Increases in Influenza-Infected Epithelial Cells
We investigated whether the IMS response could be seen in epithelial cells, the target cell population for influenza replication. Indeed, in three independent datasets of 154 samples, the IMS scores increased significantly in cell lines of epithelial origin (Calu-3 and A549) after influenza infection (Figure S6).
Next, we compared the IMS scores in primary human bronchial epithelial cells (HBECs) after infection with influenza or treatment with relevant ligands (Shapira et al., 2009). Shapira et al. (2009) (GSE: GSE19392) used four different strategies to highlight distinct components of the host response to influenza infection: (1) infection with wild-type A/PR/8/34 (PR8) influenza virus that can mount a complete replicative cycle; (2) transfection with viral RNA (vRNA) isolated from influenza particles, which does not result in the production of viral proteins or particles; (3) treatment with interferon beta (IFN-β) to identify responses mediated through type 1 interferons (IFNs); and (4) infection with a PR8 mutant virus lacking the NS1 gene (ΔNS1), which normally inhibits vRNA- or IFN-β-induced pathways in host cells.
IMS scores increased significantly for all strategies (Figure S7), where the kinetics and magnitude of the increase in the IMS score reflected the hierarchy of influenza virus detection in the epithelial cells and host response. For example, at 18 hr after infection, IMS scores were the lowest after infection with wild-type PR8 influenza virus. However, the magnitude of the change in the average IMS score at 18 hr after infection was higher for cells exposed to influenza viral RNA, infected with PR8 influenza virus lacking NS1 protein, or treated with IFN-β and is probably due to the absence of the NS1 protein that inhibits vRNA- or IFN-β-induced pathways in each of these conditions.
Furthermore, the increase in the IMS score was fastest in cells treated with IFN-β (1.5 hr), slowest for infection with wild-type PR8 influenza virus with or without NS1 protein (6 hr), and intermediate for transfection with only viral RNA (4 hr). Taken together, these differences in the timing of when the average IMS score begins to rise suggests that the IMS consists of host response genes that are directly or indirectly targeted by the influenza virus to suppress the host response, evade detection, and dampen the effects of IFN-β.
Discussion
The goal of this study was to integrate gene expression data from multiple heterogeneous sources to define both a conserved host response to respiratory viral infection and one specific to influenza. These dual goals could be useful in multiple settings, such as determining (1) the likely prognosis in a viral infection, (2) the diagnosis of a specific viral infection, (3) identifying vaccine responders, and (4) uncovering new biological pathways and possible drug targets for further study.
Using our previously described multi-cohort analysis framework, we analyzed 26 independent cohorts from 18 datasets consisting of 2,939 samples that were collected in 7 countries and represented infections from 7 viruses and 4 bacteria in whole blood, PBMCs, and epithelial cells to identify the MVS and IMS that are robustly and consistently differentially expressed across age, gender, clinical time course, and illness severity. Our results showed that the MVS is able to (1) distinguish virus-infected patients from those with bacterial infections or healthy controls with high accuracy and (2) identify influenza-infected subjects at risk for being ill before onset of clinical symptoms. Although the clinical utility of the MVS might be limited due to the large number of genes in the signature, it provides a potential starting point for simpler diagnostic gene sets. Importantly, it has identified novel pathways to better understand viral host response and identify targets for novel anti-viral therapies. We also showed that the IMS (1) is specific to influenza infection, (2) is able to distinguish influenza infection from bacterial infections, (3) is predictive of clinical illness in challenge studies, (4) is prognostic in influenza pneumonia patients, (5) is correlated with gene expression changes in subjects responding to influenza vaccination, and (6) showed different dynamics in males and females after influenza vaccination.
Although several groups have proposed influenza infection-specific gene sets, there is little overlap among them. For instance, although the total number of genes reported by these studies ranged from 25 to 615, none of the IMS genes were reported by all studies. For example, the most reported gene, MX1, was reported as being overexpressed by only seven datasets, whereas CD38 was reported as overexpressed by only one dataset. These observed discrepancies in the reported results of influenza infection across multiple studies also underscore the importance and advantage of using our rigorous integrated multi-cohort analysis approach. It enables the researchers to leverage the large amounts of publicly available datasets and the biological and technological heterogeneity present among them to create “Big Data” from multiple “Small Data” that are better representative of the heterogeneity observed in the real-world population. This approach has been previously found to be effective at uncovering genes with consistent expression profiles that are mechanistic, diagnostic, and therapeutic (Chen et al., 2014, Khatri et al., 2013, Li et al., 2015, Mazur et al., 2014, Sweeney et al., 2015).
Identification and validation of these robust and reproducible signatures across multiple independent cohorts could in turn enable further systematic global analyses. For instance, none of the existing studies of influenza infection and vaccination have explored how their proposed signatures relate infection and vaccination to each other (Furman et al., 2014, Hu et al., 2013, Li et al., 2014, Nakaya et al., 2011, Parnell et al., 2011, Parnell et al., 2012, Ramilo et al., 2007, Tsang et al., 2014, Woods et al., 2013). In contrast, we explored the IMS in three influenza vaccination studies to show that the same signature also correlates with influenza vaccine response, thereby bridging the gap between influenza infection and vaccination because the IMS is diagnostic and prognostic in influenza-infected patients and correlates with vaccine response.
Compelling clinical data have shown that men and women differ in their innate, humoral, and cell-mediated response to viral vaccines (Klein et al., 2010). For example, testosterone can have immunosuppressive role in response to influenza vaccination (Furman et al., 2014). In line with this, the IMS scores changed significantly in responders on day 1 for males and on day 3 for females after vaccination. These results suggest that the effect of sex differences on immune response might be more nuanced in that males respond sooner to vaccination compared to females and that sex differences might also have an effect on the dynamics of an immune response, not just its magnitude. These results further suggest that future vaccination studies should be designed to sample subjects more frequently in the first 3 days after vaccination to further understand sex differences in immune response after vaccination.
Jenner and Young (2005) previously carried out an analysis of 785 samples in 32 studies. However, contrary to our integrated multi-cohort analysis, which focused on identifying a robust signature of viral infection, their analysis focused on identifying a common transcriptional response to pathogenic infections irrespective of source (bacterial or viral). Furthermore, their analysis relied on a clustering of gene expression patterns across all studies to identify a “common host response” of 511 genes, where these genes were not ubiquitously expressed across all datasets. Instead, we employed an established statistical framework to account for the variability of gene expression, array types, and the number of samples within each study. In addition, all samples used in our analysis for the identification of MVS and IMS were peripheral blood samples, whereas Jenner and Young (2005) used samples from various sources including cell lines, sorted immune cells, and other tissues (liver, skin, astrocytes, fibroblasts, etc.). Because of these important differences in study design, many genes identified by Jenner and Young (2005) as those mediating inflammation were not significantly differentially expressed in our analysis, because they are probably overexpressed during both bacterial and viral infections compared to uninfected individuals. However, a number of the IFN-inducible genes they identified were also overexpressed in our analysis, suggesting that although these genes are overexpressed during bacterial and viral infections, they have greater changes in their expression after influenza infection than bacterial infections.
Many type-1-interferon-stimulated genes (ISGs) have been reported in the literature. However, depending on the cell type, IFN dose, and time of treatment, the number of ISGs can vary from 50 to 1,000 (Schoggins and Rice, 2011). Thus, it has remained unclear which of these genes were most relevant to influenza infection. Herein we identified a concise influenza response signature with many ISGs. Indeed, IFN-β treatment of HBECs suggests that some or all of the genes in the IMS are stimulated by IFN. This finding indicates that whereas ISGs are a common element among signatures, a concise and specific gene subset features most importantly in response to influenza.
Although the IMS was identified using whole blood or PBMC samples, most of the IMS genes were also overexpressed after influenza infection in primary human bronchial epithelial cells and lung adenocarcinoma cell lines A549 and Calu-3. These results provide further evidence of the conservation of the response to influenza infection and identify a correlation between the systemic immune response in blood and epithelium at the onset of infection as has been observed before (Ioannidis et al., 2012). However, not all IMS genes were overexpressed in these in vitro studies. For example, as expected, CD38 was not differentially expressed, since its expression is specific to lymphocytes (Moreno-García et al., 2005, Sandoval-Montes and Santos-Argumedo, 2005). These results suggest that the IMS response to influenza infection is present in both circulating blood and in separate tissue types.
This paper has some limitations. First, the MVS gene set might include too many genes to allow for a simple clinical test; further optimization might be needed before reduction to practice. Second, the IMS signature was specific to influenza against the other viral infections for which data were available, but there are other clinical infections similar to influenza infection (i.e., parainfluenza infection) that would need further testing. Finally, prospective testing will be needed for both gene sets.
The commonality of the MVS across multiple viral infections and specificity of the IMS for influenza infection suggest a number of potential applications. First, the MVS could serve as a starting point to identify broad host factor targets for developing broad-spectrum anti-viral drugs. Most of the existing anti-viral drugs are based on the one-drug-one-bug philosophy, which is inefficient and ineffective because viruses often mutate to develop drug resistance and novel viral strains can emerge from animal carriers. Instead, developing anti-viral drugs that target host factors involved in multiple respiratory viral infections could provide protection against a broader group of viruses and be more effective against novel strains. Second, the IMS score can be used clinically to distinguish influenza infections, regardless of whether there is concurrent bacterial pneumonia, which could help clinicians to determine whether to initiate antiviral treatments in patients with potential co-infections. Third, the IMS score can be used as a more objective measure of a vaccination response. Our results in influenza-vaccinated individuals show that the IMS score increases significantly in responders but remains unchanged in non-responders.
Finally, the specificity of IMS to influenza compared to other respiratory viruses enables identification of a virus-specific immune metric that can be applied to both vaccination and natural infection studies, and might be a starting place for studies of differential biology of influenza infection. In general, the ability to define metrics for the immune system, such as we have done here, goes beyond the concept of biomarkers and provides a means to measure and understand functional and, potentially, mechanistic pathophysiological relationships for other disease-specific clinical cohorts.
Experimental Procedures
Data Collection and Pre-processing
Our entire analysis was performed with publicly available data. We downloaded 18 microarray gene expression datasets from the NCBI GEO comprising 2,939 samples derived from whole blood, PBMCs, epithelial cells, or cell lines (Table S1). The samples in these datasets represented different biological conditions including viral infections (influenza, RSV, HRV, SARS, adenovirus, enterovirus, HHV6), bacterial infections (E. coli, S. aureus, S. pneumonie, Salmonella), non-pathogenic systematic inflammatory response, and healthy controls. We incorporated technical heterogeneity in our analysis by choosing datasets that were profiled using microarrays from different manufacturers. All datasets, except one (GSE: GSE19392), are whole blood or PBMC samples obtained from patients with or without a viral infection over wide range of ages (from fewer than 2 months to more than 60 years). Furthermore, the samples were independently collected and profiled at 14 centers in 7 countries. Supplemental Experimental Procedures provide brief description of each of these datasets.
For all datasets, we verified that the expression was normalized and log2-transformed. For each study, we used the sample phenotypes as defined by the primary publication of a source study. Microarray probes in each dataset were mapped to Entrez Gene identifiers (IDs) to facilitate integrated analysis. If a probe matched more than one gene, the expression data for that probe were expanded to add one record for each mapped gene (Ramasamy et al., 2008).
Integrating Discovery Cohorts by Meta-analysis
We applied two meta-analysis methods as described in our previous publications (Figure S1; Tables S1 and S2): (1) combining effect sizes and (2) combining p values (Chen et al., 2014, Khatri et al., 2013, Sweeney et al., 2015). We estimated the effect size for each gene in each dataset as Hedges’ adjusted g. If multiple probes mapped to a gene, the effect size for each gene was summarized via the fixed effect inverse-variance model. The study-specific effect sizes for each gene were then combined into a single meta effect-size using a linear combination of study-specific effect sizes, f i, where each study-specific effect size was weighted by inverse of the variance in the corresponding study. After computing meta effect-size, p values were corrected for multiple hypotheses testing via Benjamini-Hochberg false discovery rate (FDR) correction (Benjamini and Hochberg, 1995), and significant genes were identified via Z-statistic.
We used Fisher’s sum of logs method (Fisher, 1934) for meta-analysis by combining p values. For each gene, we summed the logarithm of the one-sided hypothesis testing p values across k studies and compared the result to a χ2-distribution with 2k degrees of freedom.
In order to avoid influence of a dataset with a large sample size on the results, we removed one dataset at a time and applied both meta-analysis methods at each iteration. We did not filter for heterogeneity between datasets when identifying MVS because different viruses can induce various genes at different levels. However, we ensured that there was no significant heterogeneity (p > 0.05) in effect size across all datasets for the IMS.
Meta Virus Signature and Influenza Meta-Signature Score
We defined the MVS score of a sample as the geometric mean of the normalized, log2-transformed expression of the 161 overexpressed genes minus that of the 235 underexpressed genes. We defined the IMS score of a sample as the geometric mean of the normalized log2-transformed expression of the 11 overexpressed genes. We scaled and centered the MVS or IMS scores of all samples in a given dataset (mean = 0, standard deviation = 1) to enable comparisons between datasets. We used the Wilcoxon-Mann-Whitney (Wilcoxon, 1945) or ANOVA to test whether there was a statistically significant difference between the MVS or IMS scores of two groups.
If a dataset contained negative values, computing a geometric mean is not possible. In these datasets, we used mean of the normalized log2-transformed expression values to compute the MVS and IMS scores.
Pathway Analysis
We performed functional pathway analysis via iPathwayGuide (Draghici et al., 2007, Khatri et al., 2008, Tarca et al., 2009). Meta-effect size across all viral infections was used as fold change in iPathwayGuide to identify significant pathways. We used FDR ≤ 10% as a threshold for identifying significant pathways. We performed network analysis of influenza-specific genes by IPA with an option to include only “direct relationship” to avoid spurious connections caused by “indirect relations.” Direct relationships in IPA result from publications citing experimental evidence for an interaction.
Author Contributions
P.K. conceived the study. M.A.-T. and P.K. collected, curated, and analyzed the data. E.B. and Y.P. assisted with data collection, curation, and organization. M.A.-T., H.M.M., T.E.S., C.M.T., and P.K. interpreted analysis results. P.K., M.A.-T., and H.M.M. wrote the manuscript. T.E.S. and C.M.T. critically revised the manuscript.
Acknowledgments
We thank Mark M. Davis for helpful discussion. M.A.-T. is funded by “La Caixa” Foundation. H.M.M. is funded by a NHMRC early career fellowship. T.E.S. is funded by a Stanford Child Health Research Institute Young Investigator Award (through the Institute for Immunity, Transplantation and Infection) and a Society for University Surgeons Resident Scholar award. P.K. is funded by NIAID grants 1U19AI109662, U19AI057229, U54I117925, and U01AI089859 and the Bill and Melinda Gates Foundation.
Published: December 15, 2015
Footnotes
Supplemental Information includes seven figures, five tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2015.11.003.
Supplemental Information
Refers to data collection and preprocessing in the Experimental Procedures.
Effect size for each MVS gene in discovery and validation cohorts are provided. Summary effect size for each gene in discovery and validation cohorts are also provided. Refers to Figure 1.
Refers to Figure 1.
Refers to Figure 3.
Refers to Figure 3.
References
- Babcock H.M., Merz L.R., Dubberke E.R., Fraser V.J. Case-control study of clinical features of influenza in hospitalized patients. Infect. Control Hosp. Epidemiol. 2008;29:921–926. doi: 10.1086/590663. [DOI] [PubMed] [Google Scholar]
- Barrett T., Suzek T.O., Troup D.B., Wilhite S.E., Ngau W.-C., Ledoux P., Rudnev D., Lash A.E., Fujibuchi W., Edgar R. NCBI GEO: mining millions of expression profiles--database and tools. Nucleic Acids Res. 2005;33:D562–D566. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Statist. Soc., B. 1995;57:289–300. [Google Scholar]
- Bermejo-Martin J.F., Martin-Loeches I., Rello J., Antón A., Almansa R., Xu L., Lopez-Campos G., Pumarola T., Ran L., Ramirez P. Host adaptive immunity deficiency in severe pandemic influenza. Crit. Care. 2010;14:R167. doi: 10.1186/cc9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Khatri P., Mazur P.K., Polin M., Zheng Y., Vaka D., Hoang C.D., Shrager J., Xu Y., Vicent S. A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res. 2014;74:2892–2902. doi: 10.1158/0008-5472.CAN-13-2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S., Khatri P., Tarca A.L., Amin K., Done A., Voichita C., Georgescu C., Romero R. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–1545. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R.A. Oliver & Boyd; New York: 1934. Statistical Methods for Research Workers. [Google Scholar]
- Franco L.M., Bucasas K.L., Wells J.M., Niño D., Wang X., Zapata G.E., Arden N., Renwick A., Yu P., Quarles J.M. Integrative genomic analysis of the human immune response to influenza vaccination. eLife. 2013;2:e00299. doi: 10.7554/eLife.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman D., Jojic V., Kidd B., Shen-Orr S., Price J., Jarrell J., Tse T., Huang H., Lund P., Maecker H.T. Apoptosis and other immune biomarkers predict influenza vaccine responsiveness. Mol. Syst. Biol. 2013;9:659. doi: 10.1038/msb.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman D., Hejblum B.P., Simon N., Jojic V., Dekker C.L., Thiébaut R., Tibshirani R.J., Davis M.M. Systems analysis of sex differences reveals an immunosuppressive role for testosterone in the response to influenza vaccination. Proc. Natl. Acad. Sci. USA. 2014;111:869–874. doi: 10.1073/pnas.1321060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herberg J.A., Kaforou M., Gormley S., Sumner E.R., Patel S., Jones K.D.J., Paulus S., Fink C., Martinon-Torres F., Montana G. Transcriptomic profiling in childhood H1N1/09 influenza reveals reduced expression of protein synthesis genes. J. Infect. Dis. 2013;208:1664–1668. doi: 10.1093/infdis/jit348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Yu J., Crosby S.D., Storch G.A. Gene expression profiles in febrile children with defined viral and bacterial infection. Proc. Natl. Acad. Sci. USA. 2013;110:12792–12797. doi: 10.1073/pnas.1302968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis I., McNally B., Willette M., Peeples M.E., Chaussabel D., Durbin J.E., Ramilo O., Mejias A., Flaño E. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J. Virol. 2012;86:5422–5436. doi: 10.1128/JVI.06757-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison M.G., Lee N. Influenza 2010-2011: lessons from the 2009 pandemic. Cleve. Clin. J. Med. 2010;77:812–820. doi: 10.3949/ccjm.77a.10135. [DOI] [PubMed] [Google Scholar]
- Jenner R.G., Young R.A. Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 2005;3:281–294. doi: 10.1038/nrmicro1126. [DOI] [PubMed] [Google Scholar]
- Khatri P., Draghici S., Tarca A.L., Hassan S.S., Romero R. Progress in Pattern Recognition, Image Analysis and Applications. Springer; Berlin: 2008. A System Biology Approach for the Steady-State Analysis of Gene Signaling Networks; pp. 32–41. [Google Scholar]
- Khatri P., Roedder S., Kimura N., De Vusser K., Morgan A.A., Gong Y., Fischbein M.P., Robbins R.C., Naesens M., Butte A.J., Sarwal M.M. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J. Exp. Med. 2013;210:2205–2221. doi: 10.1084/jem.20122709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S.L., Jedlicka A., Pekosz A. The Xs and Y of immune responses to viral vaccines. Lancet Infect. Dis. 2010;10:338–349. doi: 10.1016/S1473-3099(10)70049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Bankhead A., 3rd, Eisfeld A.J., Hatta Y., Jeng S., Chang J.H., Aicher L.D., Proll S., Ellis A.L., Law G.L. Host regulatory network response to infection with highly pathogenic H5N1 avian influenza virus. J. Virol. 2011;85:10955–10967. doi: 10.1128/JVI.05792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Rouphael N., Duraisingham S., Romero-Steiner S., Presnell S., Davis C., Schmidt D.S., Johnson S.E., Milton A., Rajam G. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 2014;15:195–204. doi: 10.1038/ni.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.D., Burns T.C., Morgan A.A., Khatri P. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol. Commun. 2015;2:93. doi: 10.1186/s40478-014-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locarnini S., Warner N. Major causes of antiviral drug resistance and implications for treatment of hepatitis B virus monoinfection and coinfection with HIV. Antivir. Ther. 2007;12(Suppl 3):H15–H23. [PubMed] [Google Scholar]
- Loveday E.K., Svinti V., Diederich S., Pasick J., Jean F. Temporal- and strain-specific host microRNA molecular signatures associated with swine-origin H1N1 and avian-origin H7N7 influenza A virus infection. J. Virol. 2012;86:6109–6122. doi: 10.1128/JVI.06892-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur P.K., Reynoird N., Khatri P., Jansen P.W.T.C., Wilkinson A.W., Liu S., Barbash O., Van Aller G.S., Huddleston M., Dhanak D. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014;510:283–287. doi: 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejias A., Dimo B., Suarez N.M., Garcia C., Suarez-Arrabal M.C., Jartti T., Blankenship D., Jordan-Villegas A., Ardura M.I., Xu Z. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med. 2013;10:e1001549. doi: 10.1371/journal.pmed.1001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-García M.E., López-Bojórques L.N., Zentella A., Humphries L.A., Rawlings D.J., Santos-Argumedo L. CD38 signaling regulates B lymphocyte activation via a phospholipase C (PLC)-gamma 2-independent, protein kinase C, phosphatidylcholine-PLC, and phospholipase D-dependent signaling cascade. J. Immunol. 2005;174:2687–2695. doi: 10.4049/jimmunol.174.5.2687. [DOI] [PubMed] [Google Scholar]
- Nakaya H.I., Wrammert J., Lee E.K., Racioppi L., Marie-Kunze S., Haining W.N., Means A.R., Kasturi S.P., Khan N., Li G.-M. Systems biology of vaccination for seasonal influenza in humans. Nat. Immunol. 2011;12:786–795. doi: 10.1038/ni.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell G., McLean A., Booth D., Huang S., Nalos M., Tang B. Aberrant cell cycle and apoptotic changes characterise severe influenza A infection--a meta-analysis of genomic signatures in circulating leukocytes. PLoS ONE. 2011;6:e17186. doi: 10.1371/journal.pone.0017186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell G.P., McLean A.S., Booth D.R., Armstrong N.J., Nalos M., Huang S.J., Manak J., Tang W., Tam O.-Y., Chan S., Tang B.M. A distinct influenza infection signature in the blood transcriptome of patients with severe community-acquired pneumonia. Crit. Care. 2012;16:R157. doi: 10.1186/cc11477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy A., Mondry A., Holmes C.C., Altman D.G. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008;5:e184. doi: 10.1371/journal.pmed.0050184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilo O., Allman W., Chung W., Mejias A., Ardura M., Glaser C., Wittkowski K.M., Piqueras B., Banchereau J., Palucka A.K., Chaussabel D. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood. 2007;109:2066–2077. doi: 10.1182/blood-2006-02-002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reghunathan R., Jayapal M., Hsu L.-Y., Chng H.-H., Tai D., Leung B.P., Melendez A.J. Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman D.D., Morton S.C., Wrin T., Hellmann N., Berry S., Shapiro M.F., Bozzette S.A. The prevalence of antiretroviral drug resistance in the United States. AIDS. 2004;18:1393–1401. doi: 10.1097/01.aids.0000131310.52526.c7. [DOI] [PubMed] [Google Scholar]
- Sandoval-Montes C., Santos-Argumedo L. CD38 is expressed selectively during the activation of a subset of mature T cells with reduced proliferation but improved potential to produce cytokines. J. Leukoc. Biol. 2005;77:513–521. doi: 10.1189/jlb.0404262. [DOI] [PubMed] [Google Scholar]
- Schoggins J.W., Rice C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011;1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira S.D., Gat-Viks I., Shum B.O.V., Dricot A., de Grace M.M., Wu L., Gupta P.B., Hao T., Silver S.J., Root D.E. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutejo R., Yeo D.S., Myaing M.Z., Hui C., Xia J., Ko D., Cheung P.C.F., Tan B.-H., Sugrue R.J. Activation of type I and III interferon signalling pathways occurs in lung epithelial cells infected with low pathogenic avian influenza viruses. PLoS ONE. 2012;7:e33732. doi: 10.1371/journal.pone.0033732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney T.E., Shidham A., Wong H.R., Khatri P. A comprehensive time-course-based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci. Transl. Med. 2015;7:287ra71. doi: 10.1126/scitranslmed.aaa5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca A.L., Draghici S., Khatri P., Hassan S.S., Mittal P., Kim J.-S., Kim C.J., Kusanovic J.P., Romero R. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang J.S., Schwartzberg P.L., Kotliarov Y., Biancotto A., Xie Z., Germain R.N., Wang E., Olnes M.J., Narayanan M., Golding H., Baylor HIPC Center. CHI Consortium Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell. 2014;157:499–513. doi: 10.1016/j.cell.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcoxon F. Individual comparisons by ranking methods on JSTOR. Biometrics. 1945;3:119–122. [PubMed] [Google Scholar]
- Woods C.W., McClain M.T., Chen M., Zaas A.K., Nicholson B.P., Varkey J., Veldman T., Kingsmore S.F., Huang Y., Lambkin-Williams R. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS ONE. 2013;8:e52198. doi: 10.1371/journal.pone.0052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaas A.K., Chen M., Varkey J., Veldman T., Hero A.O., 3rd, Lucas J., Huang Y., Turner R., Gilbert A., Lambkin-Williams R. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe. 2009;6:207–217. doi: 10.1016/j.chom.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Refers to data collection and preprocessing in the Experimental Procedures.
Effect size for each MVS gene in discovery and validation cohorts are provided. Summary effect size for each gene in discovery and validation cohorts are also provided. Refers to Figure 1.
Refers to Figure 1.
Refers to Figure 3.
Refers to Figure 3.