

Abstract
We report on 16 patients with relapsed or refractory B cell acute lymphoblastic leukemia (B-ALL) that we treated with autologous T cells expressing the 19-28z chimeric antigen receptor (CAR) specific to the CD19 antigen. The overall complete response rate was 88%, which allowed us to transition most of these patients to a standard-of-care allogeneic hematopoietic stem cell transplant (allo-SCT). This therapy was as effective in high-risk patients with Philadelphia chromosome–positive (Ph+) disease as in those with relapsed disease after previous allo-SCT. Through systematic analysis of clinical data and serum cytokine levels over the first 21 days after T cell infusion, we have defined diagnostic criteria for a severe cytokine release syndrome (sCRS), with the goal of better identifying the subset of patients who will likely require therapeutic intervention with corticosteroids or interleukin-6 receptor blockade to curb the sCRS. Additionally, we found that serum C-reactive protein, a readily available laboratory study, can serve as a reliable indicator for the severity of the CRS. Together, our data provide strong support for conducting a multicenter phase 2 study to further evaluate 19-28z CAR T cells in B-ALL and a road map for patient management at centers now contemplating the use of CAR T cell therapy.
INTRODUCTION
T cell therapy with tumor-targeted chimeric antigen receptor (CAR)–modified T cells has recently transitioned from the laboratory to the clinic and yielded outcomes that support the tremendous potential of this approach to cancer therapy (1–3). CARs are artificial receptors that redirect antigen specificity, activate T cells, and further enhance T cell function through their costimulatory component (4, 5). Three groups, including our own, have reported objective tumor responses when infusing autologous T cells genetically modified with CD19-targeted CARs into patients with chronic lymphocytic leukemia (CLL) and other indolent non-Hodgkin’s lymphomas (3, 6, 7). We next demonstrated potent antitumor benefit after infusing CD19-targeted 19-28z CAR T cells into five adults with relapsed or refractory B cell acute lymphoblastic leukemia (B-ALL) (1). In adults, relapsed B-ALL has a markedly poor prognosis with an expected median survival of less than 6 months (8, 9). In this setting of highly chemotherapy-resistant, rapidly progressive disease, therapy with CD19-targeted CAR T cells resulted in complete molecular remissions (CRm), as assessed by immunoglobulin heavy chain (IgH) deep sequencing, in five of five treated patients. Achieving CRm in this chemotherapy-refractory population allowed for subsequent allogeneic stem cell transplants (allo-SCT) in clinically eligible subjects, the standard of care in adults for this disease after relapse (8). These promising clinical outcomes were confirmed by investigators from the Children’s Hospital of Pennsylvania in a case report of two pediatric patients with relapsed B-ALL treated with a similar CD19 CAR T cell therapy (2). We have now treated an additional 11 patients with relapsed or refractory B-ALL. The clinical outcomes in these CD19-targeted CAR T cell–treated patients confirm the clinical efficacy of this approach seen with our initial results; 19-28z CAR T cells induced complete remissions (CRs) in the vast majority of patients, enabling many to transition to an allo-SCT.
Infusion of CD19 CAR T cells can be associated with toxicities including high-grade fevers, hypotension, hypoxia, and neurologic disturbances that may require aggressive medical support (1–3). This syndrome of toxicities has been described as a cytokine release syndrome (CRS) likely related to a progressive systemic inflammatory process initiated and maintained by the infused CAR T cells activated in vivo upon encounter with the targeted CD19 antigen. However, the clinical and laboratory evaluation of this syndrome has been limited to data derived from only a few patients in case reports (1–3). The paucity of published results from which to define or understand the CRS markedly limits the clinical investigator’s ability to either predict the likelihood or anticipate the severity of this associated spectrum of CAR T cell–mediated toxicities.
By analyzing all 16 adults with relapsed or refractory B-ALL treated at our center, we have established laboratory and clinical criteria for the diagnosis of the CAR T cell–related severe CRS (sCRS). Using these criteria, we established guidelines for infusion of CAR T cells and the subsequent clinical management, part of which includes the serial monitoring of C-reactive protein (CRP). We have found that daily monitoring of CRP in combination with simple clinical parameters allows us to identify patients in need of intensive medical monitoring and potentially pharmacologic management. These codified guidelines will be useful as the CAR technology, developed and currently used in only a few specialized centers, is adapted to a larger number of medical centers less experienced with this technology. On the basis of the remarkably robust clinical results and our toxicity management algorithm, we will soon open a multicenter phase 2 clinical trial to further evaluate the efficacy of 19-28z CAR T cells and prospectively validate our proposed CRS monitoring and intervention guidelines in patients with B-ALL.
RESULTS
Clinical trial
We have treated 16 patients on our 19-28z CAR T cell phase 1 trial (1). This trial is open to adults with B-ALL either in CR1 or with relapsed or refractory disease; however, patients are treated with 19-28z T cells only if they develop relapsed disease. Most patients treated to date have been enrolled under the relapsed arm (fig. S1). Enrolled patients undergo leukapheresis, and those with relapsed or refractory B-ALL receive “physician’s choice” salvage chemotherapy. This is followed, regardless of disease response, by cyclophosphamide conditioning chemotherapy and infusion of 19-28z CAR T cells. The median age of our treated patients is 50 years (Table 1). Additional poor-risk factors in our treatment cohort include Philadelphia chromosome–positive (Ph+) disease (n = 4) as well as relapse after allo-SCT (n = 4), ruling out selective “good risk” patient enrollment on this trial as a potential factor to confound clinical outcomes. Consistent with chemotherapy-resistant disease in these patients is the high rate of residual disease after salvage therapy and before the time of T cell infusion (88%, Table 1).
Table 1.
Patient demographics and disease characteristics. CNS, central nervous system.
| Characteristics | No. of patients (N = 16) | % |
|---|---|---|
| Sex | ||
| Male | 12 | 75 |
| Female | 4 | 25 |
| Age (years) | ||
| Median | 50 | |
| Range | ||
| 18–29 | 4 | 25 |
| 30–59 | 7 | 44 |
| ≥60 | 5 | 31 |
| Baseline BM cytogenetics | ||
| Unfavorable | 7 | 44 |
| Ph+ | 4 | 25 |
| Intermediate | 9 | 56 |
| Previous allo-SCT | ||
| Yes | 4 | 25 |
| No | 12 | 75 |
| Extramedullary disease | ||
| CNS | 2 | 12 |
| Other | 1 | 6 |
| None | 13 | 81 |
| Duration of CR1 (months) | ||
| Median | 8 | |
| Range | ||
| <6 | 5 | 31 |
| 6–24 | 7 | 44 |
| >24 | 4 | 25 |
| Number of salvage regimens | ||
| 1 | 9 | 56 |
| 2 | 4 | 25 |
| ≥3 | 3 | 19 |
| Refractory to immediate previous therapy | ||
| Yes | 14 | 88 |
| No | 2 | 12 |
| B-ALL tumor burden in the BM before CAR T cell infusion (n = 15)* | ||
| MRD− | 2 | 13 |
| MRD+ | 5 | 33 |
| <50% blasts | 2 | 13 |
| ≥50% | 6 | 40 |
One patient had only gross extramedullary disease (no detectable disease in the BM).
CAR T cell products were successfully generated at the dose of 3 × 106 CAR T cells/kg in 15 of 16 patients despite low T cell numbers (as low as 3.7%) in the leukapheresis product of these heavily pretreated patients (table S1). MSK-ALL09 only received 16% of the prescribed T cell dose, despite two independent production attempts. Both attempts showed low gene transfer efficiency and poor T cell expansion, possibly due to the quality of the starting T cell product because dose production was successful from a later pheresis collection. There was no other patient enrolled on this trial that did not have an adequate dose production, and this dose was not a requirement for 19-28z CAR T cell treatment. γ-Retroviral 19-28z CAR gene transfer was overall robust, with 5 to 60% (mean, 24%) 19-28z CAR expression in end-of-production T cells.
Clinical outcomes
Infusion of 19-28z CAR T cells after salvage chemotherapy markedly enhanced the overall complete response rate, composed of both patients with a CR and a CR with incomplete count recovery (CRi), to 88%. This is a higher CR rate than that expected with salvage chemotherapy alone [Table 2 and (8–10)]. After 19-28z CAR T cell infusion, the overall CR rate was 78% in the nine patients with gross morphologic residual leukemia after salvage chemotherapy. Further analyses of CR status included studies to detect minimal residual disease (MRD) by flow cytometry, quantitative polymerase chain reaction (qPCR) for the bcr-abl transcript in patients with Ph+ B-ALL, and, whenever feasible, deep sequencing for IgH rearrangements (11) associated with malignant clones (Table 2). Overall, 75% of treated patients achieved an MRD-negative (MRD−) or CRm disease status based on one or more of the above MRD assays (Table 2). These CR and CRm rates, obtained in a very poor prognostic patient population, far exceed expectations based on historical data of relapsed adult B-ALL (8–10) and are consistent with a profound antitumor effect mediated by 19-28z CAR T cells (Table 2). Furthermore, in patients wherein the malignant tumor clone could be monitored in bone marrow (BM) by deep sequencing, we found rapid elimination of the malignant B-ALL tumor clone after 19-28z CAR T cell infusion (table S2). Concomitant monitoring for 19-28z CAR T cell persistence in the BM revealed that nearly all patients had a peak of CAR T cells within 1 to 2 weeks after the infusion and that these numbers decreased to low or undetectable by 2 to 3 months (table S2). Analysis for CAR T cell persistence was limited in patients subsequently treated with allo-SCT. We further observed that in the four patients treated after a post–allo-SCT relapse, there was no clinical evidence of graft-versus-host disease despite the fact that the infused 19-28z CAR T cells were of donor origin.
Table 2.
Summary of clinical outcomes.
| Characteristics | No. of patients (N = 16) | % |
|---|---|---|
| Overall complete response to salvage chemotherapy* | 7 | 44 |
| Overall complete response to 19–28z CAR T cells | 14† | 88 |
| In patients with morphologic residual leukemia (n = 9) | 7 | 78 |
| Complete remission (CR) | 10 | 63 |
| Complete remission with incomplete count recovery (CRi) | 4 | 25 |
| Molecular complete remission (CRm)‡ | 12† | 75 |
| Median time to CR/CRi (days) | 24.5 | |
| Post-CAR T allo-SCT (n = 10 eligible patients)§ | 7 | 70 |
Overall complete response = CR + CRi (determined without regard to CRm status).
Includes two patients who were in CRm before CAR T cell infusion.
CRm or MRD− as determined by flow cytometry and/or deep sequencing for the index IgH clonotype and/or qPCR for the bcr-abl transcript.
Three patients had medical contraindication to allo-SCT, two patients in CR have declined potential allo-SCT, and one is being evaluated for an allo-SCT.
The CAR T cell–associated CRS
CAR-engineered T cells can induce in some patients a clinical syndrome of fevers, hypotension, hypoxia, and neurologic changes associated with marked elevations of serum cytokines (1–3). This spectrum of clinical and laboratory findings has been termed a CRS, which, given the anecdotal nature of this phenomenon, has remained largely undefined. We therefore analyzed our cohort to search for clinical or laboratory results that might serve as diagnostic indicators for a clinically meaningful or severe CRS, predictably requiring additional therapeutic intervention. To this end, we identified a set of criteria for the diagnosis of an sCRS based on the presence of fevers, elevation of characteristic cytokines, and clinical toxicities (Table 3, table S3, and Fig. 1). Patients with evidence of CRS typically have fevers that start about 24 hours after infusion with 19-28z CAR T cells and can persist for several days (Fig. 1A). Fevers are, however, not always the harbinger for more clinically relevant toxicities. We therefore evaluated cytokine increases to discern between patients with sCRS associated with clinical deterioration and patients whose fevers and discomfort would resolve spontaneously or with minimal support. The importance of this distinction is to avoid premature intervention that may diminish T cell persistence or efficacy.
Table 3.
Diagnostic criteria for sCRS secondary to CAR T cells.
| Criteria for sCRS |
|---|
| Fevers for at least three consecutive days |
| Two cytokine max fold changes of at least 75 or one cytokine max fold change of at least 250 |
| At least one clinical sign of toxicity such as hypotension (requiring at least one intravenous vasoactive pressor) or, |
| Hypoxia (PO2 < 90%) or, |
| Neurologic disorders (including mental status changes, obtundation, and seizures) |
Fig. 1. Characteristics of the CRS.
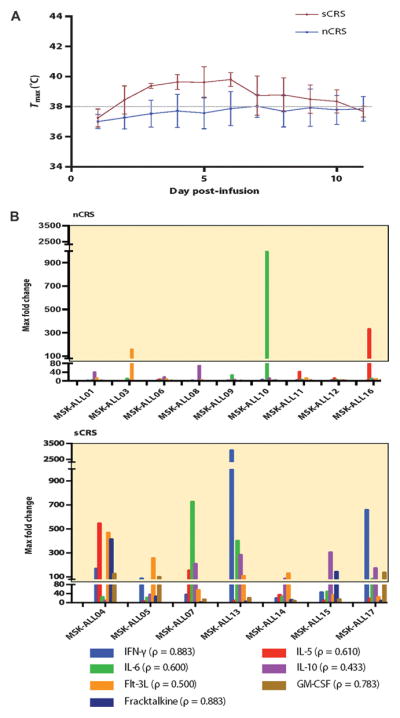
(A) Average max temperatures on days 1 to 11 after CAR T cell infusion in patients with sCRS compared to nCRS patients. Error bars represent the SD. The dashed line is at 38°C to indicate the threshold for fevers. Two-way analysis of variance (ANOVA) analysis between the sCRS and nCRS groups revealed a P = 0.019 (n = 22). (B) Max fold changes of seven inflammatory cytokines selected for their consistent pattern of elevation during sCRS. Depicted are the max fold changes relative to pretreatment values over days 1 to 21 after CAR T cell infusion. The highlighted box represents changes 75-fold and above. Correlation was assessed for pretreatment tumor burden and cytokine elevations for patients diagnosed with sCRS. The Spearman rank correlation coefficient was calculated with pretreatment tumor burden, measured by deep sequencing, and cytokine concentration (pg/ml), and is listed next to each cytokine. IFN-γ, interferon-γ; GM-CSF, granulocyte-macrophage colony-stimulating factor.
We have previously correlated cytokine levels to pretreatment B-ALL tumor burden (1), albeit in a small sample size (n = 5), which precluded more in-depth analyses. With a larger cohort of patients, we not only confirmed this correlation but also identified 7 cytokines of 39 measured, whose elevation correlated (r = 0.43 to 0.88) to pretreatment tumor burden (Fig. 1B) and also to an sCRS (table S4). Within this panel of seven cytokines, we observed that patients with CRS requiring intensive medical intervention had a 75-fold increase over pretreatment baseline levels in two of the selected seven cytokines (Table 3). Furthermore, those patients with sCRS universally exhibited at least one of the following clinical manifestations: hypoxia, hypotension, and/or neurologic changes. Thus, on the basis of the combined clinical and cytokine data, we could accurately define an sCRS in those patients with the triad of persistent fevers (38°C) for more than 3 days, selected cytokine elevations, and additional clinical evidence of toxicity. Application of these criteria enables stratification of patients into the sCRS group, which requires closer observation and is likely to require medical and pharmacologic intervention, and another group (nCRS) made up of patients who tolerate therapy and only require routine observation and management. This latter nCRS cohort includes patients with a mild CRS, characterized by low-grade fever and mild cytokine increases, or absent CRS, defined as no fevers and/or no significant cytokine elevations (Fig. 1). This is a clinically meaningful stratification because sCRS patients are in the hospital for an average of 56.7 days (SD, 28.6; range, 20 to 104), whereas nCRS patients are in the hospital for an average of 15.1 days (SD, 18.8; range, 4 to 61).
Management of the CRS
CRS-associated toxicities, when severe, require intensive medical management including support with vasoactive pressors, mechanical ventilation, antiepileptics, and antipyretics. However, although these toxicities are concerning, they are a by-product of 19-28z CAR T cell function and, to date, have been fully reversible. We treated our initial three sCRS patients with lymphotoxic high-dose steroids, >100 mg daily of prednisone equivalent, which rapidly reversed symptoms but concurrently ablated 19-28z CAR T cells (Fig. 2). The interleukin-6 receptor (IL-6R)–blocking monoclonal antibody (mAb) tocilizumab may also ameliorate sCRS, as initially reported by Grupp et al. (2) who demonstrated rapid resolution of sCRS after IL-6R blockade. We therefore treated our next three sCRS patients (presenting 27- to 400-fold increases in serum IL-6) with tocilizumab alone (Fig. 2), which reduced patients’ fevers and sCRS symptoms within 1 to 3 days similar to steroid therapy, but did not result in dampened expansion of the 19-28z CAR T cells measured in the peripheral blood. Similar results were noted in the BM by deep sequencing (Fig. 3). In aggregate, we detected a fivefold decrease in 19-28z CAR T cells in the BM of sCRS patients treated with high-dose steroids relative to sCRS patients treated either conservatively or with tocilizumab alone (Fig. 3).
Fig. 2. The effect of steroids and/or tocilizumab on the expansion of CAR T cells in patients with sCRS.

The number of CAR T cells per microliter of whole blood, detected by qPCR, was measured in samples drawn before treatment and from days 1 to 22 after CAR T cell infusion. Max temperatures on days 1 to 11 are also depicted. In addition, the days when steroids or tocilizumab was administered to manage sCRS are shown. The red dashed line represents the duration of steroid treatment, and the gray dashed line is at the 38°C fever threshold.
Fig. 3. CAR T cells detected in the BM by deep sequencing.
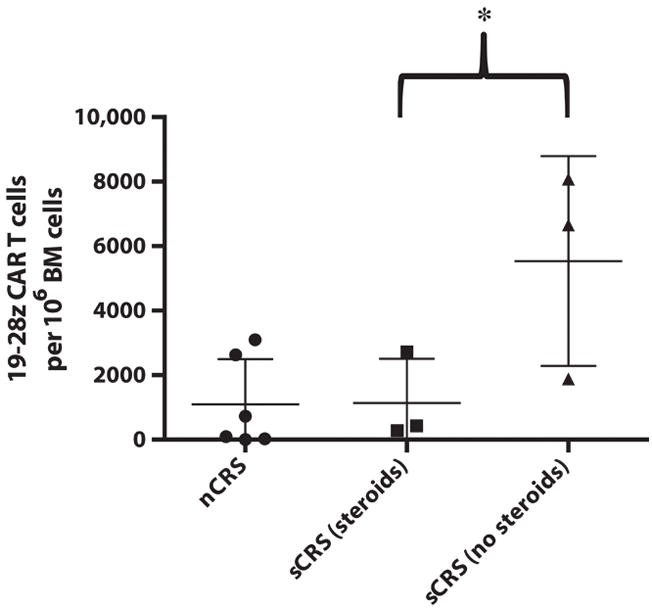
BM was isolated from patients and submitted to Adaptive Biotechnologies for deep sequencing of the IgH rearrangement associated with the 19-28z CAR. The max number of CAR T cells in the BM within 6 weeks of CAR T cell infusion is shown. The mean and SD are depicted for the patient groups stratified on the basis of CRS and its management. *P = 0.048, one-way t test between these two groups (n = 6).
Suppression of CAR T cell expansion presumably has a negative impact on antileukemia control. Indeed, deep sequencing for the IgH rearrangement associated with the malignant B-ALL clone revealed that the three sCRS patients treated with high-dose steroids all experienced a recurrence of disease despite initially achieving a CRm after 19-28z CAR T cell infusion (table S2). Unfortunately, two of these patients did not undergo the recommended allo-SCT while MRD−, because of either medical contraindications (MSK-ALL04) or having declined further therapy (MSK-ALL07), and are now deceased. MSK-ALL05 had a very low level of detectable recurrent disease in the BM by deep sequencing and achieved a CRm after allo-SCT (table S2).
CAR T cell–mediated sCRS-associated neurologic toxicities
Patients with sCRS may also develop reversible neurologic complications including delirium and seizure-like activity. Patients may develop a gradual progression of confusion, word-finding difficulty, and aphasia and ultimately become obtunded. In three cases, these neurologic complications required intubation and mechanical ventilation for airway protection (table S4). Patients with neurologic complications were evaluated with computed tomography and magnetic resonance imaging of the brain, which was nonrevealing, as well as electroencephalograms (EEGs) and lumbar punctures. The EEGs confirmed seizure-like activity, which resolved after antiepileptic treatment. Analysis of cerebrospinal fluid (CSF) obtained by lumbar puncture in three patients at the time of overt neurologic complications revealed a lymphocytosis, which, by further qPCR analyses, was found to be composed of, at least in part, 19-28z CAR T cells (table S5). One of these patients (MSK-ALL14) had a previous history of CNS disease, which had resolved at the time of CAR T cell infusion. The other two patients did not have any previous or subsequent diagnosis of CNS disease. Although CSF samples were obtained only in a subset of patients and only in the setting of clinical neurologic complications, 19-28z CAR T cells were not detected in the CSF of all patients experiencing neurologic toxicities. For example, CSF from MSK-ALL16, obtained at a time of fevers and delirium, showed no evidence of CAR T cells by direct microscopic examination or more sensitive qPCR (table S5).
CRP as an indicator of the CRS
We have observed that patients with morphologic disease at the time of 19-28z CAR T cell infusion have a greater risk for developing sCRS (Figs. 1 and 2 and table S4). Unfortunately, rapid and daily real-time analysis of serum cytokines, which could guide clinical decision-making before the onset of severe CAR T cell–associated toxicities, is not feasible because of technological limitations with cytokine measurements. Therefore, we sought a laboratory indicator for CRS severity that could be used as a surrogate for cytokine elevation. We focused on the acute-phase reactant, CRP, based on the well-documented association between serum IL-6 and CRP levels (12) and the clinical amelioration of the sCRS afforded by IL-6R blockade [Fig. 2 and (2)].
We retrospectively analyzed serum samples from all patients treated on this trial and determined that only those patients who met the criteria for sCRS had a CRP level of ≥20 mg/dl. We observed a clear difference between the CRP levels of patients with an sCRS versus patients classified as having either mild or no CRS (Fig. 4 and fig. S2). Patients treated with high-dose steroids were excluded from this analysis given the inverse correlation between high-dose steroid treatment and serum CRP. Furthermore, receiver operating characteristic (ROC) curve analysis suggests CRP as an excellent indicator for sCRS (fig. S3). Maximum value of the CRP before sCRS has an area under the curve of 0.968. We observed that patients whose CRP exceeds the threshold are particularly at high risk for CRS (sensitivity, 86%; specificity, 100%).
Fig. 4. CRP levels in patients infused with 19-28z CAR T cells.
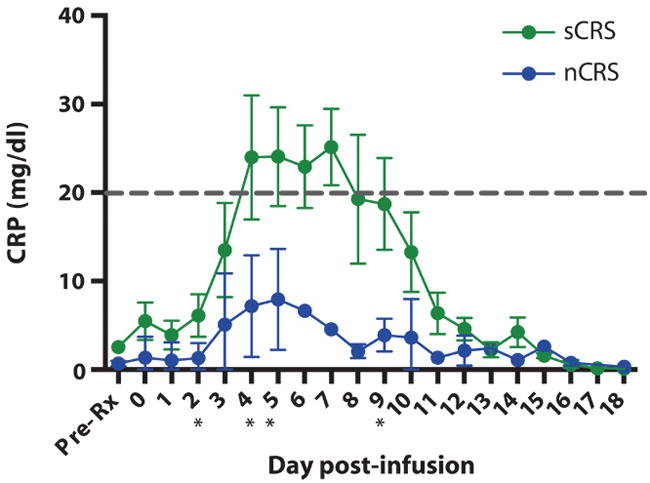
CRP was measured before treatment and from days 1 to 18 after CAR T cell infusion. The green lines represent CRP levels from patients who met the diagnostic criteria for sCRS (n = 4) and were treated with either tocilizumab or nothing. The CRP levels of patients classified as nCRS are illustrated in blue. Error bars are the SEM. The gray dashed line is at 20 mg/dl, which indicates the threshold where patients are at high risk for clinical complications secondary to sCRS. *P < 0.05, unpaired t tests at the corresponding time point. Specific P values for the time points are as follows: day 2, P = 0.035 (n = 13); day 4, P = 0.025 (n = 12); day 5, P = 0.019 (n = 11); and day 9, P = 0.01 (n = 8).
CAR T cells as a bridge to allo-SCT
As per the current standard of care for adults with relapsed or refractory B-ALL, the initial primary aim of therapy is to reinduce a CR (8–10). This, in turn, renders the patient eligible for an allo-SCT, which is, at present, the only therapeutic modality with curative potential. Of the 16 patients treated on this protocol, 3 were ineligible for allo-SCT because of a failure to achieve a CR despite 19-28z CAR T cell infusion, 3 patients in CR were ineligible because of medical comorbidities that pre-existed 19-28z CAR T cell therapy, and 2 patients in CR were eligible for allo-SCT but declined further therapy (Tables 2 and 4). One patient is currently being evaluated for a potential allo-SCT. To date, 7 of the 16 (44%) treated patients have successfully undergone an allo-SCT post-CAR T cell therapy with no relapses.
Table 4. Individual patient outcomes.
DS, deep sequencing for IgH rearrangement; DUCBT, double umbilical cord blood transplant; FC, flow cytometry; HUCT, haplo-umbilical cord transplant; MTX, methotrexate; Peg, pegaspariginase; Pred, prednisone; qPCR, quantitative PCR for bcr-abl transcript; RD, related donor; TCD, T cell–depleted; UD, unrelated donor; Vinc, vincristine; NA, not available.
| Patient ID | Age | Cytogenetics at diagnosis | Salvage therapy | Disease response to salvage therapy | Disease response to CAR T cells | Allo-SCT |
|---|---|---|---|---|---|---|
| MSK-ALL01 | 66 | Normal karyotype | Vinc/Pred/Peg | MRD+ by DS | MRD− by DS | 10/10 TCD RD at 64 days post |
| MSK-ALL03 | 56 | Normal karyotype | Vinc/Pred/Peg | MRD− by FC | MRD− by FC | 10/10 TCD MRD at 43 days post |
| MSK-ALL04 | 59 | t(9;11), 9p21 deletion | Vinc/Pred | Refractory disease, 63% blasts in BM | MRD− by DS | Ineligible because of medical contraindications |
| MSK-ALL05 | 58 | 9p21 deletion | High-dose cytarabine/mitoxantrone | Refractory disease, 70% blasts in BM | MRD− by DS | TCD DUCBT at 69 days post |
| MSK-ALL06 | 23 | Normal karyotype | Modified NYII Consolidation I (27) |
MRD+ | MRD− by DS | 8/10 TCD UD at 121 days post |
| MSK-ALL07 | 30 | 9q isochrome, 12p13 deletion | Vinc/Pred/Peg | Refractory disease, 5–10% blasts in BM | MRD− by DS | Declined |
| MSK-ALL08 | 74 | Complex including 11q23 deletion | MTX | BM-negative, +extramedullary disease | No response | No response |
| MSK-ALL09 | 23 | NA | Modified NYII Consolidation I |
MRD+ by FC | MRD− by FC, MRD+ by DS | Ineligible because of medical contraindications |
| MSK-ALL10 | 27 | Normal | Modified NYII Consolidation I |
MRD− by FC | MRD− by FC | Ineligible because of medical contraindications |
| MSK-ALL11 | 32 | Ph+ | Vinc/Peg | MRD+ by qPCR | MRD− by qPCR | 10/10 UD at ~90 days post |
| MSK-ALL12* | 42 | Ph+ | Clofarabine | Refractory disease, 97% blasts in BM | No response | No response |
| MSK-ALL13* | 36 | Ph+ | Inotuzumab | Refractory disease, 60% blasts in BM | MRD− by DS and qPCR | Declined |
| MSK ALL14 | 60 | NA | Vinc/Pred/Peg | Refractory disease, 52% blasts in BM | MRD− by FC | HUCT at ~60 days post |
| MSK ALL15* | 27 | t(2;12), monosomy 7 | L20 (28) | Refractory disease, 23% blasts in BM | MRD− by DS | 10/10 UD at 49 days post |
| MSK- ALL16* | 63 | Ph+, 11q deletion | POMP (29) | MRD+ by qPCR | MRD+ by qPCR | No response |
| MSK-ALL17 | 59 | Complex | Vinc/Pred | Refractory disease, 85% blasts in BM | MRD− by FC | Awaiting allo-SCT evaluation |
Enrolled and treated after an allo-SCT.
DISCUSSION
Our results strongly support the therapeutic potential for our first-in-class CD19-specific 19-28z CAR T cell therapy. Although these results were obtained in a single-center phase 1 study, they support further evaluation of 19-28z CAR T cell therapy for this very poor prognosis population in a multicenter phase 2 clinical trial.
Patients with relapsed B-ALL have few treatment options and a historical remission rate with “standard-of-care” salvage chemotherapy of about 30% (8–10). Consistent with this overall poor-risk prognosis, nearly all of our patients (88%) were refractory to the physician’s choice salvage therapy given before CAR T cell infusion (Table 1). In contrast, patients treated with 19-28z CD19 CAR T cells had a very high overall complete response rate (88%), with 86% of the patients from this CR group further classified (12 of 14 patients) as MRD− (CRm) (Tables 2 and 4). Subjects with MRD or overt morphologic residual leukemia after salvage chemotherapy were enrolled in our study. We observed similarly high CR rates in both groups after 19-28z CAR T cell infusion (Table 2).
We were able to successfully transition seven patients (44% of all patients) to standard-of-care therapy with an allo-SCT (Tables 2 and 4). This is especially meaningful when compared to the reported historically low frequency (5%) of relapsed or refractory adult B-ALL patients who ultimately transition to allo-SCT after salvage chemotherapy (13). Thus, 19-28z CAR T cell therapy may represent an effective “bridge” to allo-SCT. Because most of our patients underwent allo-SCT in the setting of a CRm, we hypothesize that transplants performed under these optimal conditions will markedly, if not completely, reduce the historical 30% disease relapse rate of B-ALL patients after allo-SCT (14, 15). To this end, there have been no relapses in the seven patients treated with an allo-SCT after 19-28z CAR T cell therapy (post–allo-SCT follow-up ranges from 2 to 24 months), although two of the patients died while in a CRm from allo-SCT complications. The patients who did not transition to an allo-SCT after T cell therapy did so for a variety of reasons, including suboptimal response to CAR T cell therapy (n = 3), declining allo-SCT despite achieving CRm after 19-28z CAR T cell therapy (n = 2), and preexisting medical contraindications to an allo-SCT in patients with a CR or CRm after 19-28z CAR T cell therapy (n = 3). One recently treated patient is pending an evaluation for an allo-SCT at this time. To date, no patient was precluded from allo-SCT because of toxicities associated with 19-28z CAR T cell therapy.
The design of this phase 1 clinical trial stipulated that after enrollment and leukapheresis, patients were given reinduction salvage chemotherapy and later infused with autologous 19-28z CAR T cells (fig. S1). The low efficacy and prolonged duration of myelosuppression, as well as numerous other toxic side effects, associated with salvage chemotherapy result in many patients having morbidity and/or mortality, which precludes further treatment and may even disqualify some patients from an allo-SCT (8, 9, 15). In contrast to our initial expectations that salvage chemotherapy would enhance CAR T cell antitumor efficacy, we observed similar clinical outcomes in patients who achieved a CR after salvage therapy as well as those patients who did not (Tables 2 and 4). Considering the toxicities associated with salvage reinduction chemotherapy, and the overall high complete response rates to 19-28z CAR T cell therapy, one may question the utility or wisdom of giving patients toxic high-dose chemotherapies before 19-28z CAR T cell infusions.
Consistent with our previous reports (1, 6), the persistence of the 19-28z CAR T cells in ALL patients is about 3 months (table S2). This is in contrast to at least one B-ALL pediatric patient and several CLL patients reported by the University of Pennsylvania (2, 7, 16), who exhibited CAR T cell persistence and persisting B cell aplasia for several months to even more than a year. We hypothesize that the 19-28z CAR T cell expansion and subsequent contraction are CD19 antigen–dependent, resulting in T cell clearance upon elimination of normal and malignant and B cells (1, 6), as seen in a normal T cell immune response to antigen. Accordingly, the persistence of CD19-targeted CARs incorporating a 4-1BB moiety rather than CD28 as used by the University of Pennsylvania may be due, at least in part, to antigen-independent signaling through the 4-1BB CAR, as previously demonstrated in preclinical studies (17). Additional or alternative mechanisms may apply. We are currently developing a human anti-mouse antibody assay to determine whether immune-mediated rejection might be a contributing factor to limiting 19-28z CAR T cell persistence. The CRS symptoms appear earlier with CD28-containing CD19-targeted CAR T cells, reported by both the National Cancer Institute (NCI) and Memorial Sloan-Kettering Cancer Center (MSKCC), compared to 4-1BB–containing CD19-targeted CAR T cells. We believe that this is due to a more rapid T cell expansion in the former group than in the latter, on the basis of this current report and previously published observations (1–3, 6, 7, 16, 18).
Although the overall complete response rate was dramatic, there were also examples of failure to reinduce a CR in patients with morphologic residual disease or failure to induce a CRm in patients with MRD. Two such treatment failures occurred in patients with MRD at the time of CAR T cell infusion, whereas the other two patients had overt morphologic residual leukemia at the time of CAR T cell infusion. One patient (MSK-ALL09) received a low 19-28z T cell dose (16%, table S1) without subsequent evidence of post-infusion in vivo CAR T cell expansion (table S2). Both MRD treatment failures had low to no detectable 19-28z CAR T cells in the BM after infusion, in contrast to those patients with treatment responses, pointing to limited T cell expansion in these patients as one mechanism contributing to treatment failure (Fig. 3 and table S2). The lack of response in MSK-ALL08, who had disease involvement only within a large abdominal lymph node mass, may be due to limited T cell trafficking or immunosuppression of CAR T cells within this extramedullary tumor microenvironment.
The toxicities associated with the infusion of 19-28z CAR T cells are summarized in table S4. Unlike the toxicities associated with conventional salvage chemotherapy, those associated with infused CAR T cells are related to large-scale, synchronous T cell activation upon targeting of CD19+ leukemia cells. Systematic serum cytokine analyses allowed us to select a small panel of cytokines that are strongly associated with an sCRS (Fig. 1B). Identification of these seven cytokines, commonly elevated with the sCRS, allowed us to develop laboratory and clinical criteria for the formal diagnosis of an sCRS (Table 3). On the basis of our analysis, patients with fevers alone and/or elevated serum cytokines in the absence of additional clinically apparent toxicities are unlikely to require anything more than observation or modest medical intervention. In contrast, patients who meet the sCRS criteria are likely to require more intensive observation and aggressive medical management. The diagnostic criteria we established will be useful to normalize evaluation of these toxicities across multiple trials at different medical centers, for developing preclinical models to understand the mechanism behind the CRS (19), and to further optimize the clinical management of this syndrome.
Our initial attempts to manage sCRS have included treating patients with high-dose steroids and/or tocilizumab, an IL-6R–blocking mAb. We have found that the manner of treating sCRS may affect clinical outcomes. Administration of high lymphotoxic doses of steroids as a treatment of sCRS in patients MSK-ALL04, MSK-ALL05, and MSK-ALL07 resulted in a rapid reversal of their fevers, cytokines, and other clinical symptoms but abrogated 19-28z CAR T cell expansion and persistence (Fig. 2). In contrast, administration of tocilizumab as a first-line therapy for sCRS in patients MSK-ALL13, MSK-ALL14, and MSK-ALL17 similarly reduced fevers and ameliorated clinical symptoms without apparent effect on 19-28z CAR T cell expansion and persistence (Fig. 2). Two patients, MSK-ALL13 and MSK-ALL14, had expansion of 19-28z CAR T cells after their first treatment with tocilizumab, with MSK-ALL13 demonstrating an almost 7000-fold in vivo expansion after treatment. The lymphotoxic effect of steroids appears to affect not only the in vivo expansion of the infused CAR T cells but also the clinical outcome of sCRS patients. All three patients treated with high-dose steroids relapsed despite previously achieving a CRm (MRD to morphologic relapse), whereas untreated patients or those treated with tocilizumab alone had no evidence of recurrent disease after achieving a CRm. We do not know if lower doses of steroids might be as effective at decreasing sCRS but without CAR T cell toxicity. These results strongly suggest that tocilizumab should be used in the first-line treatment of sCRS, with high-dose steroids being reserved for those patients with severe life-threatening CRS unresponsive to tocilizumab.
We, as well as others (1–3), have observed a number of clinically alarming neurologic changes associated with the sCRS (table S4). Because of similar published neurologic changes after blinatumomab infusion or CD28 mAb ligation (20, 21), which, in both cases, resulted in robust T cell activation, we speculate that these neurologic toxicities arise from a generalized T cell–mediated inflammatory state rather than direct toxicity mediated by 19-28z CAR T cells on CNS tissues. Indeed, no detectable 19-28z CAR T cells were found in the CSF of MSK-ALL16, despite the clinically evident and persistent delirium at the time of CSF collection (table S5). Understanding the mechanisms underlying the neurologic complications seen with CAR T cell therapy in the setting of an sCRS, as well as more efficient management of these toxicities, will require more intensive clinical and preclinical investigation (19). Fortunately, these complications have been medically manageable and fully reversible in our patient cohort.
We have identified CRP as a potential laboratory indicator for the sCRS, considering that cytokine monitoring is unlikely to be performed daily because of cost and time constraints. Retrospective review of patient serum CRP levels over time (fig. S2) revealed that patients with sCRS who received steroids or tocilizumab were treated at or near their peak serum CRP. Additionally, we found that patients with sCRS treated with steroids and/or tocilizumab exhibited a rapid drop in serum CRP, consistent with clinical resolution of the sCRS (fig. S2). We therefore propose that any patient who has fevers and a CRP ≥20 mg/dl should be managed as if they have sCRS and be considered at high risk for clinical complications (Table 3), a guideline that we plan to validate prospectively. Post hoc cytokine monitoring will still be useful to confirm sCRS and for further research into the biology behind the sCRS. On the basis of our experience, we have developed clinical guidelines for the management of patients being treated with CAR T cells (fig. S4).
Grouping patients according to their CRS status, sCRS (n = 7) versus mild or no CRS (n = 9), aligns significantly with the pretreatment blast burden before 19-28z CAR T cell infusion (P < 0.05, Table 4). Thus, all seven patients who developed sCRS had morphologic residual leukemia and achieved a CRm, whereas the nine patients with mild or no CRS included seven patients with MRD and two with morphologic residual disease, but no treatment response (Table 4). In our previous report (1), we observed a correlation between tumor burden and cytokine elevation but not between tumor burden and outcome, indicating that treatment-associated toxicity was not requisite for an efficient 19-28z CAR T cell–mediated antileukemia effect. With our larger cohort, we continue to report no differences in the clinical outcomes of patients with MRD versus those patients with overt morphologic residual leukemia. However, patients with sCRS, and therefore with morphologic residual leukemia before 19-28z CAR T cell infusion, have greater expansion of CAR T cells in their BM and blood. This may be related to abundant CD19 expression on residual leukemia in these patients, in contrast to low or absent levels of CD19 in patients with MRD (22), or a dampening effect of normal B cells, which may predominate in patients with MRD. Together, our ability to anticipate and manage toxicities in patients treated with 19-28z CAR T cells will greatly enhance the implementation of multicenter phase 2 studies, which the findings reported herein strongly support.
MATERIALS AND METHODS
Clinical protocol design
This is a phase 1 protocol (ClinicalTrials.gov #NCT01044069) that has been described in detail, and the protocol is available as supplemental material [fig. S1 and (1)]. Briefly, it is open to adults with B-ALL in their first CR or with relapsed/refractory disease. However, only patients with relapsed or refractory B-ALL are eligible for infusion with 19-28z CAR T cells. The patients are given a conditioning chemotherapy agent, cyclophosphamide (1.5 to 3.0 g/m2), followed by a fractionated dose (⅓ dose on day 1 and ⅔ dose on the following day) of 19-28z CAR T cells. The dose under evaluation is 3 × 106 CAR T cells/kg. Patients are treated in the inpatient setting to manage potential toxicities after 19-28z CAR T cell infusion. Patients achieving a CR after CAR T cell therapy were referred to the MSKCC BM transplantation service for evaluation of additional therapy with an allo-SCT. The Institutional Review Board at MSKCC reviewed and approved this trial. All patients enrolled and treated on this trial gave written informed consent before participation. All clinical investigation was conducted according to the Declaration of Helsinki principles.
Generation of 19-28z CAR-modified T cells
19-28z CAR T cells were harvested, transduced, formulated, and released as previously described (1, 6, 23).
Analysis of cytokines and CRP after 19-28z CAR T cell infusion
Patient serum samples were analyzed with the Luminex IS100 system and commercially available 39-plex cytokine detection assays as described (1, 6). The Department of Laboratory Medicine at MSKCC used serum to measure high-sensitivity CRP with the Siemens High Sensitivity CRP reagent kit on the ADVIA 1800, also manufactured by Siemens.
Molecular studies of whole blood, BM, and CSF
The malignant IgH rearrangement was detected from a diagnostic relapsed sample. Follow-up BM aspirates were processed to extract 7.5 μg of genomic DNA and submitted for deep sequencing at Adaptive Bio-technologies. Sequences for the malignant IgH rearrangement were then used to interrogate the high-throughput sequencing output to identify residual disease. In some patients, monitoring for the malignant IgH rearrangement was not possible because no relapsed sample was available or no IgH rearrangement was detected because the patients’ IgH locus was germ line.
19-28z CAR T cells were detected by qPCR and/or deep sequencing assays. For qPCR, genomic DNA was isolated from the appropriate tissue, and a portion of the 19-28z CAR construct was amplified as described (6). The deep sequencing process is initiated with a multiplex PCR assay that uses multiple, degenerate VH and JH family primers (11). As a consequence of the degenerate nature of the primers, the high-throughput sequencing output also included sequences for the mouse anti-CD19 IgH rearrangement associated with the 19-28z CAR. Therefore, we were able to monitor for 19-28z CAR T cell persistence in the BM by interrogating the high-throughput sequencing output with the IgH rearrangement associated with the 19-28z CAR. For all deep sequencing data, if <7.5 μg of genomic DNA was submitted, results were normalized to the output expected from 7.5 μg of genomic DNA.
Relapsed or refractory B-ALL diagnosis and clinical outcomes
B-ALL diagnoses were confirmed by pathologists at MSKCC on the basis of BM cell morphology, flow cytometry, and/or genetics. Using standard criteria (24, 25), we classified patient outcomes after CAR T cell infusion as CR, molecular CR (CRm), CR with incomplete platelet or neutrophil recovery (CRi), MRD, or morphologic residual disease. These classifications are further defined below. CR is defined as the disappearance of clinical and cellular evidence of leukemia. There should be restoration of normal hematopoiesis with a neutrophil count ≥1000 × 106/liter and a platelet count ≥100,000 × 106/liter. Blasts should be <5% in a posttreatment BM differential. In contrast, CRi is defined as meeting the criteria for CR but not having adequate platelet or neutrophil recovery. MRD is defined as patients meeting the criteria for CR or CRi, but with residual disease detected by qPCR, flow cytometry, or deep sequencing for the malignant clonal IgH rearrangement. In contrast, CRm corresponds to patients in a CR or CRi but also confirmed to have no MRD, that is, MRD−, as determined by flow cytometry and/or deep sequencing and/or qPCR for the bcr-abl transcript. Morphologic residual disease is defined as ≥5% blasts in a BM differential.
Statistics
Quantitative data were analyzed with t tests and ANOVA, when appropriate. The total number of samples, the statistical test, and P values are detailed in the figure legends. The 7 cytokines associated with sCRS were identified by screening 39 cytokines for strong Spearman rank order correlations (ρ ≥ 0.4) between cytokine max fold change and pretreatment leukemia burden. Among these, we then selected only those cytokines that were elevated in more than one patient. Last, we developed the threshold barrier by identifying the lowest max fold change among the strongly correlated cytokines. The ROC curves for CRP were constructed using the empirical method, and the best cut point was identified via the Youden index (26).
Supplementary Material
Acknowledgments
Funding: NCI (M.L.D., R.B., I.R., and M. Sadelain), Terry Fox Foundation (R.B.), American Society of Hematology–Amos Medical Faculty Development Program (M.L.D.), Alliance for Cancer Gene Therapy (M. Sadelain), Mallah Foundation (M. Sadelain), Majors Foundation (M. Sadelain, R.B., and I.R.), The Damon Runyon Cancer Research Foundation (R.B.), the Carson Family Charitable Trust (R.B.), the William Lawrence and Blanche Hughes Foundation (R.B.), Kate’s Team, and Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Cancer Foundation for Research and the Experimental Therapeutics Center of MSKCC (M. Sadelain, R.B., and I.R.).
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/6/224/224ra25/DC1
Fig. S1. Trial scheme.
Fig. S2. CRP levels in patients with sCRS.
Fig. S3. ROC curve for CRP.
Fig. S4. Management scheme for patients treated with CAR T cells.
Table S1. Apheresis and T cell production characteristics.
Table S2. Detection of B cells and CAR T cells in the BM by deep sequencing.
Table S3. Absolute maximum cytokine values after CAR T cell infusion.
Table S4. Adverse events.
Table S5. CAR T cells in the CSF of patients with neurologic changes.
Author contributions: R.B., M. Sadelain, M.L.D., and I.R. wrote and edited the manuscript. M.L.D., M. Sadelain, R.B., and I.R. conceptualized the overall strategy and developed its clinical translation and implementation. R.B. and M.L.D. designed and amended the phase 1 clinical protocol. M.L.D. is the principal investigator of the protocol. Manufacturing of T cells, release testing, and qPCR acquisition of clinical samples were performed by S.B., J.S., O.B.-O., M.O., J.Q., T.W., Q.H., M.F., H.S., M.Y., M. Satter, Y.W., and J.S.; supervised by X.W.; and directed by I.R. Data from manufacturing, flow cytometry, and qPCR monitoring were analyzed by X.W. and I.R. R.B., M. Sadelain, I.R., M.L.D., and X.W. discussed and interpreted the results. R.B., M.L.D., J.P., K.C., D.D., S.S.C., G.J.R., H.Q., E.H., S.G., and M.G.F. enrolled patients to the protocol and/or managed the leukemia patients. M.E.A. designed and performed molecular assays to identify the malignant IgH clonotype associated with the leukemia cells of enrolled and treated patients. P.M. evaluated all pre- and posttreatment BM aspirates for evidence of leukemia. M.G. designed and performed statistical analyses. Y.B. is the research study assistant for the protocol and assisted with enrollment, sample acquisition, and data safety monitoring of patients. D.C.G.B. is the data assistant for the protocol and arranged collection and presentation of the data for the study leaders.
Competing interests: M. Sadelain and R.B. are co-holders of U.S. Patent 7,446,190, which covers the 19-28z receptor and was licensed to Juno Therapeutics in November 2013. M. Sadelain, R.B., and I.R. are co-founders of Juno Therapeutics. The other authors declare no competing interests.
REFERENCES AND NOTES
- 1.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davila ML, Brentjens R, Wang X, Rivière I, Sadelain M. How do CARs work?: Early insights from recent clinical studies targeting CD19. Oncoimmunology. 2012;1:1577–1583. doi: 10.4161/onci.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, Durrant IJ, Luger SM, Marks DI, Franklin IM, McMillan AK, Tallman MS, Rowe JM, Goldstone AH Medical Research Council of the United Kingdom Adult ALL Working Party; Eastern Cooperative Oncology Group. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109:944–950. doi: 10.1182/blood-2006-05-018192. [DOI] [PubMed] [Google Scholar]
- 9.Gökbuget N, Stanze D, Beck J, Diedrich H, Horst HA, Hüttmann A, Kobbe G, Kreuzer KA, Leimer L, Reichle A, Schaich M, Schwartz S, Serve H, Starck M, Stelljes M, Stuhlmann R, Viardot A, Wendelin K, Freund M, Hoelzer D German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120:2032–2041. doi: 10.1182/blood-2011-12-399287. [DOI] [PubMed] [Google Scholar]
- 10.O’Brien S, Schiller G, Lister J, Damon L, Goldberg S, Aulitzky W, Ben-Yehuda D, Stock W, Coutre S, Douer D, Heffner LT, Larson M, Seiter K, Smith S, Assouline S, Kuriakose P, Maness L, Nagler A, Rowe J, Schaich M, Shpilberg O, Yee K, Schmieder G, Silverman JA, Thomas D, Deitcher SR, Kantarjian H. High-dose vincristine sulfate liposome injection for advanced, relapsed, and refractory adult Philadelphia chromosome-negative acute lymphoblastic leukemia. J Clin Oncol. 2013;31:676–683. doi: 10.1200/JCO.2012.46.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robins H. Immunosequencing: Applications of immune repertoire deep sequencing. Curr Opin Immunol. 2013;25:646–652. doi: 10.1016/j.coi.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 12.Pepys MB, Hirschfield GM. C-reactive protein: A critical update. J Clin Invest. 2003;111:1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas DA, Kantarjian H, Smith TL, Koller C, Cortes J, O’Brien S, Giles FJ, Gajewski J, Pierce S, Keating MJ. Primary refractory and relapsed adult acute lymphoblastic leukemia: Characteristics, treatment results, and prognosis with salvage therapy. Cancer. 1999;86:1216–1230. doi: 10.1002/(sici)1097-0142(19991001)86:7<1216::aid-cncr17>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg JD, Linker A, Kuk D, Ratan R, Jurcic J, Barker JN, Castro-Malaspina H, Giralt S, Hsu K, Jakubowski AA, Jenq R, Koehne G, Papadopoulos EB, van den Brink MR, Young JW, Boulad F, Kernan NA, O’Reilly RJ, Prockop SE, Yahalom J, Heller G, Perales MA. T cell–depleted stem cell transplantation for adults with high-risk acute lymphoblastic leukemia: Long-term survival for patients in first complete remission with a decreased risk of graft-versus-host disease. Biol Blood Marrow Transplant. 2013;19:208–213. doi: 10.1016/j.bbmt.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oriol A, Vives S, Hernández-Rivas JM, Tormo M, Heras I, Rivas C, Bethencourt C, Moscardó F, Bueno J, Grande C, del Potro E, Guardia R, Brunet S, Bergua J, Bernal T, Moreno MJ, Calvo C, Bastida P, Feliu E, Ribera JM Programa Español de Tratamiento en Hematologia Group. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica. 2010;95:589–596. doi: 10.3324/haematol.2009.014274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Stegen SJ, Davies DM, Wilkie S, Foster J, Sosabowski JK, Burnet J, Whilding LM, Petrovic RM, Ghaem-Maghami S, Mather S, Jeannon JP, Parente-Pereira AC, Maher J. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: Identifying a window of therapeutic opportunity? J Immunol. 2013;191:4589–4598. doi: 10.4049/jimmunol.1301523. [DOI] [PubMed] [Google Scholar]
- 20.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, Fitzgerald JC, Aplenc R, Gore L, Grupp SA. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–5157. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 22.Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLOS One. 2013;8:e61338. doi: 10.1371/journal.pone.0061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, Yeh R, Capacio V, Olszewska M, Hosey J, Sadelain M, Brentjens RJ, Rivière I. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32:169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheson BD, Bennett JM, Kopecky KJ, üchner TB, Willman CL, Estey EH, Schiffer CA, Doehner H, Tallman MS, Lister TA, Lo-Coco F, Willemze R, Biondi A, Hiddemann W, Larson RA, Löwenberg B, Sanz MA, Head DR, Ohno R, Bloomfield CD. International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia, Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21:4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 25.Appelbaum FR, Rosenblum D, Arceci RJ, Carroll WL, Breitfeld PP, Forman SJ, Larson RA, Lee SJ, Murphy SB, O’Brien S, Radich J, Scher NS, Smith FO, Stone RM, Tallman MS. End points to establish the efficacy of new agents in the treatment of acute leukemia. Blood. 2007;109:1810–1816. doi: 10.1182/blood-2006-08-041152. [DOI] [PubMed] [Google Scholar]
- 26.Gönen M. Analyzing Receiver Operating Characteristic Curves with SAS. SAS Institute; Cary, NC: 2007. [Google Scholar]
- 27.Steinherz PG, Redner A, Steinherz L, Meyers P, Tan C, Heller G. Development of a new intensive therapy for acute lymphoblastic leukemia in children at increased risk of early relapse. The Memorial Sloan-Kettering-New York-II protocol. Cancer. 1993;72:3120–3130. doi: 10.1002/1097-0142(19931115)72:10<3120::aid-cncr2820721038>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 28.Baccarani M, Corbelli G, Amadori S, Drenthe-Schonk A, Willemze R, Meloni G, Cardozo PL, Haanen C, Mandelli F, Tura S. Adolescent and adult acute lymphoblastic leukemia: Prognostic features and outcome of therapy. A study of 293 patients. Blood. 1982;60:677–684. [PubMed] [Google Scholar]
- 29.Rodriguez V, Hart JS, Freireich EJ, Bodey GP, McCredie KB, Whitecar JP, Jr, Coltman CA., Jr Pomp combination chemotherapy of adult acute leukemia. Cancer. 1973;32:69–75. doi: 10.1002/1097-0142(197307)32:1<69::aid-cncr2820320109>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.