

Abstract
T cells reactive to β-cell antigens are critical players in the development of autoimmune type 1 diabetes (T1D). Using a panel of diabetogenic CD4 T cell clones derived from the non-obese diabetic (NOD) mouse, we recently identified the β-cell secretory granule protein, chromogranin A (ChgA), as a new autoantigen in T1D. CD4 T cells reactive to ChgA are pathogenic and rapidly transfer diabetes into young NOD recipients. We report here that NOD.ChgA−/− mice do not develop diabetes and show little evidence of autoimmunity in the pancreatic islets. Using tetramer analysis, we demonstrate that ChgA-reactive T cells are present in these mice but remain naïve. In contrast, in NOD.ChgA+/+ mice, a majority of the ChgA-reactive T cells are antigen-experienced. Our results suggest that the presence of ChgA and subsequent activation of ChgA-reactive T cells is essential for the initiation and development of autoimmune diabetes in NOD mice.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease characterized by the destruction of pancreatic islet β-cells and orchestrated by T cells responding to -cell antigens. Identifying the autoantigens that trigger the autoimmune process is therefore key to preventing or halting disease development. We have published recently on the discovery of a new autoantigen in T1D, chromogranin A (ChgA), in both NOD mice (1) and in human T1D patients (2). ChgA, like insulin, is a protein present in the secretory granules of β-cells. We demonstrated that a peptide ligand from ChgA is antigenic for the diabetogenic CD4 T cell clones BDC-2.5, BDC-10.1 and BDC-9.46 (1, 3) and for CD4 T cells from BDC-2.5 TCR transgenic (TCR-Tg) NOD mice (3).
One method for assessing the relative importance of β-cell autoantigens in the progression of T1D is through elimination of the antigen in NOD mice. Several antigen-deficient NOD models have been described and include mice deficient in islet amyloid polypeptide (IAPP) (4), glutamic acid decarboxylase (GAD) (5), and islet antigen-2 (IA-2) (6). With the exception of insulin 1 and insulin 2 (7), knock-out of these antigens had little or no effect on the development of the disease. We report here that in the absence of ChgA, NOD mice do not develop diabetes, a result that indicates a critical role for this protein as an autoantigen in T1D.
Materials and Methods
Mice
NOD and NOD.scid breeding mice were acquired from The Jackson Laboratory and were bred and housed in specific pathogen-free conditions at National Jewish Health. NOD.ChgA−/− mice were bred in our colony by backcrossing C57BL/6.129.ChgA−/− mice (8) onto the NOD background. Mice were backcrossed to the NOD for 10 generations and then intercrossed to generate homozygous knock-out mice, verifying genotype by PCR. For the microsatellite analysis of NOD.ChgA−/− mice, a low-density panel of 73 microsatellite markers, equally spaced throughout the genome, (~30cM intervals) were used to differentiate between the genetic background of the originating/donor (B6/129) and target/recipient (NOD/ShiLtJ) mouse strains. NOD.BDC-2.5 TCR-Tg (9) and NOD.BDC-6.9 TCR-Tg (10) mice were generated as previously described. Breeding mice and experimental animals were monitored for development of disease by urine glucose testing (Diastix, Bayer) and hyperglycemia confirmed by blood glucose testing using OneTouch Ultra glucometer (LifeScan). Mice were considered diabetic when blood glucose levels were >15 mmol/l (270 mg/dl) for two consecutive readings. All experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee.
Culture of T cell clones
T cell clones (BDC-2.5, BDC-10.1, BDC-5.2.9 and BDC-4.38) were restimulated every two weeks as previously described (11). For expansion of cell numbers before transfer, T cells were subcultured for 4 days after restimulation with additional IL-2.
Ex vivo analysis of pancreas
Single cell suspensions from pancreata were prepared as previously described (4) before being stained with an appropriate master mix of antibodies and tetramers.
Histology
Pancreas were fixed in 10% formalin and embedded in paraffin. Sections were stained with H&E. For the pancreas, insulitis was determined by scoring the mononuclear infiltrate in NOD, NOD.ChgA−/−, and BALB/c islets. Islets were scored as follows: 0: no infiltrate; 1: 10–50% infiltrated; 2: 50–75% infiltrated; 3: islet is completely infiltrated.
Adoptive transfer
T cell clones were expanded in secondary cultures and 1 × 107 cells were injected i.p. into young (< 14 days-old) NOD, NOD.ChgA−/− or NOD.scid pups. CD4+ cells from BDC-2.5 and BDC-6.9 TCR-Tg mice were isolated by negative selection using a kit from StemCell Technologies (Vancouver, Canada). Purified CD4 populations were then stained with a vital dye: CFSE (Invitrogen) or Violet Proliferation Dye 450 (VPD; BD biosciences) and 5 × 106 from each population were co-transferred i.v. into adult (> 8 weeks old) wt NOD or NOD.ChgA−/− mouse. In repeat experiments dyes were switched.
Antigen assay
T cell responses were assessed as described previously (4). Forms of antigens used included islet cell suspensions (1 × 104 cells) from either BALB/c, NOD, NOD.ChgA−/− or NOD.IAPP−/− mice, or synthetic peptides: ChgA peptide WE14 (WSRMDQLAKELTAE; 200, 400 g/ml), Insulin B9-23 (SHLVEALYLVCGERG; 25, 100 μg/ml) or the IAPP peptide KS20 (KCNTATCATQRLANFLVRSS; 25, 100 μg/ml). After 18 h, IFN-γ concentrations were determined in culture supernatants by ELISA.
Tissue harvest
After sacrifice of experimental transfer mice, islets were isolated and dissociated as previously described (12). In some experiments, blood was collected by cardiocentesis. Inguinal lymph nodes, pancreatic lymph nodes, and spleen were also harvested for analysis of T cells.
Salivary gland digestion
The submaximal salivary gland was isolated from the mouse following careful removal of nearby lymph nodes. The salivary glands were digested using Collagenase D (Roche), with some of the collagenase solution directly injected into the salivary gland, for 30 min at 37°C and pressed through a 40 μm filter to yield a single cell suspension.
Flow cytometry
Antibodies were used conjugated with the following fluorophores: FITC, PE, PerCP-Cy5.5, Allophycocyanin (APC), APC-Cy7, Pacific Blue (PB), Brilliant Violet 421 (BV421), Brilliant Violet 510 (BV510) and Brilliant Violet 711 (BV711). Gating strategies are indicated in each figure; the lymphocyte gate was based on FSC/SSC properties and the singlets gate was based on the Pulse Width parameter. Samples were run on a Cyan (Beckman Coulter) or BD Fortessa flow cytometers and data were analyzed using the FlowJo software (Tree Star, USA). Tetramer staining was performed as previously described (4).
Statistical analysis
Statistical analysis included Wilcoxon rank-sum test and Student’s t-test. Statistical significance was defined as p <0.05.
Results and Discussion
The antigen for ChgA-reactive CD4 T cells is not present in NOD.ChgA−/− mice
C57BL/6.129.ChgA−/− mice (8) were backcrossed onto the wildtype (wt) NOD background for 10 generations and then intercrossed to generate NOD.ChgA−/− mice. To verify that the backcross was complete, microsatellite analysis was performed and all markers were congenic for NOD except for one marker situated close to the CHGA gene on Chromosome 12. Importantly, all NOD insulin-dependent diabetes mellitus (iddm) loci were present in the NOD.ChgA−/−, indicating that the backcross was complete.
To establish that the antigen for ChgA-reactive T cell clone BDC-2.5 was not present in the islets of NOD.ChgA−/− mice, we tested reactivity of T cells to ChgA-deficient islets in vitro. CD4 T cell clones from the BDC panel were cultured with pancreatic islet cells from different mouse strains (BALB/c, NOD, NOD.ChgA−/− and NOD.IAPP−/−), and IFN-γ secretion was used as a measure of T cell activation. The BDC T cell clones BDC-2.5 and BDC-10.1, which express different TCRs, are reactive with ChgA (1). Islets from NOD.ChgA−/− mice did not stimulate BDC-2.5 or the second ChgA-reactive T cell clone BDC-10.1, indicating that the ligand for these clones was absent from NOD.ChgA−/− islets (Fig. 1). BDC-2.5 and BDC-10.1 did respond to positive control antigens such as islets from NOD and NOD.IAPP−/− mice and the ChgA peptide WE14. NOD.ChgA−/− islets elicited responses from BDC clones reactive to other antigens, including an IAPP-reactive T cell clone BDC-5.2.9 and an insulin-reactive T cell clone BDC-4.38.
FIGURE 1. ChgA-reactive T cell clones are not activated by ChgA-deficient islets.
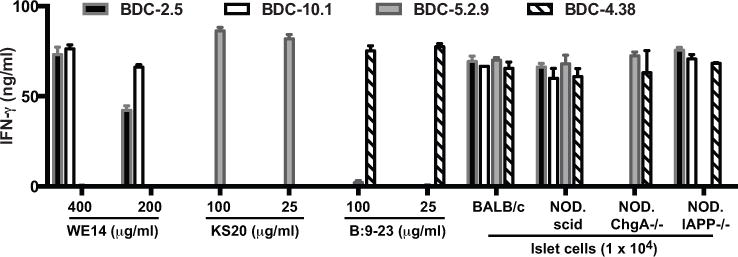
BDC T cell clones (2 × 104) were challenged with islets or peptides, in the presence of peritoneal exudate cells (2.5 × 104) as antigen-presenting cells. After 24 h of culture, supernatants from duplicate wells were harvested and IFN-γ was detected by ELISA. Data is representative of two independent experiments.
In a second set of experiments, T cell transfer was used to test for the presence of antigen in vivo in different NOD mouse variants. T cell clones from the BDC panel rapidly transfer diabetes into young (< two weeks old) wt NOD recipient mice. The BDC-2.5 T cell clone was adoptively transferred into NOD or NOD.ChgA−/− and the recipients were subsequently monitored for diabetes (Supplemental Fig. 1A). NOD mice became diabetic within a few weeks after transfer, but BDC-2.5 failed to transfer diabetes to the NOD.ChgA−/− mouse, confirming the absence of the antigen for BDC-2.5 in this mouse. As a control, the insulin-reactive T cell clone BDC-4.38 was transferred into NOD or NOD.ChgA−/− mice and all recipients became diabetic within 3 weeks following transfer (Supplemental Fig. 1A).
In a third set of experiments, CD4 T cells isolated from BDC-2.5 and BDC-6.9 TCR-Tg NOD mice (9, 10) were co-transferred into wt NOD or NOD.ChgA−/− mice. The BDC-6.9 TCR-Tg mouse was derived from a diabetogenic BDC T cell clone that is not reactive with ChgA. Each population was labeled with a vital dye (CFSE or VPD) which allowed us to distinguish between host cells, BDC-2.5 TCR-Tg cells, or BDC-6.9 TCR-Tg cells, and monitor T cell proliferation as an indicator of T cell activation (see gating strategy in Supplemental Fig. 1B). T cells from BDC-2.5 TCR-Tg mice proliferated and accumulated in the islets and pancreatic LN of wt NOD mice (Supplemental Fig. 1C & D). In NOD.ChgA−/− mice, BDC-2.5 TCR-Tg T cells were present but fewer in number and did not proliferate in the islets and pancreatic LN. In contrast, we observed that BDC-6.9 TCR-Tg cells could readily proliferate and accumulate in the islets and pancreatic LN of both wt NOD and NOD.ChgA−/− mice (Supplemental Fig. 1C& D). Results obtained with transgenic CD4 T cells provide further confirmation that ChgA-deficient NOD mice lack the antigen for BDC-2.5.
NOD mice deficient in chromogranin A do not develop diabetes
To determine the effect of ChgA deficiency in the islets on the development of autoimmunity in the islets, we followed cohorts of wt NOD and NOD.ChgA−/− mice in parallel, males and females, for up to 12 months. Mice were monitored for disease by checking weekly urine glucose levels as a sign of hyperglycemia and were considered diabetic when blood glucose was above 15 mM for two consecutive days. Cumulative incidence in our wt NOD colony indicates that about 90% of female mice become diabetic at 52 weeks. In contrast, during the same time period, while males were completely protected from disease, only three female NOD.ChgA−/− mice (of 118 total) became diabetic, demonstrating that in the absence of ChgA, incidence of autoimmune diabetes is greatly reduced (Fig. 2).
FIGURE 2. ChgA-deficient NOD mice are protected from diabetes.
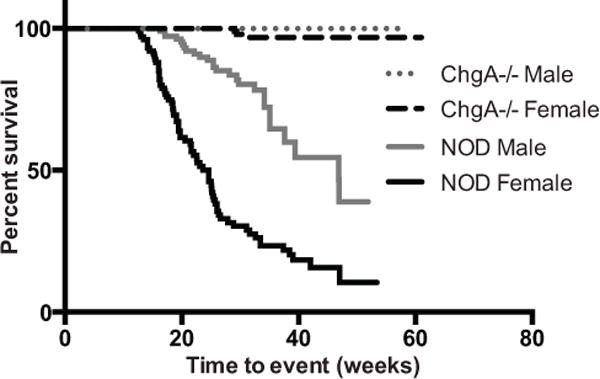
Male or female wt NOD (female n=101; male n=110) or NOD.ChgA−/− mice (female n=118; male n=109) were followed for the development of diabetes by monitoring urine and blood glucose levels. The impact of the microbiota was not assessed and littermate controls were not monitored for disease incidence. Statistical significance was assessed by using the Wilcoxon rank-sum test.
NOD.ChgA−/− mice are protected from insulitis but not sialitis
Histological analysis was performed on sections from the pancreas of BALB/c, NOD or NOD.ChgA−/− mice. Tissue from BALB/c mice was used as a negative control as these mice do not develop autoimmunity and the pancreas contains no infiltrate (Fig. 3A). In pancreatic sections from NOD.ChgA−/− mice, we observed that over 80% of the islets were completely protected from insulitis (Fig. 3A), with no difference between younger and older mice. In contrast, in wt NOD mice, we observed a clear progression of insulitis as the mice aged and in older mice, up to 50% of the islets had a score of 3, indicating extensive infiltration. The histological differences between the pancreatic islets of wt NOD and NOD.ChgA−/− indicate that the autoimmune process is markedly reduced in the pancreas of mice lacking ChgA.
FIGURE 3. Chromogranin A-deficient mice are protected from insulitis but not sialitis.
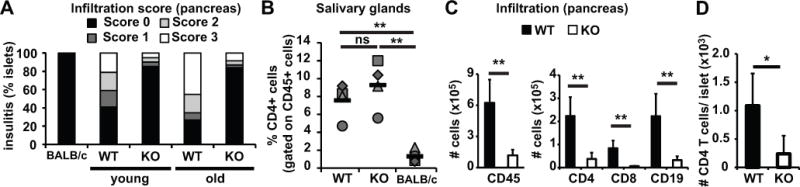
(A) Pancreatic sections from BALB/c, NOD (WT) or NOD.ChgA−/− (KO) mice were stained with H&E and analyzed for the presence of mononuclear infiltrate in the islets. Thirty-six islets were scored for BALB/c mice. The average age for younger NOD mice was 14.6 wks (n=6, 66 islets scored) and 13.25 weeks for NOD.ChgA−/− (n=4, 61 islets scored); for older mice, the average age was 42 weeks for NOD (n=13, 119 islets scored) and 52 weeks for NOD.ChgA−/− (n=14, 275 islets scored). (B) Submaximal salivary glands from 6–10 month-old wt NOD, NOD.ChgA−/− or BALB/c mice were analyzed by flow cytometry. The percentages of CD4 cells present in submaximal salivary glands are indicated. Gates were set on the lymphocyte gate, singlets and on CD45+ and dump- (CD8- CD11b- CD11c- CD19-). Each symbol represents an individual mouse. Data is representative of two independent experiments with 2 mice per group per experiment. Averages are indicated as black horizontal bars. (C) We also investigated islet infiltration by flow cytometric analysis of cells in the pancreas. Single cell suspension from pancreata from 12–16 week-old NOD (WT) or NOD.ChgA−/− (KO) female mice were stained with anti-CD45 (BV421), anti-CD4 (FITC), anti-CD8 (APC-Cy7) and anti-CD19 (PerCP-Cy5.5) and analyzed by flow cytometry. Data are a summary of four independent experiments with 2 mice per group per experiment (n = 8). (D) Islets of wt NOD or NOD.ChgA−/− mice were hand picked, counted, dissociated into single cell suspensions and stained with appropriate antibodies. Gates were set on the lymphocyte gate, singlets, CD8-, CD4+. Data is a summary of 2 independent experiments with 2 mice per group per experiment (n = 4). Black columns represent NOD mice (WT) and white columns represent NOD.ChgA−/− mice (KO). Significance: ns: no significance (P > 0.05); ** = P < 0.01; * = P < 0.05.
NOD mice often exhibit signs of autoimmunity in sites other than the pancreas and for example, provide a model of Sjogren’s syndrome (13). We examined the salivary glands to determine whether ChgA−/− mice develop inflammatory infiltrates outside the pancreas. Sialitis is the characteristic lymphocytic infiltrate of the salivary glands previously described in NOD mice (13). We observed a focal infiltrate in the salivary glands of both wt NOD and NOD.ChgA−/− mice (data not shown). Flow cytometric analysis of the salivary glands showed that in the CD45+ lymphoid cell population, CD4+ cells were significantly increased in wt NOD and NOD.ChgA−/− mice compared to BALB/c mice (Fig. 3B), indicating that in NOD mice, the autoimmune process in the salivary was intact, either in the presence or absence of ChgA. Although the antigens driving Sjögren’s syndrome are not clearly defined, ChgA is not expressed in the salivary glands (14). These data indicate that NOD.ChgA−/− mice are still prone to autoimmunity. Additionally, our findings suggest that ChgA is necessary for development of the infiltrate present in the pancreas of NOD mice but apparently is not a target for the autoreactive CD4 T cells responsible for sialitis.
CD4 T cells do not accumulate in the pancreas of NOD.ChgA−/− mice
To gain further insight into the cells present in the pancreas of NOD.ChgA−/− mice, we performed flow cytometric analysis of the pancreatic infiltrate. Compared to wt NOD mice, the total number of CD45+ cells present in the pancreas of NOD.ChgA−/− was significantly reduced (Fig. 2C). Numbers of CD4, CD8, and CD19+ were also less (Fig. 3C). We evaluated the overall number of CD4 cells per islet by flow cytometry and our data indicate that there was a significant increase in the number of CD4 T cells per islet in wt NOD mice compared to NOD.ChgA−/− mice (Fig. 3D). These data confirm and expand our observations shown in Fig. 3A that overall there are decreased leukocyte numbers present in the pancreas of NOD.ChgA−/− mice.
We used tetramer analysis to further investigate antigen specificity of T cells present in the spleen and the pancreas of NOD.ChgA−/− mice. BDC-2.5-like cells were monitored by using the 2.5mi tetramer (15), which contains a BDC-2.5 mimotope peptide (AHHPIWARMDA). As a negative control, we used a tetramer loaded with a peptide from hen egg lysosome (HEL12–25). In the pancreas of NOD.ChgA−/− mice, 2.5mi tetramer-positive cells were virtually absent (Fig. 4A and Supplemental Fig. 2C), indicating that in the absence of ChgA, 2.5mi tetramer-positive cells do not migrate and accumulate in the pancreas. In contrast, the pancreas of wt NOD mice contained large populations of 2.5mi tet+ cells. We observed 2.5mi tet+ cells in the spleen of both wt NOD and NOD.ChgA−/− mice and examined the activation status of these cells by looking at markers of T cell memory (CD44hi CD62Llo) (Supplemental Fig. 2A). In the spleen of NOD.ChgA−/− mice, 2.5mi tetramer-positive cells were present but remained mostly CD44lo CD62Lhi. Both memory and naïve populations of 2.5mi tetramer-positive cells were present in the spleen of wt NOD mice (Fig. 4B and Supplemental Fig. 2 B). These data indicate that 2.5mi tetramer-positive cells are present in NOD.ChgA−/− mice, but remain naïve.
FIGURE 4. ChgA-reactive T cells develop in the absence of ChgA but remain naïve.
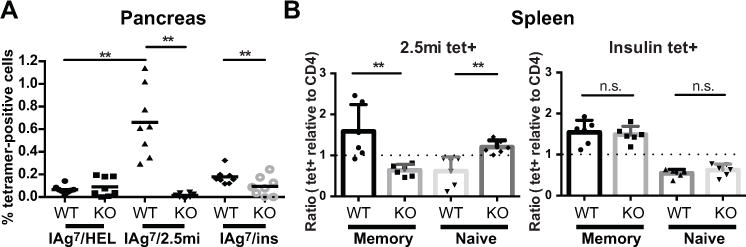
Single cell suspensions from pancreas (A) and spleen (B) were stained with tetramers specific for T cells reactive to HEL, ChgA (2.5mi), or insulin (Insp8G), and a master mix of antibodies. Gates were set on the lymphocyte gate, singlets, CD45, dump- (CD8, CD11b, CD11c, CD19, F4/80 and 7AAD) and CD4+. (A) Data shows the percentage of tetramer-positive cells in the pancreas of 12–20 week-old NOD (WT) or NOD.ChgA−/− (KO) female mice (n = 8). Each symbol represents an individual mouse and averages are indicated as a black horizontal bar. (B) Memory versus naïve cells in spleens of NOD (WT) or NOD.ChgA−/− (KO) female mice (n = 6). Data is expressed as ratios of percentages of tetramer-positive cells relative to percentages of polyclonal CD4 population, for memory vs naïve populations. Memory CD4 T cells were defined as CD44hiCD62Llo and naïve as CD44loCD62Lhi; each symbol represents an individual mouse. The dotted line indicates a ratio of 1. Data are from four (A) and three (B) independent experiments with 2 mice per group per experiment. Significance: n.s.: no significance (P > 0.05); ** = P < 0.01; * = P < 0.05.
We also analyzed wt NOD and NOD.ChgA−/− mice for the presence of insulin-reactive T cells using a recently described insulin tetramer (16) that stains one of the first insulin-reactive CD4 T cell clones described, PD12.4.4 (17). Insulin tetramer-positive cells were significantly increased in the pancreas of wt NOD mice compared to NOD.ChgA−/− mice (Fig. 4A). We observed that insulin tetramer-positive cells were also present in the spleen of both wt NOD and NOD.ChgA−/− mice, but there were no significant differences between the 2 mouse strains.
The ultimate goal of this study was to investigate whether the lack of ChgA had any impact on disease incidence in NOD mice. Because knock-out of other antigens such as GAD65 and IAPP does not affect development of diabetes in NOD mice (4), the lack of disease development in NOD.ChgA−/− mice was unexpected and may signify a different, more critical role for this secretory granule protein. In other attempts to pinpoint the importance of specific T cell epitopes, investigators have mutated peptide sequences from islet-specific glucose 6 phosphatase (IGRP) or insulin and then determined the effect on disease development. Whereas mutating IGRP had no effect (19), a single amino acid substitution at position 16 in the B9-23 peptide of the insulin B chain did protect mice from the development of diabetes (7). We did observe some infiltrate in the pancreatic islets and in the salivary gland of NOD.ChgA−/− mice suggesting that there is not a complete absence of autoimmunity, but with the rare exception, these animals do not develop diabetes. Our data thus provide evidence that ChgA is required for spontaneous disease development in NOD mice and underscore the importance of ChgA in the initiation and overall pathogenesis of the disease.
Our results suggesting that the presence of ChgA is necessary for the development of autoimmune diabetes in NOD mice indicate new future directions for the study of this autoantigen. Although the peptide WE14 from ChgA has been found to elicit responses in T cells from human T1D patients (2), it is not clear that this is the only, or optimal, ChgA ligand for autoreactive T cells in humans. It is therefore of great importance to determine the natural peptide ligands of ChgA, both from the standpoint of determining the role of ChgA in pathogenesis and also, because better reagents are needed to identify autoreactive T cells as biomarkers. For example, can peptide sequences from ChgA be used to design better tetramers for monitoring the ongoing autoimmune process directed towards ChgA and identify subjects at risk of developing T1D? Another important area of interest concerns the question of whether peptides from ChgA could be used in strategies to induce antigen-specific tolerance. Trials are in progress to test the ability of myelin peptides to suppress multiple sclerosis in humans (20). It may be that peptides from ChgA could contribute to controlling autoimmunity in T1D.
Supplementary Material
Acknowledgments
We would like to thank the NIH Tetramer Core for providing all tetramers used in this study.
This study was supported by NIH research grant R01DK081166 (KH), ADA Junior Faculty Award 1-15-JF-04 (RLB), JDRF 2-2012-197 (RSF), and CRI AWD-112499 (RSL).
References
- 1.Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, Marrack P, Mahata SK, Kappler JW, Haskins K. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gottlieb PA, Delong T, Baker RL, Fitzgerald-Miller L, Wagner R, Cook G, Rewers MR, Michels A, Haskins K. Chromogranin A is a T cell antigen in human type 1 diabetes. J Autoimmun. 2014;50:38–41. doi: 10.1016/j.jaut.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes. 2012;61:3239–3246. doi: 10.2337/db12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker RL, Delong T, Barbour G, Bradley B, Nakayama M, Haskins K. Cutting edge: CD4 T cells reactive to an islet amyloid polypeptide peptide accumulate in the pancreas and contribute to disease pathogenesis in nonobese diabetic mice. J Immunol. 2013;191:3990–3994. doi: 10.4049/jimmunol.1301480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamoto T, Yamato E, Tashiro F, Sato T, Noso S, Ikegami H, Tamura S, Yanagawa Y, Miyazaki JI. Development of autoimmune diabetes in glutamic acid decarboxylase 65 (GAD65) knockout NOD mice. Diabetologia. 2004;47:221–224. doi: 10.1007/s00125-003-1296-0. [DOI] [PubMed] [Google Scholar]
- 6.Kubosaki a, Miura J, Notkins AL. IA-2 is not required for the development of diabetes in NOD mice. Diabetologia. 2004;47:149–150. doi: 10.1007/s00125-003-1252-z. [DOI] [PubMed] [Google Scholar]
- 7.Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahapatra NR, O’Connor DT, Vaingankar SM, Hikim APS, Mahata M, Ray S, Staite E, Wu H, Gu Y, Dalton N, Kennedy BP, Ziegler MG, Ross J, Mahata SK. Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J Clin Invest. 2005;115:1942–1952. doi: 10.1172/JCI24354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 10.Pauza ME, Dobbs CM, He J, Patterson T, Wagner S, Anobile BS, Bradley BJ, Lo D, Haskins K. T-cell receptor transgenic response to an endogenous polymorphic autoantigen determines susceptibility to diabetes. Diabetes. 2004;53:978–988. doi: 10.2337/diabetes.53.4.978. [DOI] [PubMed] [Google Scholar]
- 11.Haskins K, Portas M, Bergman B, Lafferty K, Bradley B. Pancreatic islet-specific T-cell clones from nonobese diabetic mice. Proc Natl Acad Sci U S A. 1989;86:8000–8004. doi: 10.1073/pnas.86.20.8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman RS, Lindsay RS, Lilly JK, Nguyen V, Sorensen CM, Jacobelli J, Krummel MF. An evolving autoimmune microenvironment regulates the quality of effector T cell restimulation and function. Proc Natl Acad Sci U S A. 2014;111:9223–9228. doi: 10.1073/pnas.1322193111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soyfoo MS, Steinfeld S, Delporte C. Usefulness of mouse models to study the pathogenesis of Sjögren’s syndrome. Oral Dis. 2007;13:366–375. doi: 10.1111/j.1601-0825.2007.01376.x. [DOI] [PubMed] [Google Scholar]
- 14.Wu C, Macleod I, Su AI. BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res. 2013;41:D561–D565. doi: 10.1093/nar/gks1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stratmann T, Martin-Orozco N, Mallet-Designe V, Poirot L, McGavern D, Losyev G, Dobbs CM, Oldstone MBA, Yoshida K, Kikutani H, Mathis D, Benoist C, Haskins K, Teyton L. Susceptible MHC alleles, not background genes, select an autoimmune T cell reactivity. J Clin Invest. 2003;112:902–914. doi: 10.1172/JCI18337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, Marrack P, Eisenbarth G, Kappler JW. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A. 2011;108:16729–16734. doi: 10.1073/pnas.1113954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol. 1995;25:1056–1062. doi: 10.1002/eji.1830250430. [DOI] [PubMed] [Google Scholar]
- 18.Pauken KE, Linehan JL, Spanier JA, Sahli NL, Kalekar LA, Binstadt BA, Moon JJ, Mueller DL, Jenkins MK, Fife BT. Cutting edge: type 1 diabetes occurs despite robust anergy among endogenous insulin-specific CD4 T cells in NOD mice. J Immunol. 2013;191:4913–4917. doi: 10.4049/jimmunol.1301927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alkemade GM, Clemente-Casares X, Yu Z, Xu BY, Wang J, Tsai S, Wright JR, Roep BO, Santamaria P. Local autoantigen expression as essential gatekeeper of memory T-cell recruitment to islet grafts in diabetic hosts. Diabetes. 2013;62:905–911. doi: 10.2337/db12-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lutterotti A, Yousef S, Sputtek A, Stürner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, Schippling S, Miller SD, Sospedra M, Martin R. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med. 2013;5:188ra75. doi: 10.1126/scitranslmed.3006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.