

Abstract
Macrophage phagocytosis of particles and pathogens is an essential aspect of innate host defense. Phagocytic function requires cytoskeletal rearrangements that depend on the interaction between macrophage surface receptors, particulates/pathogens and the extracellular matrix. In the present report we determine the role of a mechanosensitive ion channel, transient receptor potential vanilloid 4 (TRPV4), in integrating the lipopolysaccharide (LPS) and matrix stiffness signals to control macrophage phenotypic change for host defense and resolution from lung injury. We demonstrate that active TRPV4 mediates LPS-stimulated murine macrophage phagocytosis of non-opsonized particles (E. coli) in vitro and opsonized particles (IgG-coated latex beads) in vitro and in vivo in intact mice. Intriguingly, matrix stiffness in the range seen in inflamed or fibrotic lung is required to sensitize the TRPV4 channel to mediate the LPS-induced increment in macrophage phagocytosis. Furthermore, TRPV4 is required for the LPS induction of anti-inflammatory/pro-resolution cytokines. These findings suggest that signaling through TRPV4, triggered by changes in extracellular matrix stiffness, cooperates with LPS-induced signals to mediate macrophage phagocytic function and lung injury resolution. These mechanisms are likely to be important in regulating macrophage function in the context of pulmonary infection and fibrosis.
Introduction
Macrophage phagocytosis (particle engulfment) is a complex, multistep physiologic process that determines the host’s capacity to defend against foreign particulates, pathogens, or apoptotic cells, and mediates resolution of inflammation and tissue homeostasis (1-5). Phagocytosis requires a coordinated interaction among macrophage surface receptors, particles, and the surrounding matrix, which ultimately drive the cytoskeletal rearrangements required for efficient engulfment (6-10). In fact, the phagocytic function of the macrophage depends on the biophysical properties of the matrix itself (9,10). For example, studies with pre-patterned matrix substrates reveal that matrix stiffness results in cell shape changes that can influence macrophage phenotypic properties (9-13). The mechanism by which macrophages sense extracellular matrix stiffness remains unknown.
Calcium is known to be an essential second messenger in many physiologic cell processes including phagocytosis (14-16). Many studies show that macrophage phagocytic function depends on a finely tuned orchestration of the intracellular calcium signal and the actin cytoskeleton (17). For example, studies show that particle binding to macrophages induces calcium transients, and calcium appears to be required for both FcR-dependent and -independent phagocytosis (18-20). Intracellular calcium is tightly regulated in a spatio-temporal manner through a system of ion channels and membrane pumps (21). One such channel is the transient receptor potential vanilloid 4 (TRPV4). TRPV4 is a ubiquitously-expressed, plasma membrane-based, calcium-permeable cation channel that is sensitized and activated by both chemical (5,6-Epoxyeicosatrienoic acid (EET), 4 alpha-phorbol 12,13-didecanoate (4-αPDD)) and physical stimuli (temperature, stretch, and hypotonicity) (22-25).
In fact, TRPV4 has been implicated in lung diseases associated with lung parenchymal stretch, such as pulmonary edema due to pulmonary venous hypertension, acute lung injury due to pulmonary parenchymal overdistension, and most recently, pulmonary fibrosis (26-34). As TRPV4 can be sensitized by changes in matrix stiffness, can regulate calcium flux into the cell, and induces its effect, in part, through modulating cytoskeletal remodeling (27,35), we reasoned that TRPV4 may mediate macrophage phenotypic function. We undertook this work to determine if the TRPV4 channel modulates the LPS signal for macrophage phagocytosis and cytokine release in a matrix stiffness-dependent manner. This work is potentially applicable to lung host defense, resolution of inflammation, infection, and fibrosis.
Materials and Methods
Antibodies and reagents
Primary antibodies to intracellular TRPV4 (Alomone Labs, Jerusalem, Israel), GAPDH (Fitzgerald Industries International, Acton, MA), α-CD45 (BD Biosciences), and purified rabbit IgG from mouse serum (Sigma, St. Louis, MO) were purchased. Secondary antibody to rabbit was obtained from Jackson Laboratories and rat Alexa Fluor-594 was obtained from Life Technologies (NY, USA). HC067047 (HC) was obtained from EMD Millipore and GSK1016790A (GSK101 or GSK) was obtained from Sigma-Aldrich (St. Louis, MO). Escherichia coli lipopolysaccharide 0111:B4 (LPS) for the in vitro experiments and Escherichia coli LPS 055:B5 for the in vivo experiments was obtained from Sigma (St. Louis, MO).
Cell culture, cell area, transfection, western blot analysis, and cytokine measurement
All animal protocols were performed as approved by the Cleveland Clinic Institutional Animal Care and Use Committee (IACUC). Primary murine bone marrow derived macrophages (BMDMs) and alveolar macrophages were harvested from 8-12 week old C57BL/6 wild type or TRPV4 null mice. BMDMs were differentiated in recombinant mouse macrophage colony stimulating factor (MCSF, 50 ng/mL, R&D Systems) as previously published (36). BMDMs and alveolar macrophages were plated on fibronectin-coated (10 μg/ml) glass or polyacrylamide hydrogels with varying stiffness (1, 8, and 25 kPa) (Matrigen, Brea, CA). Cells were treated with LPS (100 ng/mL) alone or with LPS and pretreated for 1 h with TRPV4 inhibitor (HC) for a total of 6-24 h. Primary isolates of alveolar macrophages obtained from bronchoalveolar lung lavage (BAL) were purified by adherence and cultured in DMEM/10% FBS as previously described (37). TRPV4 expression was downregulated by transfecting BMDMs with TRPV4-specific mouse siRNA duplexes or scrambled siRNA controls (Origene technologies) using electroporation, as previously published (36). Immunoblotting was performed for the indicated proteins as previously published (38). ELISAs (IL-1β, TNFα, IL-10 from R&D systems) were run on conditioned media from WT BMDMs ± LPS ± HC and TRPV4 KO BMDMs.
In vitro phagocytosis assays
BMDMs were stimulated in vitro ± LPS (100 ng/mL, 24 h) ± Ca2+ [1.802 mM (200 mg/mL)]) in DMEM (stimulation phase). In order to measure phagocytic function, the media was replaced with fluorescently labeled-heat inactivated E. coli ± Ca2+ [1.261 mM (140 mg/mL)] for 2 h, per manufacturer’s instructions (phagocytic phase) (K-12 strain, Vybrant Phagocytosis Assay Kit, V-6694, Molecular Probes). In selected conditions, TRPV4 inhibitor (HC) was added 1 h before LPS stimulation. Non-opsonized phagocytosis was measured as fluorescence intensity in the FlexStation system (Molecular Devices). Preliminary experiments determined that 50 µL of E. coli particles/well was the optimal concentration. A dose response of TRPV4 inhibitor (HC) determined its maximal effects were noted at 30 µM. Opsonized phagocytosis was measured by uptake of IgG-coated latex beads (Molecular probes, F8853 2 µm beads) and imaged via confocal (Leica DM6000 CFS SP5) microscopy per a previously published protocol (39). Fluorescence intensity was measured as image integrated pixel intensity per cell using Image J software. Alveolar macrophages were maintained in RPMI containing Ca2+ [0.427 mM (100 mg/mL)] and phagocytosis was measured.
In vivo phagocytosis assay
LPS-stimulated phagocytosis of IgG-coated latex beads in intact TRPV4 null mice and age matched 8-12 week old female congenic WT C57BL/6 mice was performed by intratracheal (IT) instillation of LPS (3 µg/g) or phosphate-buffered saline per previously published protocols (40,41). Sixteen hours after LPS or saline injection, 1.5 × 108 IgG-coated latex bead particles in 40 µL saline was IT-instilled for 6 h. Lung lavage was performed to determine total white blood cell (WBC) counts or cell differentials as described previously (36). Cytospin preparations were performed for each lavage and images were taken via confocal microscope as per a previously published protocol (39). Neutrophils and macrophages were distinguished by nuclear shape (PMN: multilobular nucleus; macrophage: single concentric nucleus). Phagocytosis was analyzed by quantifying the number of beads per cell using Image J software. All animal protocols were performed according to guidelines approved by the Cleveland Clinic IACUC.
Measurement of intracellular calcium
The calcium response to increasing concentrations of a TRPV4 agonist (GSK) was analyzed using fluorescent Calcium 5 dye (Molecular Devices) treated cells in a microplate reader as previously published (27). Cytosolic calcium increases (Ca2+ influx) are presented as Max-Min (RFU) as published previously (27).
Statistical analysis
All data are presented as means ± SEM, unless otherwise specified. Comparison of data from two groups was performed with the Student’s t-test. Comparing change scores of more than two groups was performed via ANOVA followed by Dunnett’s test or Student-Newman-Keuls. Significance was accepted at the p ≤ 0.05 level.
Results
Differentiated macrophages express functional TRPV4
To determine whether BMDMs express TRPV4 ± LPS, immunoblots for TRPV4 were performed in WT BMDMs, TRPV4 KO BMDMs, and TRPV4-specific siRNA treated BMDMs (serving as negative controls). TRPV4 protein expression was unchanged ± LPS in WT BMDMs and absent in TRPV4 KO whole cell lysates (Figure 1A). Downregulation of TRPV4 protein with TRPV4-specific siRNA resulted in an 80-85% reduction of TRPV4 protein (Figure 1A). To determine whether BMDMs express functionally active TRPV4, varying doses of a TRPV4-specific agonist (GSK) were examined for their ability to induce calcium influx (Figure 1B, EC50 = 50 nM). Downregulation of TRPV4 by siRNA reduces the maximal calcium influx in response to the TRPV4 agonist by 50%, when compared to scrambled siRNA-transfected controls (Figure 1B, *p < 0.05). Concordantly, genetic deletion of functional TRPV4 (BMDMs from TRPV4 KO mice) completely abrogates agonist-induced (GSK) calcium influx (Figure 1B, +p < 0.001). Blockade of TRPV4 with a selective small molecule inhibitor of TRPV4 (HC) reduces calcium influx in a dose-dependent manner (Figure 1C, IC50 = 7µM). Taken together, these data clearly demonstrate that TRPV4 is expressed in a functionally active, non-redundant manner in murine BMDMs.
Figure 1. Functional TRPV4 is expressed in murine BMDMs.

(A) WT or TRPV4 KO BMDMs (differentiated bone marrow derived macrophages) were incubated ± LPS (100 ng/mL, 24 h). Immunoblot reveals that TRPV4 protein is expressed and unchanged ± LPS in BMDMs. TRPV4 protein is deleted in BMDMs from TRPV4 KO cells and decreased by 80-85% in BMDMs treated with TRPV4-specific siRNA compared to control (CNTL) siRNA after 3 or 4 days (3d, 4d) (panels cut from the same blot and exposure). (B) Calcium influx measured in the presence of TRPV4 agonist (GSK) using fluorescent dye-treated BMDMs from WT mice with TRPV4 downregulation (siRNA) or TRPV4 KO mice. BMDMs have a decreased or absent TRPV4 agonist (GSK)-induced calcium signal in siRNA transfected cells, or in TRPV4 KO BMDMs on glass substrates (* denotes p < 0.05 TRPV4 siRNA vs CNTL, + denotes p < 0.001 TRPV4 KO vs WT). RFU - Fluorescence intensity reflects intracellular calcium concentration, WT - Wild type cells, CNTL siRNA - Non-targeting siRNA, TRPV4 siRNA - TRPV4-specific siRNA, TRPV4 KO - genetic deletion of TRPV4. (C) Small molecule inhibition of TRPV4 (HC) induces a dose-dependent decrease in the TRPV4 agonist (GSK)-induced calcium signal (IC50 = 7 µM). n ≥ 3 times in quadruplicate.
LPS-stimulated macrophage phagocytosis of E. coli is dependent on TRPV4
We and others have shown that TRPV4 can play a role in force-dependent cytoskeletal changes in other systems/cell types, thus it was hypothesized that TRPV4 may play a role in macrophage phagocytosis (27). LPS stimulates phagocytosis of E. coli particles in the presence of calcium by 151 ± 3% in murine BMDMs (Figure 2A, +p < 0.001). For LPS to induce macrophage phagocytosis of E. coli particles, extracellular calcium is required during both the LPS-stimulation and E. coli phagocytic phases (Figure 2A, *p < 0.05). LPS-stimulation of phagocytosis is completely abrogated in BMDMs from TRPV4 KO mice (Figure 2B, *p < 0.001) and decreased by 67 ± 9% upon downregulation of TRPV4 with siRNA (Figure 2B, *p = 0.002). Lastly, the small molecule TRPV4 inhibitor (HC) completely abrogates the LPS stimulation of phagocytosis in a dose-dependent manner (Figure 2C, p < 0.001), with an IC50 (8 µM) that is comparable to the TRPV4 inhibitor’s effect on calcium influx (7 µM, Figure 1C). In contrast, TRPV4 inhibition has no effect on basal phagocytosis (Data not shown). LPS-stimulated phagocytosis was similarly dependent on TRPV4 in both a murine macrophage cell line (RAW 267.4, Data not shown) and in freshly-isolated murine alveolar macrophages (AM) (Figure 2D, *,+p < 0.05). Collectively, these results demonstrate that LPS-stimulated phagocytosis of non-opsonized E. coli bacteria requires TRPV4.
Figure 2. TRPV4 mediates LPS-stimulated macrophage phagocytosis of E. coli particles.

BMDMs or freshly-isolated alveolar macrophages were incubated ± LPS (100 ng/mL, 24 h), in DMEM ± Ca2+ [1.802 mM (200 mg/mL)] followed by an incubation with fluorescently-labeled E. coli particles (2 h in HBSS ± Ca2+ [1.261 mM (140 mg/mL)]). Phagocytosis was measured as fluorescence intensity per cell. (A) Optimal macrophage phagocytosis requires extracellular calcium during both the LPS incubation (LPS, stimulation - 24 h) and subsequent period of E. coli particle incubation (E. coli, phagocytic phase - 2 h). +Ca denotes the presence of Ca2+ during both the LPS (stimulation phase) and E. coli incubations (phagocytic phase); No Ca LPS denotes absence of Ca2+ during the LPS incubation; No Ca E. coli denotes absence of Ca2+ during the E. coli incubation; No Ca denotes absence of Ca2+ during both LPS and E. coli incubation periods. Phagocytosis is quantified as % of induction by LPS (*,+p < 0.05). (B) LPS stimulates macrophage phagocytosis in WT BMDMs (+p < 0.001) that is abrogated upon TRPV4 deletion (KO) BMDMs (*p = 0.002) and TRPV4 downregulation (siRNA) BMDMs (*p < 0.001). UT - untreated cells, LPS - lipopolysaccharide treated cells. (C) TRPV4 inhibitor (HC) blocks LPS-stimulated phagocytosis in BMDMs in a concentration-dependent manner (IC50 = 8 µM, p < 0.001). [HC] ≥ 30 µM completely inhibited LPS-induced phagocytosis (Figure 2C) to a level comparable to that seen in the TRPV4 KO BMDMs (as in Figure 2B). Quantified as % of induction by LPS. (D) TRPV4-dependent phagocytic defect seen in TRPV4 KO primary murine alveolar macrophages (*,+p < 0.05). AM - alveolar macrophages. + denotes the increase by LPS vs UT, * denotes difference in LPS response from WT mice (B, D) or ± Ca2+ (A) under the indicated conditions. n ≥ 3 times in duplicate.
LPS-stimulated macrophage phagocytosis of IgG-coated latex beads is dependent on TRPV4
To determine if TRPV4 mediates specific receptor-initiated phagocytosis, we evaluated FcR dependent phagocytosis by incubating macrophages with IgG-coated latex beads. LPS stimulates uptake of IgG-coated beads by 4-fold, as compared to untreated BMDMs (Figure 3A and 3B, +p < 0.05). The LPS-stimulated increment in uptake is reduced by 44 ± 16% upon inhibition of TRPV4 (HC) (Figure 3A and 3B, *p < 0.05). Similarly, LPS-stimulated macrophage phagocytosis is abrogated upon either deletion (TRPV4 KO) or downregulation (TRPV4 siRNA) of TRPV4 (Figure 3C and 3D, *,+p < 0.05). LPS-stimulated phagocytosis was similarly dependent on TRPV4 in freshly-isolated murine alveolar macrophages (AM) (Figure 3E and 3F, *,+p < 0.05). The LPS-stimulated cell-spreading response is also completely abrogated upon inhibition, deletion or downregulation of TRPV4 (Data not shown). Overall, these data demonstrate that the LPS-stimulated, FcR-dependent phagocytosis, and the cell spreading response, are mediated by TRPV4.
Figure 3. TRPV4 mediates LPS-stimulated macrophage phagocytosis of IgG-coated latex beads in vitro.

BMDMs or freshly-isolated alveolar macrophages were incubated ± LPS (100 ng/mL, 6-24 h) ± other indicated molecules and then incubated with IgG-coated latex beads (1.5 h). Phagocytosis was measured as signal intensity/cell. (A) Representative photomicrographs of BMDMs given IgG-coated beads ± LPS ± 30 µM HC. Green- beads, Blue- nuclei, Red- CD45 to show plasma membrane (B) Quantification of signal intensity from panel A (*,+p < 0.05). (C) Representative photomicrographs of BMDMs given IgG-coated beads ± LPS in TRPV4 siRNA-treated and TRPV4 KO cells. (D) Quantification of signal intensity from panel C (*,+p < 0.05). (E) Representative photomicrographs of alveolar macrophages (AM) given IgG-coated beads ± LPS ± HC. (F) Quantification of signal intensity from panel E (*,+p < 0.05). WT denotes WT cells in the absence of HC, WT+HC denotes WT cells in the presence of the TRPV4 inhibitor (HC). + denotes the increase by LPS vs UT, * denotes difference in LPS response as compared to WT (D) ± TRPV4 inhibitor (B,F; HC). n ≥ 3 times in at least duplicate. All photomicrograph panels, 40X Orig. Mag., scale bar 30 µm.
LPS-stimulated macrophage phagocytosis is dependent on TRPV4 under conditions of pathophysiologic range matrix stiffness
As lung inflammation causes changes in matrix stiffness and TRPV4 is a mechanosensitive ion channel, we sought to determine if the LPS response is altered by matrix stiffness-sensing through TRPV4. We first noted that LPS itself upregulates TRPV4 activity (calcium influx; Figure 4A; 62 ± 4%; *,+p < 0.05) on a supraphysiologically stiff substrate (glass as used in standard culture conditions, 50 × 106 kPa). Importantly, this LPS-inducing effect increases directly with the matrix stiffness over the pathophysiologic range seen in inflamed or fibrotic lungs (1 - 25 kPa) (Figure 4B, +p < 0.05). The matrix stiffness dependency was also demonstrable for LPS-stimulated phagocytosis over the same pathophysiologic range (Figure 4C, +p < 0.05). The LPS-stimulated phagocytic response on fibrotic range matrix stiffness (25 kPa) was lost upon either deletion (TRPV4 KO, 55 ± 5%), or pharmacologic inhibition (HC, 63 ± 3%), of TRPV4 (Figure 4D, #p < 0.05). No stiffness effect was noted in either calcium influx or phagocytic function in the absence of LPS (Data not shown). In summary, these data show that TRPV4 cooperates with LPS to effect calcium influx and phagocytosis in a matrix stiffness-dependent manner.
Figure 4. TRPV4 mediates the stiffness induction effect on LPS-stimulated calcium influx and macrophage phagocytosis.

BMDMs were treated ± LPS while attached to fibronectin-coated glass (50 × 106 GPa) (A) or polyacrylamide hydrogels of indicated stiffnesses (B-D). (A) Calcium influx was measured as in Figure 1B in WT BMDMs ± LPS ± HC versus KO BMDMs on glass substrate. LPS stimulated an increase in calcium influx that was abrogated with inhibition of TRPV4 (HC) or deletion of TRPV4 (KO) (*,+p < 0.05). (B) Calcium influx and (C) LPS-stimulated phagocytosis of E. coli particles, measured as % induction by LPS, were dependent on pathophysiologic range stiffness (>8-25 kPa) (+p < 0.05 ± LPS). (D) LPS-enhanced phagocytosis in WT BMDMs was decreased 4-fold upon deletion of TRPV4 (KO) or inhibition of TRPV4 (HC) on pathophysiologic range stiffness (25 kPa) (#p < 0.05). * denotes difference as compared to ± LPS, + denotes the increase in LPS vs UT (A) or difference consistent with 1 kPa (B-C), # denotes difference as compared to LPS treated WT ± HC (WT-No HC). n ≥ 3 times in quadruplicate.
LPS-stimulated cytokine release is dependent on TRPV4 under conditions of pathophysiologic range matrix stiffness
As TRPV4 is sensitized by matrix stiffness and increases its activity in response to LPS, we examined the role of TRPV4 on other LPS-induced macrophage functions related to lung inflammation or infection. As expected, LPS induces macrophage cytokine production of IL-1β and IL-10. Upon deletion of TRPV4 (KO), the LPS induction of IL-1β increased by 50 ± 3% and IL-10 decreased by 47 ± 5% on a glass substrate (Figure 5A and 5C, *p < 0.05). Both of these effects were dependent on extracellular matrix stiffness in the pathophysiologic range in WT BMDMs (Figure 5B and 5D, +p < 0.05). These data show that TRPV4 mediates macrophage release of key anti-inflammatory/pro-resolution cytokines (↓ IL-1β, ↑ IL-10) in response to LPS, in a stiffness-dependent manner.
Figure 5. TRPV4 modulates the cytokine response to LPS in a manner that depends on matrix stiffness.

ELISAs (IL-1β and IL-10) were performed on macrophage conditioned media from BMDMs cultured on both glass substrate and/or pathophysiologic-range matrix stiffness ± LPS (100 ng/mL, 24 h). (A) LPS-stimulated release of IL-1β was enhanced in TRPV4 KO BMDMs (*p < 0.05) and (B) LPS-stimulated release of IL-1β was suppressed in WT BMDMs as stiffness increased over the pathophysiologic-range (+p < 0.05). (C) LPS-stimulated release of IL-10 was suppressed in TRPV4 KO BMDMs (*p < 0.05) and (D) LPS-stimulated release of IL-10 was enhanced in WT BMDMs as stiffness increased over the pathophysiologic-range (+p < 0.05). * denotes difference in LPS response compared with WT, + denotes the increase in LPS response compared with 1 kPa. n ≥ 3 times in quadruplicate.
TRPV4 mediates macrophage phagocytosis in vivo
Lastly, to examine if TRPV4 also mediates macrophage phagocytosis in vivo, we examined macrophage phagocytosis of IgG-coated latex beads intratracheally in intact mice. LPS induces a similar increment in alveolar neutrophil and macrophage numbers in the WT and TRPV4 KO mice (Data not shown). Concordant with the in vitro data, LPS induces macrophage phagocytosis in WT mice in a manner that is completely lost in the TRPV4 KO mice (Figure 6A and B, *,+p < 0.05). The data show that TRPV4 is required for LPS-stimulated macrophage phagocytosis of IgG-coated latex beads in mouse lungs in vivo.
Figure 6. TRPV4 mediates LPS-stimulated macrophage phagocytosis of IgG-coated latex beads in vivo.

WT and TRPV4 KO C57BL/6 were treated with IT LPS (3 µg/g) for 16 h followed by intratracheal (IT) IgG-coated latex beads for 6 h. Cell phagocytic analysis was performed on the BAL by microscopic analysis of cytospin preparations. (A) Representative confocal images of WT and TRPV4 KO mice given IT saline (n = 2) or LPS (n = 5) followed by IgG-coated latex beads (white arrow heads) - Green-beads, Blue-nuclei (PMN: multilobular nucleus, macrophage: single concentric nucleus), Red-CD45 to show membrane (All panels, 40X Orig. Mag.). (B) LPS treated WT mice had increased macrophage phagocytosis of IgG-coated latex beads compared to the LPS treated TRPV4 KO. The number of beads per macrophage were quantified from panel A (*,+p < 0.05). + denotes the increase in LPS vs UT, * denotes difference in LPS response between KO and WT.
Discussion
In this study, we describe a novel role for TRPV4 in macrophage function as follows: LPS-stimulated phagocytosis of both non-opsonized E. coli in vitro and FcR-dependent (IgG-coated beads) in vitro and in vivo is mediated by TRPV4. The ability of LPS to induce phagocytosis through TRPV4 is dependent on matrix stiffness in a range comparable to that seen in inflamed or fibrotic lung. Finally, TRPV4 mediates the LPS signal to release anti-inflammatory/pro-resolution cytokines in macrophages. Taken together, these data implicate the mechanosensing channel, TRPV4, in the pathogenesis of lung infection/bacterial pneumonia by integrating the LPS and the matrix stiffness signals for macrophage phagocytosis and pro-resolution cytokine release.
Our data demonstrate that LPS-stimulated macrophage phagocytosis on stiff matrices (> 8 - 25 kPa) is TRPV4 dependent. This was shown using two independent methods of inhibiting TRPV4 activity, including small molecule inhibition (HC) and TRPV4 deletion (KO). Further, the TRPV4 dependency of LPS-stimulated phagocytosis was demonstrable in BMDM’s, a macrophage-like cell line (RAW cells, Data not shown), in primary alveolar macrophages in vitro, and in live intact mice, using both opsonized and non-opsonized particles. This suggests that the TRPV4 mediation of the LPS response is fundamental to phagocytosis and is an inherent property of the macrophage cell lineage.
Studies show that LPS-stimulated macrophage phagocytosis depends on MAPK (p38) activation of small GTPases (Rac, Rho and Cdc42), and on actin polymerization (17,42). While not specifically studied in LPS-stimulated phagocytosis, TRPV channels have been shown to modulate p38 MAPK in response to thermal stimuli in neurons and osmotic stress in chondrocytes, and have been shown to modulate small GTPase/actin remodeling in keratinocytes (43-45). Future studies in our lab will identify the downstream molecular effectors of the TRPV4 mediated effect on LPS-stimulated phagocytosis.
Recent studies demonstrate that resident alveolar macrophages are relatively quiescent and capable of self-renewal under basal homeostatic conditions (2). Our data are consistent with the hypothesis that the macrophage response to LPS is down-modulated under conditions of uninflamed lung (i.e., 1 - 3 kPa), thereby supporting maintenance of tissue integrity (46-51). In contrast, during an acute inflammatory or infectious process, studies demonstrate that overall lung compliance is significantly reduced and alveolar level vessel stiffness is increased > 10-fold (from 3 to 45 kPa) after intratracheal LPS instillation in mice (53,54). Thus, bone marrow-derived monocytic lineage cells are recruited to an inflamed/infected lung that possesses enhanced rigidity, in the range noted in our study (52). Taken together with our findings, we hypothesize that TRPV4 mediates a feed-forward upregulation of phagocytosis in lung macrophages when they sense infection/injury-associated lung matrix stiffening. This concept is further supported by the observation of altered alveolar macrophage shape and function in association with increased alveolar surface tension in surfactant protein B-deficient mice (55). TRPV4 also mediates the LPS signal to secrete pro-resolution cytokines (suppressed IL-1β and enhanced IL-10) in a stiffness-dependent manner.
While we have convincingly shown, using multiple complementary methods, that TRPV4 is a key mediator of LPS stimulated macrophage phagocytosis and cytokine production, there are some limitations to our study. Although the intratracheal LPS murine model is a neutrophil-predominant inflammatory stimulus, we consistently observed > 20% macrophages in the alveolar space, and the neutrophil uptake of IgG-coated beads was less than 1/10 that of the macrophages. Therefore, macrophages are the key phagocytic cell in our in vivo assay system. Additionally, TRPV2 has previously been mechanistically-linked to macrophage phagocytosis, however, its effects were on basal phagocytosis, and were found to be calcium-independent (56). Thus, it is unlikely that TRPV2-mediated effects are confounding our results. IL-1β and IL-10 are the best studied pro-inflammatory and pro-resolution cytokines, respectively (57). However, given the complex pleiotropic, overlapping, and cell type-specific effects of cytokines, one cannot predict the biologic response based on measurements of individual or multiple cytokines with certainty (58-63).
As shown in our proposed schematic model (Figure 7), our work suggests that macrophage TRPV4 is sensitized by a stiff matrix (inflamed, infected lung) and cooperates with LPS to mediate macrophage phenotypic change. The macrophage phenotypic change results in increased phagocytosis and altered cytokine expression (decreased IL-1β and increased IL-10). The primed macrophage phenotype has increased ability for bacterial clearance and for resolution of infection-associated lung tissue injury. Thus, we envision that TRPV4 would play a significant role in the cytokine-based lung injury response to gram negative infection in the setting of underlying lung injury (ARDS), underlying lung fibrosis, and/or high volume mechanical ventilation (26,64,65). Our observations may also underlie the observed protective effect of LPS on experimental lung fibrosis (66).
Figure 7. Working model illustrating that LPS and TRPV4 signal cooperate to alter macrophage phenotypic change leading to enhanced clearance of infection and resolution of lung injury.
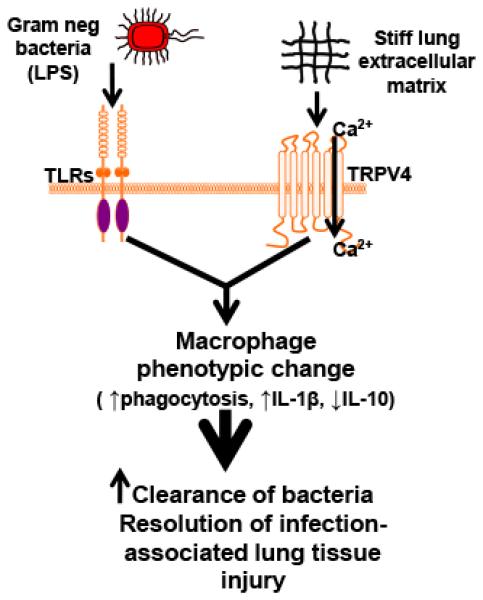
Our data suggest that TRPV4 is sensitized by extracellular matrix stiffness in the range of inflamed/fibrotic lung. Interaction between the LPS signal and the matrix stiff signal through TRPV4 promote increased TRPV4 channel activity and macrophage phenotypic change leading to increased clearance of bacteria and resolution of infection-associated lung injury.
In summary, TRPV4 is essential in LPS-stimulated macrophage phagocytosis under conditions of pathological matrix stiffness. This work reveals a novel role of TRPV4 in macrophage phagocytosis that is applicable to lung homeostasis, inflammation, and host defense.
Acknowledgements
We would like to thank Dr. Serpil Erzurum for her critical reading of this manuscript.
Grant Support: This work was supported by NIH grants (HL-103553, HL-085324, and HL-119792) to M.A. Olman.
References
- 1.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 2.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eddens T, Kolls JK. Host defenses against bacterial lower respiratory tract infection. Curr Opin Immunol. 2012;24:424–430. doi: 10.1016/j.coi.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jennings JH, Linderman DJ, Hu B, Sonstein J, Curtis JL. Monocytes recruited to the lungs of mice during immune inflammation ingest apoptotic cells poorly. Am J Respir Cell Mol Biol. 2005;32:108–117. doi: 10.1165/rcmb.2004-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barth K, Remick DG, Genco CA. Disruption of immune regulation by microbial pathogens and resulting chronic inflammation. J Cell Physiol. 2013;228:1413–1422. doi: 10.1002/jcp.24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belikoff BG, Hatfield S, Georgiev P, Ohta A, Lukashev D, Buras JA, Remick DG, Sitkovsky M. A2B adenosine receptor blockade enhances macrophage-mediated bacterial phagocytosis and improves polymicrobial sepsis survival in mice. J Immunol. 2011;186:2444–2453. doi: 10.4049/jimmunol.1001567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fazzi F, Njah J, Di Giuseppe M, Winnica DE, Go K, Sala E, St Croix CM, Watkins SC, Tyurin VA, Phinney DG, Fattman CL, Leikauf GD, Kagan VE, Ortiz LA. TNFR1/phox interaction and TNRF1 mitochondrial translocation thwart silica-induced pulmonary fibrosis. J Immunol. 2014;192:3837–3846. doi: 10.4049/jimmunol.1103516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arredouani MS, Palecanda A, Koziel H, Huang YC, Imrich A, Sulahian TH, Ning YY, Yang Z, Pikkarainen T, Sankala M, Vargas SO, Takeya M, Tryggvason K, Kobzik L. MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J Immunol. 2005;175:6058–6064. doi: 10.4049/jimmunol.175.9.6058. [DOI] [PubMed] [Google Scholar]
- 9.Blakney AK, Swartzlander MD, Bryant SJ. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. Journal of Biomedical Materials Research Part A. 2012;100A:1375–1386. doi: 10.1002/jbm.a.34104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fereol S, Fodil R, Labat B, Galiacy S, Laurent VM, Louis B, Isabey D, Planus E. Sensitivity of alveolar macrophages to substrate mechanical and adhesive properties. Cell Motil. Cytoskeleton. 2006;63:321–340. doi: 10.1002/cm.20130. [DOI] [PubMed] [Google Scholar]
- 11.Akei H, Whitsett JA, Buroker M, Ninomiya T, Tatsumi H, Weaver TE, Ikegami M. Surface tension influences cell shape and phagocytosis in alveolar macrophages. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2006;291:L572–L579. doi: 10.1152/ajplung.00060.2006. [DOI] [PubMed] [Google Scholar]
- 12.Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. The Journal of Immunology. 2010;184:1049–1061. doi: 10.4049/jimmunol.0902223. [DOI] [PubMed] [Google Scholar]
- 13.McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. PNAS. 2013;43:17253–17258. doi: 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nunes P, Demaurex N. The role of calcium signaling in phagocytosis. J. Leukoc. Biol. 2010;88:57–68. doi: 10.1189/jlb.0110028. [DOI] [PubMed] [Google Scholar]
- 15.Melendez A,TH. Phagocytosis: a repertoire of receptors and Ca2+ as a key second messenger. Biosci. Rep. 2008;28:287–298. doi: 10.1042/BSR20080082. [DOI] [PubMed] [Google Scholar]
- 16.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 17.Rougerie P, Miskolci V, Cox D. Generation of membrane structures during phagocytosis and chemotaxis of macrophages: role and regulation of the actin cytoskeleton. Immunol. Rev. 2013;256:222–239. doi: 10.1111/imr.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birmelin M, Decker K. Ca2+ flux as an initial event in phagocytosis by rat kupffer cells. Eur. J. Biochem. 1982;131:539–543. doi: 10.1111/j.1432-1033.1983.tb07295.x. [DOI] [PubMed] [Google Scholar]
- 19.Hishikawa T, Cheung JY, Yelamarty RV, Knutson DW. Calcium transients during Fc receptor-mediated and nonspecific phagocytosis by murine peritoneal macrophages. The Journal of Cell Biology. 1991;115:59–66. doi: 10.1083/jcb.115.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gronski MA, Kinchen JM, Juncadella IJ, Franc NC, Ravichandran KS. An essential role for calcium flux in phagocytes for apoptotic cell engulfment and the anti-inflammatory response. Cell Death Differ. 2009;16:1323–1331. doi: 10.1038/cdd.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang L, Gamal El-Din TM, Payandeh J, Martinez GQ, Heard TM, Scheuer T, Zheng N, Catterall WA. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature. 2014;505:56–61. doi: 10.1038/nature12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell. 2002;108:595–598. doi: 10.1016/s0092-8674(02)00670-0. [DOI] [PubMed] [Google Scholar]
- 23.Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog. Biophys. Mol. Biol. 2010;103:2–17. doi: 10.1016/j.pbiomolbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Wu L, O'Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J. Biol. Chem. 2003;278:27129–27137. doi: 10.1074/jbc.M302517200. [DOI] [PubMed] [Google Scholar]
- 25.Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Nat Acad Sci U S A. 2004;101:396–401. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamanaka K, Jian MY, Townsley MI, King JA, Liedtke W, Weber DS, Eyal FG, Clapp MM, Parker JC. TRPV4 channels augment macrophage activation and ventilator-induced lung injury. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2010;299:L353–L362. doi: 10.1152/ajplung.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahaman SO, Grove LM, Paruchuri S, Southern BD, Niese KA, Abraham S, Scheraga RG, Ghosh S, Thodeti CK, Zhang DX, Moran MM, Schilling WP, Tschumperlin DJ, Olman MA. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest. 2014;124(12):5225–5238. doi: 10.1172/JCI75331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu S, Jian M, Xu Y, Zhou C, Al-Mehdi, Liedtke W, Shin H, Townsley MI. Ca2+ entry via a1G and TRPV4 channels differentially regulated surface expression of P-selectin and barrier integrity in pulmonary capillary endothelium. Am J Physiol Lung Cell Mol Physiol. 2009;297:L650–L657. doi: 10.1152/ajplung.00015.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jian MY, King JA, Al-Mehdi AB, Liedtke W, Townsley MI. High vascular pressure induced lung injury requires P450 epoxygenase dependent activation of TRPV4. Am. J. Respir. Cell Mol. Biol. 2008;38:386–392. doi: 10.1165/rcmb.2007-0192OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4 mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ. Res. 2006;99:988–995. doi: 10.1161/01.RES.0000247065.11756.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, King JA, Liedtke W, Parker JC. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2007;293:L923–L932. doi: 10.1152/ajplung.00221.2007. [DOI] [PubMed] [Google Scholar]
- 32.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, Eidam HS, Ye G, Willette RN, Thorneloe KS, Bradshaw HB, Matalon S, Jordt SE. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2014;307:L158–L172. doi: 10.1152/ajplung.00065.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu G, Gulsvik A, Bakke P, Ghatta S, Anderson W, Lomas DA, Silverman EK, Pillai SG. Association of TRPV4 gene polymorphisms with chronic obstructive pulmonary disease. Hum. Mol. Genet. 2009;18:2053–2062. doi: 10.1093/hmg/ddp111. [DOI] [PubMed] [Google Scholar]
- 34.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, Gordon E, Lozinskaya I, Elefante L, Qin P, Matasic DS, James C, Tunstead J, Donovan B, Kallal L, Waszkiewicz A, Vaidya K, Davenport EA, Larkin J, Burgert M, Casillas LN, Marquis RW, Ye G, Eidam HS, Goodman KB, Toomey JR, Roethke TJ, Jucker BM, Schnackenberg CG, Townsley MI, Lepore JJ, Willette RN. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Science Translational Medicine. 2012;4:159ra148. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- 35.Moran MM, McAlexander MA, Biro T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10:601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- 36.Zhao C, Pavicic PG, Datta S, Sun D, Novotny M, Hamilton TA. Cellular stress amplifies TLR3/4-induced CXCL1/2 gene transcription in mononuclear phagocytes via RIPK1. The Journal of Immunology. 2014;193:879–888. doi: 10.4049/jimmunol.1303396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding Q, Cai GQ, Hu M, Yang Y, Zheng A, Tang Q, Gladson CL, Hayasaka H, Wu H, You Z, Southern BD, Grove LM, Rahaman SO, Fang H, Olman MA. FAK-related nonkinase is a multifunctional negative regulator of pulmonary fibrosis. The American Journal of Pathology. 2013;182:1572–1584. doi: 10.1016/j.ajpath.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grove LM, Southern BD, Jin TH, White KE, Paruchuri S, Harel E, Wei Y, Rahaman SO, Gladson CL, Ding Q, Craik CS, Chapman HA, Olman MA. Urokinase-type plasminogen activator receptor (uPAR) ligation induces a raft-localized integrin signaling switch that mediates the hypermotile phenotype of fibrotic fibroblasts. J. Biol. Chem. 2014;289:12791–12804. doi: 10.1074/jbc.M113.498576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das R, Ganapathy S, Settle M, Plow EF. Plasminogen promotes macrophage phagocytosis in mice. Blood. 2014;124:679–688. doi: 10.1182/blood-2014-01-549659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aggarwal NR, Alessio FR, Tsushima K, Files DC, Damarla M, Sidhaye VK, Fraig MM, Polotsky VY, King LS. Moderate oxygen augments lipopolysaccharide-induced lung injury in mice. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2010;298:L371–L381. doi: 10.1152/ajplung.00308.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Files DC, D'Alessio FR, Johnston LF, Kesari P, Aggarwal NR, Garibaldi BT, Mock JR, Simmers JL, DeGorordo A, Murdoch J, Willis MS, Patterson C, Tankersley CG, Messi ML, Liu C, Delbono O, Furlow JD, Bodine SC, Cohn RD, King LS, Crow MT. A Critical Role for Muscle Ring Finger-1 in Acute Lung Injury-Associated Skeletal Muscle Wasting. Am. J. Respir. Crit. Care Med. 2012;185:825–834. doi: 10.1164/rccm.201106-1150OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kong L, Ge BX. MyD88-independent activation of a novel actin-Cdc42/Rac pathway is required for Toll-like receptor-stimulated phagocytosis. Cell Res. 2008;18:745–755. doi: 10.1038/cr.2008.65. [DOI] [PubMed] [Google Scholar]
- 43.Sokabe T, Fukumi-Tominaga T, Yonemura S, Mizuno A, Tominaga M. The TRPV4 channel contributes to intercellular junction formation in keratinocytes. J. Biol. Chem. 2010;285:18749–18758. doi: 10.1074/jbc.M110.103606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hdud IM, Mobasheri A, Loughna PT. Effect of osmotic stress on the expression of TRPV4 and BKCa channels and possible interaction with ERK1/2 and p38 in cultured equine chondrocytes. American Journal of Physiology - Cell Physiology. 2014;306:C1050–C1057. doi: 10.1152/ajpcell.00287.2013. [DOI] [PubMed] [Google Scholar]
- 45.Holland S, Coste O, Zhang DD, Pierre SC, Geisslinger G, Scholich K. The ubiquitin ligase MYCBP2 regulates transient receptor potential vanilloid receptor 1 (TRPV1) internalization through inhibition of p38 MAPK signaling. J. Biol. Chem. 2011;286:3671–3680. doi: 10.1074/jbc.M110.154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Millonig G, Reimann FM, Friedrich S, Fonouni H, Mehrabi A, Buchler M, Seitz H, Mueller S. Extrahepatic cholestasis increases liver stiffness (fibroscan) irrespective of fibrosis. Hepatology. 2008;48:1718–1723. doi: 10.1002/hep.22577. [DOI] [PubMed] [Google Scholar]
- 47.Castera L, Vergniol J, Foucher J, Le Bail B, Chanteloup E, Haaser M, Darriet M, Couzigou P, de Ledinghen V. Prospective comparison of transient elastography, Fibrotest, APRI, and liver biopsy for the assessment of fibrosis in chronic hepatitis C. Gastroenterology. 2005;128:343–350. doi: 10.1053/j.gastro.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 48.Mink SN, Light RB, Wood LD. Effect of pneumococcal lobar pneumonia on canine lung mechanics. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 2008;50:283–291. doi: 10.1152/jappl.1981.50.2.283. [DOI] [PubMed] [Google Scholar]
- 49.Ingenito EP, Mark L, Davison B. Effects of acute lung injury on dynamic tissue properties. J. Appl. Physiol. 1994;77:2689–2697. doi: 10.1152/jappl.1994.77.6.2689. [DOI] [PubMed] [Google Scholar]
- 50.Kang I, Wang Q, Eppell SJ, Marchant RE, Doerschuk CM. Effect of Neutrophil Adhesion on the Mechanical Properties of Lung Microvascular Endothelial Cells. Am. J. Respir. Cell Mol. Biol. 2010;43:591–598. doi: 10.1165/rcmb.2006-0381OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bastard C, Bosisio M, Chabert M, Kalopissis AD, Mahrouf-Yorgov M, Gilgenkrantz H, Mueller S, Sandrin L. Transient micro-elastography: a novel non-invasive approach to measure liver stiffness in mice. World Journal Gastroenterology. 2014;17:968–975. doi: 10.3748/wjg.v17.i8.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meng F, Mambetsariev I, Tian Y, Beckham Y, Meliton A, Leff A, Gardel ML, Allen MJ, Birukov KG, Birukova AA. Attenuation of lipopolysaccharide-induced lung vascular stiffening by lipoxin reduces lung inflammation. Am. J. Respir. Cell Mol. Biol. 2014;52:152–161. doi: 10.1165/rcmb.2013-0468OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perlman CE, Lederer DJ, Bhattacharya J. Micromechanics of Alveolar Edema. Am. J. Respir. Cell Mol. Biol. 2011;44:34–39. doi: 10.1165/rcmb.2009-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meng F, Mambetsariev I, Tian Y, Beckham Y, Meliton A, Leff A, Gardel ML, Allen MJ, Birukov KG, Birukova AA. Attenuation of Lipopolysaccharide-Induced Lung Vascular Stiffening by Lipoxin Reduces Lung Inflammation. Am. J. Respir. Cell Mol. Biol. 2014;52:152–161. doi: 10.1165/rcmb.2013-0468OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akei H, Whitsett JA, Buroker M, Ninomiya T, Tatsumi H, Weaver TE, Ikegami M. Surface tension influences cell shape and phagocytosis in alveolar macrophages. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2006;291:L572–L579. doi: 10.1152/ajplung.00060.2006. [DOI] [PubMed] [Google Scholar]
- 56.Link TM, Park U, Vonakis BM, Raben DM, Soloski MJ, Caterina MJ. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat Immunol. 2010;11:232–239. doi: 10.1038/ni.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ware LB, Matthay MA. The acute respiratory distress syndrome. N. Engl. J. Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 58.Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1996;154:602–611. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- 59.Suter PM, Suter S, Girardin E, Roux-Lombard P, Grau GE, Dayer JM. High Bronchoalveolar Levels of Tumor Necrosis Factor and Its Inhibitors, Interleukin-1, Interferon, and Elastase, in Patients with Adult Respiratory Distress Syndrome after Trauma, Shock, or Sepsis. Am. Rev. Resp. Dis. 1992;145:1016–1022. doi: 10.1164/ajrccm/145.5.1016. [DOI] [PubMed] [Google Scholar]
- 60.Park W, Goodman R, Steinberg K, Ruzinski J, Radella F, Park D, Pugin J, Skerrett S, Hudson L, Martin T. Cytokine Balance in the Lungs of Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2001;164:1896–1903. doi: 10.1164/ajrccm.164.10.2104013. [DOI] [PubMed] [Google Scholar]
- 61.Donnelly SC, Strieter RM, Reid PT, Kunkel SL, Burdick MD, Armstrong I, Mackenzie A, Haslett C. The Association between Mortality Rates and Decreased Concentrations of Interleukin-10 and Interleukin-1 Receptor Antagonist in the Lung Fluids of Patients with the Adult Respiratory Distress Syndrome. Ann. Intern. Med. 1996;125:191–196. doi: 10.7326/0003-4819-125-3-199608010-00005. [DOI] [PubMed] [Google Scholar]
- 62.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. The Journal of Immunology. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 63.Bogdan C, Vodovotz Y, Nathan C. Macrophage deactivation by interleukin 10. The Journal of Experimental Medicine. 1991;174:1549–1555. doi: 10.1084/jem.174.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beck-Schimmer B, Schwendener R, Pasch T, Reyes L, Booy C, Schimmer R. Alveolar macrophages regulate neutrophil recruitment in endotoxin-induced lung injury. Respiratory Research. 2005;6:61. doi: 10.1186/1465-9921-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pugin J, Dunn I, Jolliet P, Tassaux D, Magnenat J, Nicod LP, Chevrolet J. Activation of human macrophages by mechanical ventilation in vitro. American Journal of Physiology - Lung Cellular and Molecular Physiology. 1998;275:L1040–L1050. doi: 10.1152/ajplung.1998.275.6.L1040. [DOI] [PubMed] [Google Scholar]
- 66.Phan SH, Fantone JC. Inhibition of bleomycin-induced pulmonary fibrosis by lipopolysaccharide. Lab Invest. 1984;50:587–591. [PubMed] [Google Scholar]