

Abstract
Microbe-induced receptor trafficking has emerged as an essential means to promote innate immune signal transduction. Upon detection of bacterial lipopolysaccharides (LPS), CD14 induces an inflammatory endocytosis pathway that delivers Toll-like Receptor 4 (TLR4) to endosomes. Although several regulators of CD14-dependent TLR4 endocytosis have been identified, the cargo selection mechanism during this process remains unknown. We reveal that, in contrast to classic cytosolic interactions that promoted the endocytosis of transmembrane receptors, TLR4 was selected as cargo for inflammatory endocytosis entirely through extracellular interactions. Mechanistically, the extracellular protein MD-2 bound to and dimerized TLR4 in order to promote this endocytic event. Our analysis of LPS variants from human pathogens and gut commensals revealed a common mechanism by which bacteria prevent inflammatory endocytosis. We suggest that evasion of CD14-dependent endocytosis is an attribute that transcends the concept of pathogenesis, and may be a fundamental feature of bacteria that inhabit eukaryotic hosts.
Introduction
The pattern recognition receptors (PRRs) of the innate immune system are a diverse family of structurally unrelated proteins that are grouped functionally by their ability to detect microbial products (Akira et al., 2006). Microbial detection initiates signal transduction pathways that promote the expression of inflammatory chemokines, cytokines and interferons (IFNs). Central to our understanding of PRR biology is the Toll-like Receptor (TLR) family, which consists of type I transmembrane proteins that reside at plasma or endosomal membranes, depending on the family member (Akira et al., 2006). Individual TLRs recognize conserved microbial products, such as bacterial lipopolysaccharides (LPS) and lipoproteins, flagellin subunits, double stranded RNA and others.
While TLRs are often discussed as receptors for microbial products, increasing evidence indicates that high affinity receptor-ligand interactions depend on the actions of ligand-binding proteins that work together to dimerize TLRs and promote signal transduction (Lee et al., 2012). For example, LPS-binding protein (LBP), CD14, MD-2 and TLR4 all form direct contacts with LPS, and each is necessary to promote the detection of picomolar LPS concentrations (Gioannini et al., 2004). In the absence of any of these accessory proteins, the concentrations of LPS needed to activate inflammation increase by several orders of magnitude (Gioannini and Weiss, 2007). In recent years, the idea that accessory proteins facilitate high affinity receptor-ligand interactions has been extended to include TLR2, TLR3, TLR7 and TLR9 (Lee et al., 2012). Based on these discoveries, it is generally thought that after ligand binding, all subsequent cellular responses are mediated by the TLR, not the accessory proteins.
The idea that accessory proteins cannot elicit any cellular responses independent of a TLR was challenged in recent years by the discovery that CD14 can induce cellular responses that TLR4 cannot (Zanoni et al., 2011). This finding derived from our studies of the subcellular sites of TLR4 signal transduction (Kagan et al., 2008).
The initiation of TLR4 signal transduction occurs in phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-rich regions of the plasma membrane, where a sorting adaptor called TIRAP resides (Kagan and Medzhitov, 2006). TIRAP senses active (i.e. dimerized) TLR4 through interactions between the Toll/IL-1 Receptor (TIR) domains present in each of these proteins (Kagan and Barton, 2014). These TIR-TIR interactions prompt TIRAP to assemble a higher order filamentous structure called the myddosome, which consists of the signaling adaptor MyD88 and several IRAK family kinases (Bonham et al., 2014; Lin et al., 2010). The myddosome is a supramolecular organizing center (SMOC) that initiates signaling events that activate inflammatory transcription factors such as AP-1 and NF-κB (Kagan et al., 2014; Medzhitov and Horng, 2009).
Concomitant with TLR4 signaling is the initiation of events that promote TLR4 endocytosis. Upon delivery to endosomes, TLR4 engages another sorting adaptor TRAM and the signaling adaptor TRIF, which promote the subsequent expression of IFNs and IFN stimulated genes (ISGs) (Kagan et al., 2008). The importance of the endosome-specific signaling pathway is underscored by the fact that most TLR4-inducible genes (even those activated by MyD88) are dependent on TRIF (Yamamoto et al., 2003). Thus, in the case of TLR4, a dominant site of TLR4 signal transduction (endosomes) is distinct from the site of ligand binding (plasma membrane) (Kagan and Barton, 2014).
CD14 is a crucial regulator of TLR4 endocytosis and consequently, TRIF-dependent signaling from endosomes (Zanoni et al., 2011). Several cytosolic regulators of CD14-dependent endocytosis are known, including the tyrosine kinase Syk, ITAM containing adaptors, phospholipase C gamma 2 (PLCγ2) and various second messenger molecules (Chiang et al., 2012; Zanoni et al., 2011). These factors may constitute an inflammatory endocytosis pathway that is triggered by diverse upstream receptors, such as CD14 and the immune-related receptors Dectin-1 and FcγR1 (Crowley et al., 1997; Underhill et al., 2005). The finding that CD14 promotes TLR4 endocytosis is unusual, as this protein is anchored to the plasma membrane via a C-terminal glycosylphosphatidylinositol (GPI) anchor. Few other examples exist of GPI-anchored proteins that promote transmembrane receptor endocytosis. How GPI-anchored proteins promote the endocytosis of transmembrane receptors is an under-explored area of biology.
Based on the function of CD14 as an LPS-binding protein, it is possible that this protein simply transfers LPS to TLR4, which then enlists either MyD88 or TRIF to promote endocytosis. However, signaling-deficient TLR4 mutants are fully capable of undergoing endocytosis, as are macrophages derived from mice lacking both the MyD88 and TRIF adaptors (Zanoni et al., 2011). Thus, the endocytosis pathway induced by CD14 does not proceed via TLR4. These observations reveal that an accessory protein (CD14) can have more functions than simply delivering ligands to TLRs. Rather, this protein acts to transport ligands (LPS) and receptors (TLR4) to intracellular sites where signaling can occur. Despite these advances, we lack a clear understanding of the mechanisms governing the LPS-inducible transport events.
Herein, we report that the selection of TLR4 as cargo for CD14-dependent endocytosis proceeded by an unusual mechanism, in that the cytosolic tail of TLR4 was dispensable for entry into the cell. We identified the extracellular protein MD-2 as a cargo-selection agent for endocytosis, and revealed a common mechanism by which CD14-dependent endocytosis is prevented by pathogenic and commensal bacteria of the human intestine. These discoveries establish that CD14 is not the only accessory protein that has a dual function in ligand transport and receptor transport. MD-2 also promotes ligand transport to receptor and receptor transport to endosomes.
Results
CD14 must bind to LPS directly to select TLR4 as cargo for inflammatory endocytosis
While CD14 is required to promote inflammatory endocytosis in response to LPS, the molecular interactions needed to accomplish this task are unclear. To determine whether the interaction between LPS and CD14 was required to initiate the inflammatory endocytosis pathway, we generated a CD14 mutant that could not bind LPS. Four regulatory regions within murine CD14, designated as R1 to R4 respectively, are involved in LPS recognition (Stelter et al., 1997) (Figure 1A). Mutagenizing one or two of these region(s) by alanine replacement (designated as mutants 1R and 2R; Figure 1B) resulted in a modest defect in the binding between CD14 and biotinylated LPS in vitro. Only when all four regions were substituted with alanine (mutant 4R) did we observe a severe LPS binding defect (Figure 1B). This 4R mutant could therefore be used as a tool to study the importance of LPS-binding by CD14.
Figure 1. CD14 must bind LPS to promote TLR4 endocytosis.
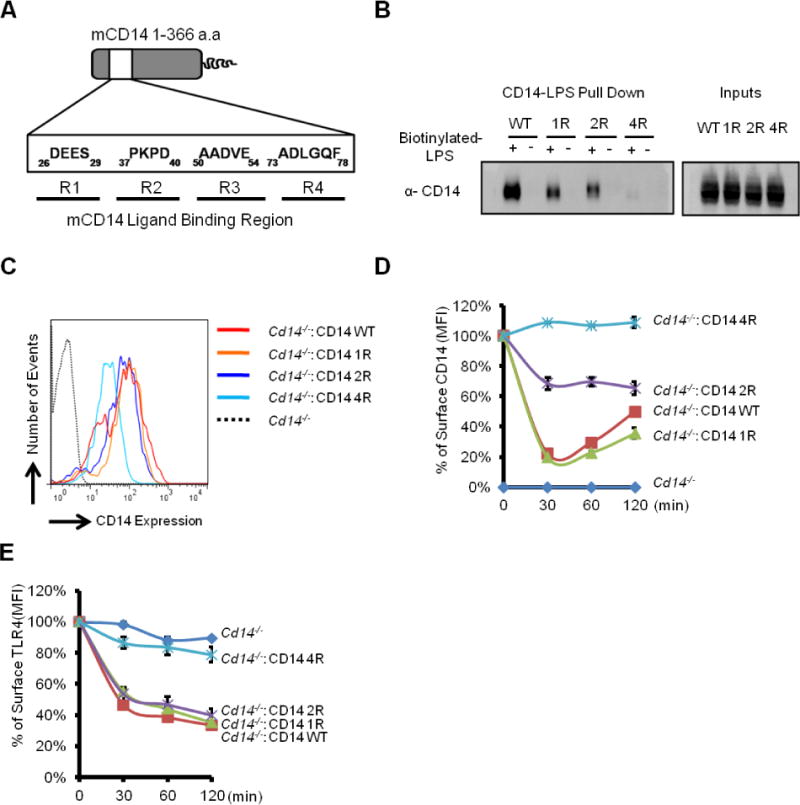
(A) Schematic of the CD14 LPS binding region. Key regulatory motifs involved in LPS binding are highlighted. The amino acids indicated were all mutated to alanine to create the mutant alleles 1R, 2R or 4R, which are used in this figure.
(B) Lysates of 293T cells expressing the indicated CD14 mutants were incubated with biotinylated LPS (5 μg). The CD14-LPS complex were then captured using neutraviden beads. The amount of indicated CD14 mutants retained by LPS was determined by western blotting.
(C) Surface level of different CD14 alleles expressed in Cd14−/− iBMDMs was determined by flow cytometry.
(D) Cells of the indicated genotypes were treated with LPS (1 μg/ml) for the times indicated. CD14 surface staining was measured by flow cytometry. Line graphs represent mean florescence intensity (MFI) of CD14 over the time course.
(E) Cells of the indicated genotypes were treated with LPS (1 μg/ml) for the times indicated. Surface level of TLR4 was measured by flow cytometry. Line graphs represent MFI of TLR4 (right) over the assay time course.
See also Figure S1.
To determine if defects in LPS binding in vitro resulted in defects in endocytosis within cells, wild type (WT) and mutant CD14 alleles were introduced into Cd14−/− immortal bone marrow derived macrophages (iBMDM) by retroviral transduction. Fluorescence activated cell sorting (FACS) was used to isolate homogenous cell populations that exhibited comparable surface CD14 staining (Figure 1C). The resulting cells were examined for their ability to promote CD14 and TLR4 endocytosis in response to LPS. Endocytosis assays were performed using a highly sensitive and quantitative flow cytometry-based assay that tracks the endogenous receptors as they are internalized from the plasma membrane into endosomes (Akashi et al., 2003; Kagan et al., 2008; Zanoni et al., 2011).
Upon LPS stimulation, rapid CD14 endocytosis was observed in cells expressing WT CD14 and the CD14 1R mutant, whereas the 2R mutant exhibited a delayed rate of CD14 internalization (Figure 1D). The CD14 4R, which is most defective for LPS-binding activity in vitro, was unable to be internalized into cells in the presence of LPS (Figure 1D). These data indicate that LPS binding by CD14 is necessary for endocytosis.
We observed a similar requirement for LPS-binding by CD14 to promote TLR4 endocytosis, with only the LPS-binding deficient CD14 mutant being defective for TLR4 endocytosis (Figure 1E). These data establish that a physical interaction between CD14 and LPS is required to select TLR4 as cargo for inflammatory endocytosis.
CD14 is on a constitutive endocytosis pathway that is accelerated by LPS
Extensive kinetic analysis of LPS-induced CD14 endocytosis revealed that this process is rapid, as 80% of the CD14 was internalized into iBMDMs within 2 minutes of LPS treatment (Figure S1A). The internalization of TLR4 occurred within the same time frame (Figure S1B). Further monitoring of surface CD14 revealed the re-appearance of CD14 at the plasma membrane at late time points (120 min) (Figure 1D). In contrast, TLR4 never reappeared at the plasma membrane after endocytosis (Figure 1E). One possible mechanism of CD14 re-appearance is through the recycling of receptors to the cell surface after endocytosis, whereas another possibility is that newly synthesized CD14 re-populates the plasma membrane. These possibilities can be distinguished through the use of translation inhibitors, which should block new CD14 synthesis, but not affect recycling of previously internalized receptors.
WT iBMDMs were treated with the translation inhibitor cycloheximide in the presence or absence of LPS, and the endocytosis of CD14 and TLR4 was monitored. LPS treatment caused the rapid endocytosis of CD14, followed by re-appearance of this receptor 120 minutes after stimulation (Figure 2A). In contrast, LPS and cycloheximide co-administration abrogated the reappearance of CD14 at the plasma membrane, but did not prevent endocytosis (Figure 2A).
Figure 2. Constitutive endocytosis is accelerated by LPS.
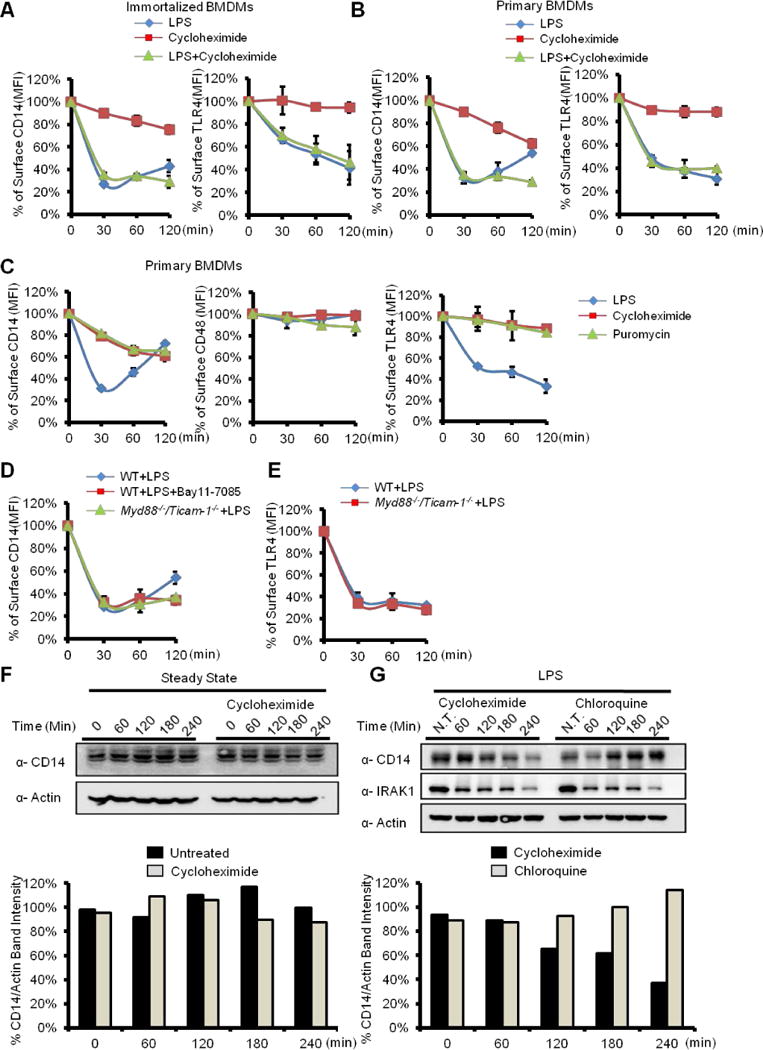
WT iBMDMs (A) and Primary BMDMs (B) were treated with LPS (1 μg/ml), cycloheximide (50 μg/ml) and LPS plus cyloheximide, as indicated. Surface level of CD14 (left) and TLR4 (right) was determined by flow cytometry.
(C) Primary BMDMs were treated with LPS (1 μg/ml), cycloheximide and puromycin (25 μg/ml) for the indicated times. Surface staining of CD14 (left), CD48 (middle), and TLR4 (right) was determined by flow cytometry.
(D) iBMDMs indicated were incubated with Bay11-7085 (5 μM) or not for 15 min, then treated with LPS (1 μg/ml) for the indicated times.
(E) WT and myd88−/−/ticam1−/− iBMDMs were treated with LPS at indicated time points. Surface level of TLR4 was determined by flow cytometry.
(F) iBMDMs were left untreated or treated with cycloheximide (50 μg/ml) for the indicated times. Cells were lysed and CD14 and Actin were detected by western blot. Densitometry analysis of the protein bands was performed using Image J (lower panel).
(G) iBMDMs were treated with cycloheximide (50μg/ML) or chloroquine (100μM) plus LPS for indicated time points. Cells were lysed and CD14, IRAK1 and Actin were detected by western blot. Densitometry analysis of the protein bands was performed using Image J (lower panel).
See also Figure S2.
Treatment of cells with cycloheximide in the absence of LPS revealed that CD14 is not immobile at the plasma membrane, but is rather undergoing a slow rate of constitutive endocytosis (Figure 2A). The constitutive rate of CD14 internalization revealed by cycloheximide was more prominent in primary cells than that observed in iBMDMs (Figure 2B). This observation is consistent with the fact that non-dividing (i.e., primary) macrophages possess higher intrinsic endocytosis activities than macrophages undergoing mitosis (i.e., iBMDM) (Berlin et al., 1978; Bonham et al., 2014). Similar results were obtained with another translation inhibitor, puromycin, where constitutive CD14 endocytosis was observed in primary BMDMs treated with this drug (Figure 2C). In contrast to the constitutive endocytosis of CD14, surface staining of TLR4 or the GPI-anchored protein CD48 did not change when protein synthesis was inhibited in iBMDMs or primary BMDMs (Figure 2A–C). These data indicate that CD14 resides specifically within a constitutive endocytosis pathway, and that new protein synthesis is required to replenish the plasma membrane with CD14. This cycling of CD14 through the cell would explain why constitutive CD14 endocytosis is only revealed by inhibition of translation.
As surface levels of CD14 in untreated cells are stable, the rate of CD14 endocytosis and resynthesis must be tightly controlled in order to maintain surface expression of this receptor. As CD14 endocytosis is accelerated by LPS, the rate of re-synthesis must also be accelerated to accommodate for the greater loss of cell surface CD14. We therefore predicted that blocking NF-κB activation should offset the balance between CD14 endocytosis and re-synthesis, and should result in the loss of surface expression of this protein. NF-κB activation was prevented through the use of Myd88−/−/Ticam-1−/− iBMDMs, or treatment of WT iBMDM with the NF-κB inhibitor Bay11-7085. Both strategies of NF-κB inhibition prevented the re-appearance of surface CD14 120 minutes after LPS treatment (Figure 2D). As expected (Zanoni et al., 2011), TLR4 endocytosis proceeded normally in Myd88−/−/Ticam-1−/− iBMDMs cells (Figure 2E). Consistent with the idea that upregulation of CD14 is responsible for the re-population of this receptor at the plasma membrane, we found that CD14 mRNA was increased upon LPS stimulation (Figure S2B), as was the inflammatory cytokine Il1b (Figure S2A). Neither Cd14 nor Il1b were expressed upon LPS stimulation of Myd88−/−/Ticam-1−/− iBMDMs (Figure S2A and B). A perfect correlation therefore exists between CD14 upregulation and the re-appearance of CD14 at the plasma membrane after LPS treatment. LPS therefore accelerates the rate of Cd14 expression in order to compensate for an accelerated rate of CD14 endocytosis.
To explain why CD14 must be re-synthesized to repopulate the plasma membrane after endocytosis, we considered that internalized CD14 is degraded in lysosomes. To address this possibility, the abundance of CD14 in cell lysates was examined. Consistent with our flow cytometry-based analysis, CD14 protein abundance remained constant in the steady state (Figure 2F). In contrast, LPS treatment in the presence of cycloheximide led to the time-dependent decrease of CD14 abundance (Figure 2G). Chloroquine treatment, which blocks lysosomal proteolysis, resulted in the accumulation of CD14 after LPS stimulation (Figure 2G). Chloroquine did not alter the LPS-induced degradation of the kinase IRAK1 (Figure 2G), which is mediated by the proteasome (Yamin and Miller, 1997). These results explain the need for CD14 re-synthesis, as the endocytosed pool of this protein is degraded in lysosomes.
Collectively, these data reveal that CD14 is present within a constitutive endocytosis pathway that is accelerated by LPS. The process of LPS-induced endocytosis acceleration results in the selection of TLR4 as cargo for entry into the cell, which effectively converts this immunologically silent entry pathway into a pathway of inflammatory endocytosis.
The cytosolic tail of TLR4 is not necessary for LPS-induced endocytosis
The observation that CD14 is present on a constitutive endocytosis pathway that is accelerated by LPS has implications for the mechanisms by which TLR4 is selected as cargo for entry into the cell. Typically, transmembrane receptors initiate the process of endocytosis de novo, by recruiting proteins to their cytosolic tails to promote their own internalization (Mellman, 1996). The fact that CD14 endocytosis occurs in resting cells suggests that the tail of TLR4 is not necessary to initiate entry events, but whether the tail of TLR4 is necessary for its own selection as cargo is unclear.
To determine if the TLR4 tail promotes endocytosis, we constructed two TLR4 alleles (Figure 3A). The first is an allele called TLR4-delta Tail, which lacks the entire cytosolic tail but contains the extracellular domain and most of the transmembrane domain (Figure 3A). For the second allele (CD4-TLR4 Cyto), the TLR4 extracellular domain was replaced with the ecto-domain of CD4, leaving the TLR4 transmembrane domain and the cytosolic tail intact (Figure 3A). As macrophages do not express CD4, it serves as a marker to monitor expression of this chimeric protein. These alleles were retrovirally transduced into Tlr4−/− iBMDMs and FACS-based isolation resulted in stable lines that permitted analysis of the domains necessary to select cargo for endocytosis. The surface expression of the TLR4 and CD4 alleles in the respective stable lines verified by flow cytometry (Figure S3A, B). Importantly, TLR4 surface staining in TLR4-delta Tail expressing cells was comparable to that of endogenous TLR4 in WT iBMDMs (Figure S3A).
Figure 3. The cytosolic tail of TLR4 is not required for endocytosis.
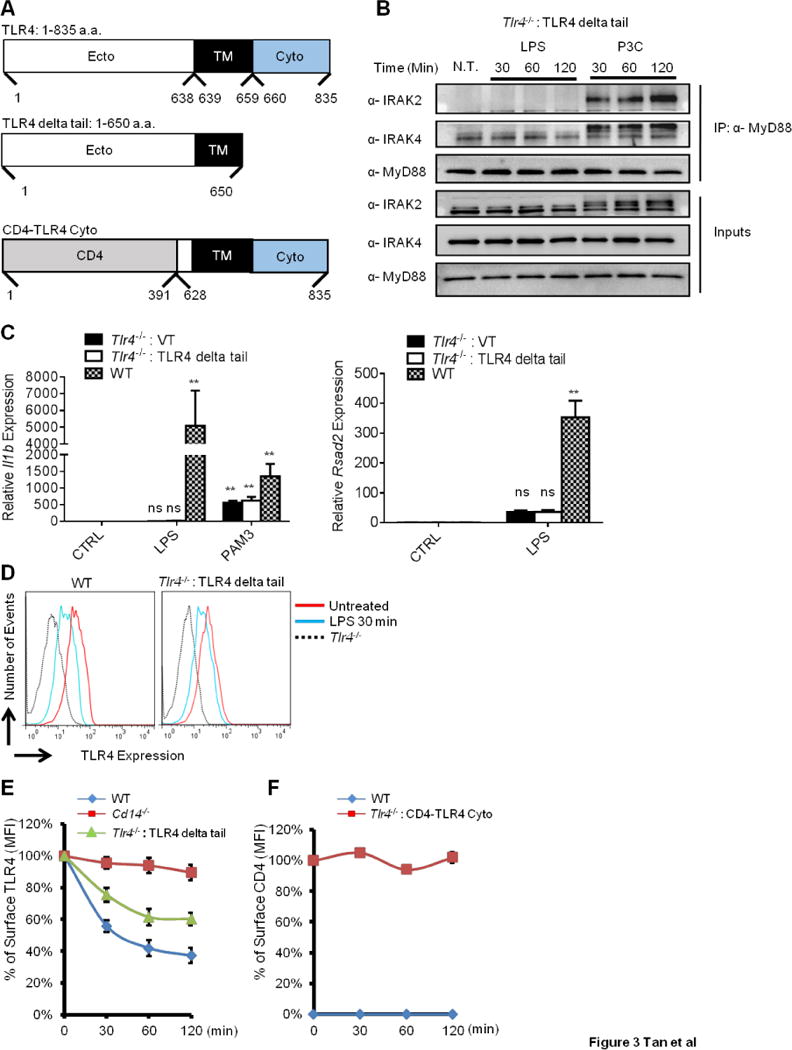
(A) Schematic of three TLR4 alleles. Amino acid designations, relative to the start codon (position 1) are indicated. Ecto: extracellular domain. TM: transmembrane domain. Cyto: cytosolic tail. CD4: extracellular domain of CD4.
(B) Cells were treated with LPS or P3C for the indicated times and lysed. MyD88 was immunoprecipitated (IP) from lysates and IRAK2 and IRAK4 were detected by western blot.
(C) Indicated iBMDM lines were treated with LPS (100 ng/ml) or P3C (100 ng/ml) for 4 hours. il1b and rsad2 expression were measured by qPCR.
(D) The iBMDMs indicated were treated with LPS (1 μg/ml) for 30 min. Surface staining of TLR4 was measured by flow cytometry.
(E) The iBMDMs indicated were treated with LPS (1 μg/ml) for the times indicated. Surface staining of TLR4 was measured by flow cytometry. Line graphs represent the MFI of TLR4 surface staining at each time point, as compared to the staining pre-stimulation.
(F) The iBMDMs indicated were treated with LPS (1 μg/ml) for the times indicated. Surface staining of CD4 was measured by flow cytometry. Line graphs represent the MFI of CD4 surface staining at each time point, as compared to the staining pre-stimulation.
Error bars represent mean SEM from triplicate readings in one experiment. **,p<0.01, ns: not significant;
See also Figure S3.
To verify the expected signaling defects of the TLR4 alleles, we used myddosome formation and Il1b expression as read-outs for signaling from the plasma membrane, and the expression of the ISG Rsad2 (Chin and Cresswell, 2001) to indicate signaling from endosomes. LPS was unable to induce myddosome assembly, Il1b or Rsad2 expression in cells expressing the TLR4-delta Tail allele (Figure 3B, C). This observation was expected since this allele lacks the TIR domain. In contrast, the TLR2 ligand Pam3CSK4 (PAM3) induced myddosome formation and Il1b expression, thus demonstrating a specific defect of TLR4-delta Tail expressing cells in responding to LPS (Figure 3B, C).
Whereas the delta Tail allele of TLR4 was completely defective for signaling, this allele retained the ability to be internalized in response to LPS. Indeed, LPS treatment resulted in the time-dependent loss of TLR4-delta Tail surface staining, albeit slightly less efficiently than that observed in WT iBMDMs (Figure 3D, E). In contrast, no endocytosis of CD4 was observed in the CD4-TLR4 Cyto expressing iBMDMs (Figure 3F). The efficiency of CD14 internalization in each cell population was similar to that observed in WT iBMDMs (Figure S3C, D). These data establish that the TLR4 cytosolic tail is neither necessary nor sufficient for its own endocytosis. Thus, unlike traditionally discussed mechanisms by which transmembrane receptors use their cytosolic tails to promote internalization, extracellular interactions likely promote the selection of TLR4 as cargo for CD14-dependent endocytosis.
The extracellular protein MD-2 is the cargo-selection agent for TLR4 endocytosis
The observation that the TLR4 ecto- and transmembrane domains are sufficient to support TLR4 endocytosis suggests that extracellular interactions are important for this process. The LPS-binding protein MD-2 was a candidate regulator of TLR4 cargo selection, as this protein interacts directly with the TLR4 ecto-domain (Shimazu et al., 1999). However, the only known function of MD-2 is to deliver LPS to TLR4, not to transport TLR4. To characterize the role of MD-2 in TLR4 endocytosis, we generated MD-2 deficient iBMDMs from the marrow of mice lacking the Ly96 gene. Consistent with prior observations using primary cells (Nagai et al., 2002), Ly96−/− iBMDMs are non-responsive to LPS, as we observed neither myddosome formation nor expression of Il1b or Rsad2 in the absence of MD-2 (Figure 4A, B). The defects in LPS signaling could be complemented by retroviral expression of an HA-tagged MD-2 in Ly96−/− iBMDMs (Figure 4A, B).
Figure 4. MD-2 selects TLR4 as cargo for LPS-induced endocytosis.
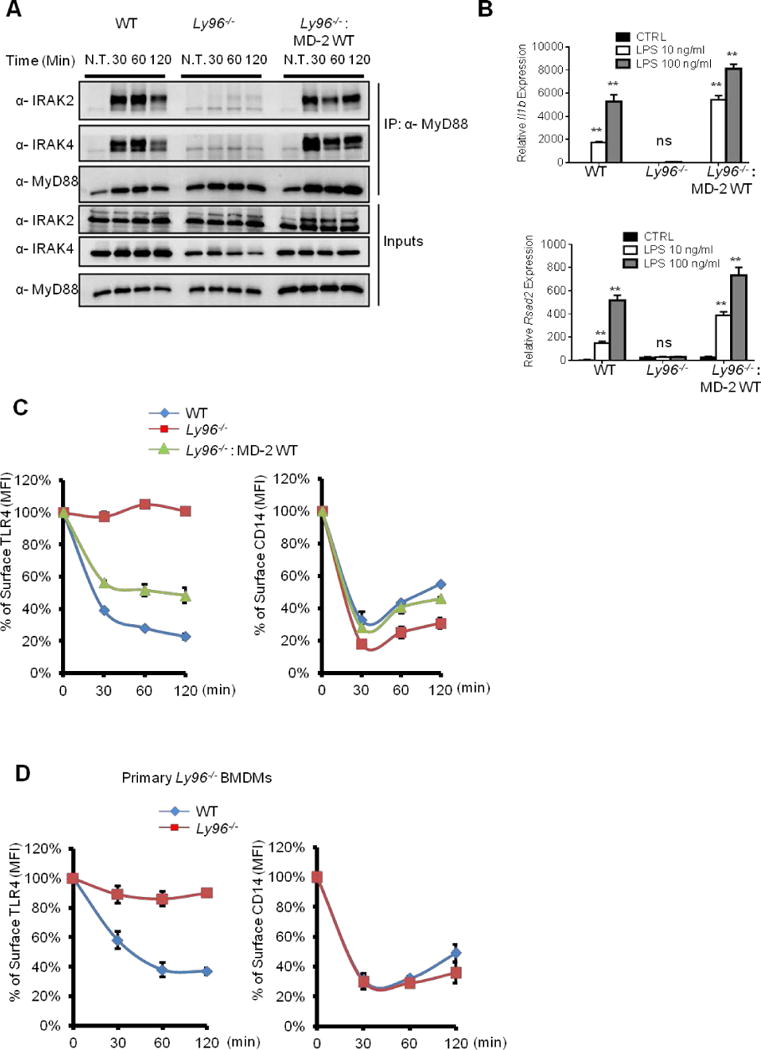
(A) The iBMDMs indicated were treated with LPS for the times indicated. Myddosome formation were then examined as described in Figure 3 (B).
(B) The iBMDMs indicated were treated with LPS at 10 ng/ml or 100 ng/ml for 4 hours. il1b and rsad2 expression was then determined by qPCR.
(C, D) iBMDMs (C) or primary BMDMs (D) indicated were treated with LPS (1 μg/ml) for the indicated times. Surface staining of CD14 and TLR4 was determined by flow cytometry. Line graphs represent the MFI of TLR4 and CD14 surface staining at given time points from each cell line.
Error bars represent mean SEM from triplicate readings in one experiment. **,p<0.01, ns: not significant.
See also Figure S5.
To examine the contribution of MD-2 to LPS-induced endocytosis, we monitored TLR4 endocytosis in Ly96−/− BMDMs. The absence of MD-2 completely abolished the internalization of TLR4 upon LPS treatment of immortal or primary Ly96−/− BMDM (Figure 4C, D). Expression of MD-2 in Ly96−/− iBMDMs restored TLR4 endocytosis (Figure 4C). Remarkably, unlike all other known regulators of TLR4 endocytosis, which are required for both CD14 and TLR4 internalization, MD-2 was not necessary for CD14 endocytosis in either primary or immortal BMDMs (Figure 4C, D). These collective data reveal that, like CD14, MD-2 has more functions than just transporting microbial ligands; this protein also transports receptors into the cell.
The mechanisms governing TLR4 cargo selection apply to all means of LPS endocytosis
LPS can exist within soluble micelles or in association with bacteria, and may enter cells through several pathways, depending on its formulation. The mechanisms of CD14 endocytosis may also be diverse, as cells defective for galectin-3, a protein that controls the internalization of several GPI-anchored proteins (Lakshminarayan et al., 2014), were not defective for CD14 or TLR4 endocytosis (Figure S5). We sought to determine if alterations in the cargo property (intact bacteria vs soluble LPS) or the route of entry (phagocytosis vs endocytosis) could influence the mechanisms of TLR4 endocytosis. iBMDMs were treated with E. coli that had been opsonized with IgG (or not) and monitored for CD14 and TLR4 endocytosis. Confocal microscopy revealed a substantial increase in uptake of opsonized E. coli into all iBMDM genotypes (WT, Cd14−/− and Ly96−/−), in comparison to their unopsonized counterparts (Figure S4A), as expected. Interestingly, the requirements of CD14 and MD-2 for TLR4 endocytosis could not be bypassed by altering the physical properties of cargo, as Cd14−/− and Ly96−/− iBMDMs were unable to internalize TLR4 under any condition examined (Figure S4B, C). CD14 internalization proceeded normally in all cells (Figure S4C). Thus, despite the fact that multiple entry routes for LPS (and perhaps CD14) exist, the means by which TLR4 is selected as cargo for endocytosis in macrophages is universal and always requires CD14 and MD-2.
Dimerization of TLR4 by MD-2 coordinates TLR4 signaling and endocytosis
MD-2 promotes TLR4 dimerization at the plasma membrane (Saitoh et al., 2004), a process necessary to initiate TLR4 signaling. As CD14-dependent endocytosis is not promoted by TLR4 signaling, it was unclear whether the ability of MD-2 to dimerize TLR4 would contribute to endocytosis. We performed mutational analysis of MD-2 to identify the mechanism by which this protein selects TLR4 as cargo for endocytosis. We focused on two MD-2 mutants. The first mutant is MD-2 C95A, which abolishes MD-2 and TLR4 interactions (Schromm et al., 2001). The second mutant is MD-2 F126A, which does not affect TLR4 binding, but impairs the LPS-induced dimerization of different TLR4/MD-2 complexes at the plasma membrane (Figure 5A) (Kawasaki et al., 2003; Schromm et al., 2001)). The behavior of these mutant MD-2 alleles (with a C-terminal HA epitope tag) was first verified biochemically in 293T cells. Compared to WT MD-2 and MD-2 F126A, TLR4 formed a weak complex with MD-2 C95A, as assessed by co-immunoprecipitation after production of each protein in 293T cells (Figure S5A). The specificity of this interaction was verified through the use of the TLR4 allele in which the ecto-domain was replaced with that of CD4. This allele of TLR4 could not form a complex with MD-2 (Figure S5A).
Figure 5. Dimerization of TLR4 by MD-2 coordinates TLR4 signaling and endocytosis.
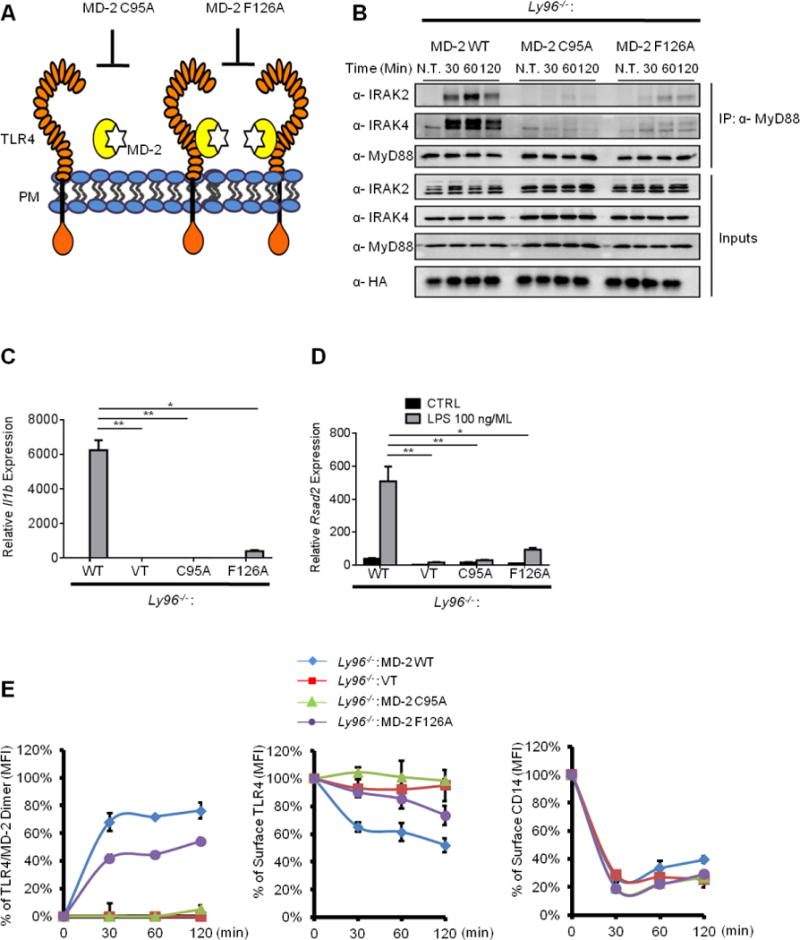
(A) Schematic of MD-2 mutants used in the study. The C95A mutation abolishes the binding between MD-2 and TLR4. The F126A mutation prevents efficient dimerization of TLR4/MD-2 complexes.
(B) iBMDMs indicated were stimulated with LPS and myddosomes were isolated from cell lysates at the times indicated.
(C, D) iBMDMs indicated were stimulated with LPS (100 ng/ml) for 4 hours, and il1b and rsad2 expression was examined RNA by qPCR.
(E) iBMDMs indicated were stimulated with LPS for times indicated. TLR4/MD-2 dimerization (left), TLR4 endocytosis (middle) and CD14 endocytosis (right) were determined by flow cytometry.
Error bars represent mean SEM from triplicate readings in one experiment. *, p<0.05, **,p<0.01, ns: not significant.
See also Figure S6.
A functional flow cytometry-based assay was also used to determine the association of these MD-2 alleles with TLR4 in living iBMDMs. We reasoned that since MD-2 is a secreted protein that binds the TLR4 ecto-domain, then the MD-2 mutant defective in TLR4 binding would be secreted directly into the cell culture medium, thereby resulting in negative surface HA staining. In contrast, MD-2 alleles that bind TLR4 should result in HA-positive cells. HA-tagged MD-2 alleles were introduced into Ly96−/− iBMDMs, and surface HA staining was examined by flow cytometry. Our prediction was correct, as Ly96−/− iBMDMs transduced with WT MD-2 and MD-2 F126 displayed positive surface HA staining (Figure S5B). In contrast, no surface HA staining could be detected from the MD-2 C95A-expressing cells (Figure S5B). Expression of WT MD-2 in tlr4−/− iBMDMs did not result in any surface HA staining (Figure S5B). Importantly, all the MD-2 mutants retained their LPS-binding capacity (Figure S5C). These MD-2 mutants were therefore used as tools to assess their influence on TLR4 signaling and endocytosis.
iBMDMs expressing various MD-2 mutants were treated with LPS, and myddosome formation, gene expression and endocytosis were assessed. Cells expressing WT MD-2 fully restored TLR4-dependent myddosome formation, Il1b and Rsad2 expression (Figure 5B–D). In contrast, Ly96−/− iBMDMs expressing MD-2 C95A or F126A remained poorly responsive to LPS, as did the cells expressing an empty retroviral vector (Figure 5B–D).
TLR4 endocytosis and TLR4/MD-2 dimerization was then assessed. Whereas TLR4 endocytosis was examined with the Sa15-21 antibody used throughout this study, TLR4 dimerization was assessed using the MTS510 antibody, which detects only TLR4 monomers (Akashi et al., 2000). Because this antibody recognizes monomeric TLR4/MD2 at the plasma membrane, loss of surface staining of MTS510 represents receptor dimerization (or clustering) induced by LPS (Akashi et al., 2000). Consistent with this idea, LPS induced the rapid dimerization of TLR4 at the plasma membrane in cells expressing WT MD-2 (Figure 5E). No change in dimerization was observed in cells expressing the MD-2 C95A mutant that cannot bind TLR4, or in cells harboring the empty vector (Figure 5E). Cells expressing MD-2 F126A displayed a modest increase in TLR4 dimers in the presence of LPS (Figure 5E). These results are consistent with residue F126 being required to efficiently dimerize TLR4. Remarkably, the exact same trend of cellular behaviors was observed when the LPS-induced endocytosis of TLR4 was examined. TLR4 was internalized rapidly into cells expressing WT MD-2, whereas no TLR4 endocytosis was observed in cells carrying the empty vector or expressing MD-2 C95A (Figure 5E). Cells expressing the dimerization-deficient F126A mutant exhibited a substantial defect in TLR4 endocytosis (Figure 5E). Indeed, at 30 minutes post-LPS treatment, cells expressing this allele were as defective as Ly96−/− cells at inducing TLR4 endocytosis. None of the MD-2 mutants alter the efficiency of CD14 endocytosis (Figure 5E). Collectively, these data reveal that, in addition to promoting transcriptional responses, TLR4 dimerization by MD-2 promotes the selection of TLR4 as cargo for endocytosis.
A therapeutic LPS variant prevents the selection of TLR4 as cargo for inflammatory endocytosis
LPS variants that contain less than 6 acyl chains are weak inducers of TLR4 dimerization (Tan and Kagan, 2014), and may therefore be less capable of promoting TLR endocytosis. To address this possibility, we compared penta-acylated LPS from Rhodobacter sphaeroides (Rs-LPS) (Strittmatter et al., 1983; Visintin et al., 2005) to its hexa-acylated counterpart from E. coli (Ec-LPS). Ec-LPS triggered robust myddosome formation, Il1b and Rsad2 expression, whereas Rs-LPS did not elicit any of these responses (Figure 6A, B). Rs-LPS was also capable of only modest dimerization of TLR4, as compared to Ec-LPS (Figure 6C). Consequently, TLR4 endocytosis proceeded very inefficiently in cells treated with Rs-LPS (Figure 6C). These data are consistent with the idea that dimerization of TLR4 is necessary for its selection as endocytosis cargo (Figure 5E). Interestingly, despite its defective ability to crosslink or activate TLR4, Rs-LPS accelerated CD14 endocytosis as efficiently as Ec-LPS (Figure 6C), indicating that CD14 does not discriminate between Rs-LPS and Ec-LPS (Delude et al., 1995). These results therefore reveal that CD14 and TLR4 endocytosis can be uncoupled in WT cells, which creates an inducible CD14-deficiency at the plasma membrane.
Figure 6. An LPS variant dissociates CD14 and TLR4 endocytosis.
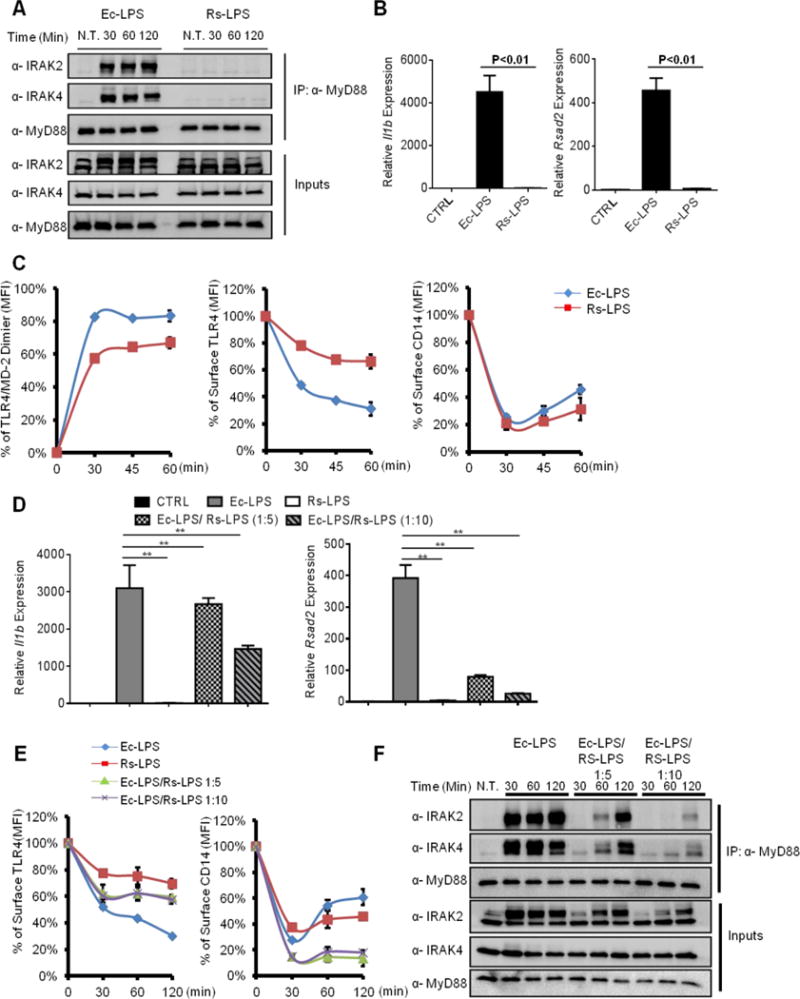
(A) iBMDMs were stimulated with E. coli LPS (Ec-LPS, 100 ng/ml) and R. sphaeroides LPS (Rs-LPS, 100 ng/ml) for indicated times. The assembly of myddosome was then examined by western blot.
(B) iBMDMs were treated with Ec-LPS (100 ng/ml) or Rs-LPS (100 ng/ml) for 4 hours, il1b and rsad2 expression were measured by qPCR.
(C) iBMDMs were stimulated with LPS for times indicated. TLR4/MD-2 dimerization (left), TLR4 endocytosis (middle) and CD14 endocytosis (right) were determined by flow cytometry.
(D–F) iBMDMs were stimulated with Ec-LPS (100 ng/ml) and mixtures of Ec-LPS and Rs-LPS at the ratio of 1:5 or 1:10 (100 ng/ml Ec-LPS plus 500 ng/ml Rs-LPS or 100 ng/ml Ec-LPS plus 1000 ng/ml Rs-LPS) for 4 hours. il1b and rsad2 expression was then measured by qPCR.
(E, F) Similar treatments as D, except surface TLR4 (left) and CD14 (right) staining was determined by flow cytometry, or myddosome assembly was examined.
Error bars represent mean SEM from triplicate readings in one experiment. **,p<0.01.
The ability of Rs-LPS to create a CD14-deficiency at the plasma membrane would be expected to render this LPS variant a TLR4 antagonist. Indeed, Rs-LPS is well-known to have inhibitory actions towards TLR4 (Mullarkey et al., 2003). We reasoned that if the CD14-deficiency induced by Rs-LPS is functionally important, then TLR4 responses most dependent on CD14 should be most inhibited by Rs-LPS. Of the known TLR4-dependent transcriptional responses, TRIF-dependent gene expression is more sensitive to the loss of CD14 than MyD88-dependent gene expression (Perera et al., 1997). Consistent with this idea, co-administration of Ec-LPS and Rs-LPS resulted in a near complete loss of TRIF-dependent Rsad2 expression, whereas MyD88-dependent Il1b expression was only modestly affected (Figure 6D). This strong defect in TRIF-dependent signaling correlated with a defect in TLR4 endocytosis (Figure 6E). Interestingly, whereas Rs-LPS antagonized TLR4 endocytosis induced by Ec-LPS, co-administration of these LPS variants potentiated CD14 endocytosis (Figure 6E). This observation supports the idea that CD14 does not differentiate Ec-LPS and Rs-LPS. Rs-LPS also prevented myddosome formation, but not to an extent that MyD88 signaling was ablated (Figure 6D, F). Thus, uncoupling of CD14 and TLR4 endocytosis can occur in WT cells and results in a functional CD14 deficiency that results in severe defects in TLR4-TRIF signal transduction.
A common strategy is used by pathogenic and commensal bacteria of the human intestine to evade CD14-dependent inflammatory endocytosis
R. sphaeroides does not naturally interact with a mammalian host. We therefore sought to examine the role of LPS variation in bacteria that naturally colonize mammals. We first chose LPS from Francisella tularensis, because of its low immuno-stimulatory effect (Gunn and Ernst, 2007). Key to the immune-evasion features of F.tularensis LPS (Ft-LPS) is the modifications on Lipid A, which renders it hypo-phosphorylated, hypo-acylated and possessing longer acyl chains in comparison to the Ec-LPS (Vinogradov et al., 2002). We treated WT iBMDMs with Ec-LPS and Ft-LPS. As compared to Ec-LPS treatment, which induced myddosome formation, Il1b and Rsad2 expression, Ft-LPS did not induce these responses (Figure 7A and B). Because of the under-acylated nature of Ft-LPS, we expected that Ft-LPS would behave similarly to Rs-LPS, and promote the endocytosis of CD14, but not TLR4. However, we found that Ft-LPS did not induce the internalization of CD14 or TLR4 (Figure 7C). Moreover, Ft-LPS was unable to induce any detectable dimerization of TLR4/MD-2 (Figure 7C). Thus, unlike Rs-LPS, Ft-LPS does not uncouple CD14 and TLR4 endocytosis, but rather evades detection by CD14 entirely.
Figure 7. LPS from pathogenic and commensal bacteria evade detection by CD14 and TLR4.
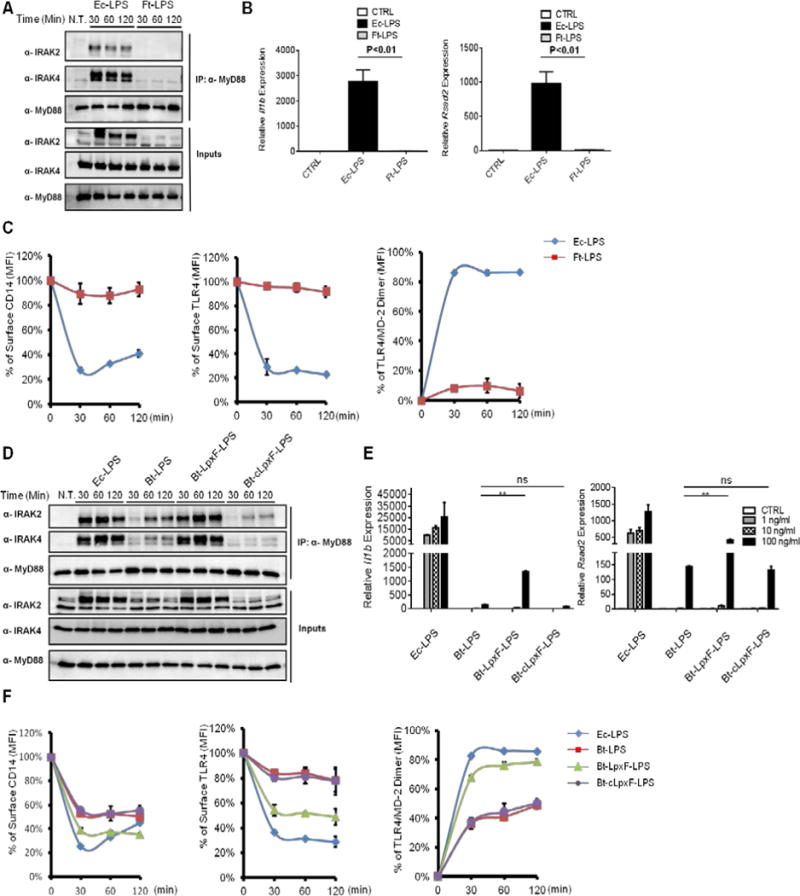
(A) iBMDMs were treated with Ec-LPS or Ft-LPS (1 μg/ml) for the indicated times, and the assembly of myddosome was examined as described in Figure 3B.
(B) iBMDMs were treated with Ec-LPS (100 ng/ml) or Ft-LPS (100 ng/ml) for 4 hours, and the expression of Il-1 beta (left) and rsad2 (right) was measured by qPCR.
(C–D) WT iBMDMs were stimulated with Ec-LPS or Ft-LPS (1 μg/ml) for the times indicated. CD14 endocytosis (left), TLR4 endocytosis (middle) and TLR4/MD-2 dimerization (right) were determined by flow cytometry (C) or myddosome was examined (D).
(E) Cells were treated with Ec-LPS and Bt-LPS species at the concentration of 1 ng/ml, 10ng/ml and 100 ng/ml for 4 hours, il1b and rsad2 expression was measured by qPCR.
(F) iBMDMs were stimulated with the LPS variants indicated (1 μg/ml). At the indicated times, CD14 endocytosis (left), TLR4 endocytosis (middle) and TLR4/MD-2 dimerization (right) were determined by flow cytometry.
Error bars represent mean SEM from triplicate readings in one experiment. **,p<0.01.
To determine if the ability to evade CD14 was a feature common to other bacteria that colonize mammals, we considered commensal bacteria of the human intestine. Like bacterial pathogens, commensal bacteria have co-evolved with the host immune system. Bacteroides, the most prominent genus in the distal human intestine, produce LPS with penta-acylated and monophosphorylated Lipid A (Xu et al., 2003). Bacteroides encode the LpxF phosphatase, which is responsible for producing monophosphorylated Lipid A (Cullen et al., 2015). To determine if the phosphorylation state of B. thetaiotaomicron LPS (Bt-LPS) influences detection by CD14 and TLR4/MD-2, we treated iBMDMs with LPS purified from three B. thetaiotaomicron strains (the wild type strain, an lpxF mutant, and a complemented strain). Monophosphorylated Bt-LPS induced delayed and weak myddosome formation (Figure 7D), and weak Il1b and Rsad2 expression, as compared to Ec-LPS. Interestingly, addition of a single phosphate group to Bt-LPS, through the use of the lpxF mutant, was sufficient to promote myddosome assembly and high levels of Il1b and Rsad2 expression, as compared to WT Bt-LPS (Figure 7D, E). Indeed, 100 ng/ml Bt-lpxF-LPS treatment induced Il1b expression to levels comparable to that of Ec-LPS (Figure 7E). Importantly, all the phenotypes associated with Bt-lpxF-LPS were reversed when using LPS derived from the complemented strain of bacteria (Bt-clpxF-LPS), thus indicating that the phosphorylation state of Bt-LPS is responsible for evasion of myddosome formation and gene expression.
Finally, we determined the effect of Bt-LPS on receptor trafficking. Interestingly. monophosphorylated Bt-LPS was less able to promote CD14 endocytosis than its diphosphorylated counterparts. Indeed, diphosphorylated Bt-LPS induced CD14 endocytosis to an extent comparable to that induced by Ec-LPS (Figure 7F). The ability to evade CD14, would be predicted to allow monophosphorylated Bt-LPS to evade all downstream events, such as TLR4 dimerization and endocytosis. This prediction was correct, as monophosphorylated Bt-LPS triggered less efficient TLR4/MD-2 dimerization and TLR4 endocytosis, as compared to their diphosphorylated counterparts, or Ec-LPS (Figure 7F). These collective data reveal a common feature of commensal and pathogenic LPS: both can evade CD14 endocytosis, perhaps by generating monophosphorylated LPS. The ability to evade CD14-dependent activities may therefore represent a common feature of bacteria that inhabit mammalian hosts.
Discussion
Emerging evidence indicates that ligand-induced TLR trafficking events can promote specific innate immune responses. The discovery of a requirement for TLR4 endocytosis to induce TRIF-dependent gene expression exemplifies the importance of these newly defined microbe-inducible transport events. While CD14 represents the first specific regulator controlling the transport of TLR4 to endosomes, the mechanism of how TLR4 is selected as endocytosis cargo is unknown. Our discovery that MD-2 is the cargo-selection agent for TLR4 endocytosis revealed a mechanism by which extracellular interactions play a determinant role in the entry process into a cell. This mechanism is highly unusual, as most other transmembrane proteins direct themselves into endosomes through interactions between their cytosolic tail and various endocytosis regulators (McMahon and Boucrot, 2011). In contrast, TLR4 alleles that contain no cytosolic tail retain the ability to be internalized by LPS treatment. We do note, however, that while the TLR4 tail is not necessary for endocytosis, our findings do not negate a role for the tail in post-endocytosis sorting of TLR4 into the lumen of multivesicular bodies (Chuang and Ulevitch, 2004; Husebye et al., 2006), which is critical for signaling downregulation.
Our analysis of the mechanism by which MD-2 promotes endocytosis revealed that direct interactions with TLR4 are not sufficient. Rather, MD-2 must dimerize TLR4 before endocytosis can be achieved. This conclusion is supported by our analysis of both MD-2 mutants that are defective for TLR4 crosslinking activity and analysis of hypo-acylated LPS variants that cannot efficiently crosslink TLR4. This finding is notable, as TLR4 dimerization has most often been considered a mechanism of promoting TLR4 signal transduction. The fact that endocytosis is not mediated by TLR4 signaling indicates that TLR4 crosslinking by MD-2 is an activity that coordinates the signaling functions of TLR4 with the endocytosis functions of CD14. Of note, in BMDMs, the cargo selection function of MD-2 for TLR4 endocytosis could not be circumvented by altering the means of uptake (e.g. Fc receptor-mediated phagocytosis). These results support a model whereby CD14 is constantly cycling through the plasma membrane, surveying the extracellular environment for most variants of LPS. Upon binding to hexa-acylated LPS, CD14 transfers LPS to MD-2, which then dimerizes TLR4. Dimerized TLR4 induces myddosome assembly and signaling at the plasma membrane, and converts the immunologically silent entry route taken by CD14 into an inflammatory endocytosis pathway.
Although host transcriptional responses are established to be influenced by LPS variation, limited information exists of how LPS variation affects receptor-proximal events. Our observations with LPS variants from the therapeutically interesting R.sphaeroides and the pathogenic F. tularensis revealed two mechanisms by which CD14-dependent effects on TLR4 signaling can be disrupted. Whereas R.sphaeroides LPS uncouples CD14 endocytosis from TLR4 endocytosis, resulting in a functional CD14 deficiency at the plasma membrane, the other bacterial LPS variants evade detection by CD14 completely. Remarkably, our analysis of B. thetaiotaomicron revealed that this bacterium behaves similarly to pathogens, in that B.thetaiotaomicron LPS evades CD14-dependent events. This discovery is notable when considering that the LPS structure of B. thetaiotaomicron is conserved across the genus Bacteroides, which is one of the most dominant taxonomic groups in all mammals. We therefore propose that the tendency to alter LPS to evade CD14 likely represents an adaptation strategy that transcends the concept of pathogenesis, and includes any type of persistent biological interaction between microbe and host.
Currently, the structural mechanism for how CD14 recognizes LPS is elusive, as the published CD14 structures are in their ligand-free conformations (Kelley et al., 2013; Kim et al., 2005). Despite this gap in our knowledge, it is clear from our observations that host-adapted pathogens and commensals are able to avoid detection by CD14 via LPS modifications, most notably, dephosphorylation of their LPS. Interestingly, LPS monophosphorylation is not only a mechanism of CD14 evasion, but is also a mechanism by which B.thetaiotaomicron evades the killing activities of anti-microbial peptides (AMPs). This finding reveals symmetry between the mechanisms of action of the antimicrobial effectors of the innate immune system (AMPs) and the receptors that detect those microbes (PRRs).
Finally, the common activities of CD14 and MD-2 in transporting ligands and receptors reminiscent of the functions of taxi motorcars in human society, which transport items and individuals to pre-determined locations for a purpose. This similarity prompts us to classify CD14 and MD-2 as Transporters Associated with the eXecution of Inflammation, or “TAXI” proteins, which transport various items throughout the cell for the purpose of initiating an inflammatory signaling cascade. While it remains to be determined if other accessory proteins involved in microbial ligand transport have the receptor-trafficking functions of TAXI proteins, our studies provide a mandate for elucidating these additional cell biological activities that control our interactions with the microbial world.
Experimental Procedures
Cell lines, transfection and retroviral transduction
iBMDMs were cultured in complete DMEM (GIBCO) with 10% FBS and antibiotics. Primary BMDMs were cultured in complete RPMI with 15% FBS, 30% L929 conditioned supernatant and antibiotics. The J2 retrovirus was used to immortalize primary BMDMs as described (Blasi et al., 1985). For generating iBMDM stable lines, retroviruses expressing the indicated alleles were produced as described (Bonham et al., 2014). Transduced cells were subjected to FACS to isolate stable lines at least 85% positive for the transgenes of interest. Transient transfections were performed with the Fugene reagent following the manufacturer’s instructions.
Antibodies and Reagents
E. coli LPS was supplied by Enzo (ALX-581-012-L002); Pam3CSK4 from Invivogen (tlrl-pms); Rs-LPS from Invivogen (tlrl-prslps). Anti-MyD88 (R&D; AF3109), anti-IRAK2 (Prosci; 3595), anti-Actin (Sigma; A 5441) and anti-CD14 (R&D; AF982), were supplied by the companies indicated. Anti-IRAK4 antibody was a gift from Shizuo Akira. The following fluorophore-conjugated antibodies were used: PE anti-TLR4 (Biolegend; clone Sa15-21; 145404), APC anti-TLR4 (Biolegend; clone Sa15-21; 145406), PE/Cy7 anti-TLR4/MD2 (Biolegend; clone MTS510; 117610), PE anti-CD48 (Biolegend; clone HM48-1; 103406), FITC anti-CD14 (ebioscience; clone Sa2-8, 11-0141), APC anti-CD14 (ebioscience; clone Sa-28, 17-0141), APC anti-hCD2 (Biolegend; clone RPA-2.10; 300214), PE anti-CD4 (Biolegend; clone GK1.5;100408), APC anti-HA (Miltenyi; 130-098-404). Anti-E. coli (ab25823), anti-EEA1 (CST, 3288S). Donkey anti-Rabbit IgG (H+L) Alexa Fluor® 568 conjugate (Lifetechologies, A10042). Cycloheximide (C1988), puromycin (P9620) and Bay11-7085 (B5681) were purchased from Sigma.
cDNAs encoding full length murine CD14, MD-2 and TLR4 were purchased from Open Biosystems (GE Dharmacon). In particular, the NotI site in the CD14 DNA sequence was abolished by site-directed mutagenesis, which introduced a neutral mutation in the nucleotides without altering amino acid composition. For expression in mammalian cells, indicated cDNAs were cloned into pcDNA3.1, pMSCV2.2-IRES-GFP (TLR4 and MD-2 alleles) or pMSCV2.2-IRES-hCD2 (CD14 alleles).
qPCR probes were purchased from Life technologies as the following: Gapdh (MM99999915_G1), Il1b (MM99999061_MH), Rsad2 (MM00491265_M1) Cd14 (Mm00438094_g1).
Myddosome formation
Myddosome formation assays were performed as described (Bonham et al., 2014)
mRNA isolation and real-time qPCR
Total RNA was isolated from samples using GeneJet RNA purification kit (Thermo; K0732). Gene expression analyses were performed using the Taqman one-step qPCR reagents (Life technologies; 4392938) with indicted probes following the manufacturer’s instructions. Gene induction fold changes were normalized to GAPDH, shown as mean and SD of three technical replicates. All qPCR graphs were representative data from at least three independent experiments.
Flow cytometry
iBMDMs, primary BMDMs (0.5×106) of the indicated genotypes were treated as indicated at 37 °C. Cells were then washed with 1 ml cold PBS and stained for appropriate antibodies on ice in the cold room for 20 to 30 minutes. 2% mouse serum or rat serum were used as the blocking reagent to reduce non-specific binding of the antibodies. The stained cells were then wash with 1ml cold PBS and resuspended in 200 μL PBS. Staining of the surface receptors was analyzed with BD FACSCanto II. The mean fluorescence intensity (MFI) of CD14, TLR4 from unstimulated or stimulated cells were recorded. The percentage of surface receptor staining at indicated time points, which is the ratio of the MFI values measured from the stimulated cells to those measured from the unstimulated cells, was plotted to reflect the efficiency of receptor endocytosis. For measuring the extent of TLR4/MD-2 dimerization, the percentage of TLR4/MD-2 dimer was calculated by 100%-the percentage of TLR4/MD-2 monomer. The percentage of the TLR4/MD-2 monomer was determined by the ratio of the MFI values (obtained from MTS510 antibody staining) of the stimulated cells to those of the unstimulated cells. Flow cytometry graphs shown in the results section were representative data from at least three independent experiments.
E. coli opsonization and infection
Overnight E. coli culture was harvested by centrifugation, and washed with PBS twice. 1 OD of bacteria were resuspended in 1 ml PBS and the E. coli-specific antibody was added at a dilution factor of 1:50. The bacteria-antibody mixture was incubated at 37 °C for 1hr, unbound antibody was rinsed off by 2× PBS wash. The opsonized bacteria were resuspended in warm PBS and used to infect iBMDMs.
iBMDMs grew on coverslips (in a 24 well plate) were infected with E. coli at an MOI of 5. After adding bacteria, the plate was centrifuged at 1500 rpm for 5 min to synchronize infection. Bacterial uptake by iBMDMs were allowed for 5 min, and unbound bacteria were rinsed off by 3× washing with pre-warmed PBS. Cells were then fixed with 4% paraformaldehyde and proceed for immunofluorescence staining.
Cells were fixed at room temperature for 30 min, permeabilized with 0.2% Triton X-100 for 5 min at room temperature and permeabilized with 2% goat serum in PBS supplemented with 50 mM ammonium chloride. Primary and secondary antibody staining were performed following product instructions. Cells were imaged by confocal microscopy.
Anaerobic culturing
Bacteroides thetaiotaomicron VPI-5482 (ATCC 29184) strains (Cullen et al., 2015) were cultured anaerobically at 37°C in liquid TYG medium (Holdeman, 1977) in a flexible anaerobic chamber (Coy Laboratory Products) containing 20% CO2, 10%H2, and 70% N2. Stationary-phase cultures were pelleted (8,000 × g, 10 minutes), resuspended in phosphate-buffered saline at an OD600 of 1.0 prior to use.
Statistical Analysis and experimental repeats
Means were compared by t-tests (two groups) or one-way ANOVA (three or more groups). Data are expressed and plotted as means ± SEM values.
All protein blots shown were representative data from at least three independent experiments. All FACS experiments were performed three times and one representative result out of three is presented. All qPCR data were representative data from at least three independent experiments.
Supplementary Material
Acknowledgments
We would like to thank the members of the Kagan lab for helpful discussions. We also appreciate Noreen Francis from the BCH IDDRC – Flow Cytometry Facility for FACS assistance, and Stefanie Vogel for providing purified F. tularensis LVS LPS. This work was supported by NIH grants AI093589 and AI113141 and an unrestricted gift from Mead Johnson & Company to J.C.K. and NIH grants GM103574 and GM105456 to A.L.G. J.C.K. and A.L.G. hold Investigators in the Pathogenesis of Infectious Disease Awards from the Burroughs Wellcome Fund. Y.T. is supported by a postdoctoral fellowship from the Jane Coffin Childs Memorial Fund for Medical Research (the Merck Fellowship).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akashi S, Saitoh S, Wakabayashi Y, Kikuchi T, Takamura N, Nagai Y, Kusumoto Y, Fukase K, Kusumoto S, Adachi Y, et al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4-MD-2: higher affinity than that with MD-2 or CD14. J Exp Med. 2003;198:1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, Miyake K. Cutting edge: cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol. 2000;164:3471–3475. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Berlin RD, Oliver JM, Walter RJ. Surface functions during Mitosis I: phagocytosis, pinocytosis and mobility of surface-bound Con A. Cell. 1978;15:327–341. doi: 10.1016/0092-8674(78)90002-8. [DOI] [PubMed] [Google Scholar]
- Blasi E, Mathieson BJ, Varesio L, Cleveland JL, Borchert PA, Rapp UR. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature. 1985;318:667–670. doi: 10.1038/318667a0. [DOI] [PubMed] [Google Scholar]
- Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, Iwasaki A, Knipe DM, Kagan JC. A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell. 2014;156:705–716. doi: 10.1016/j.cell.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CY, Veckman V, Limmer K, David M. Phospholipase Cgamma-2 and intracellular calcium are required for lipopolysaccharide-induced Toll-like receptor 4 (TLR4) endocytosis and interferon regulatory factor 3 (IRF3) activation. J Biol Chem. 2012;287:3704–3709. doi: 10.1074/jbc.C111.328559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin KC, Cresswell P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc Natl Acad Sci U S A. 2001;98:15125–15130. doi: 10.1073/pnas.011593298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol. 2004;5:495–502. doi: 10.1038/ni1066. [DOI] [PubMed] [Google Scholar]
- Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VL, DeFranco AL. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science. 2015;347:170–175. doi: 10.1126/science.1260580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delude RL, Savedra R, Jr, Zhao H, Thieringer R, Yamamoto S, Fenton MJ, Golenbock DT. CD14 enhances cellular responses to endotoxin without imparting ligand-specific recognition. Proc Natl Acad Sci U S A. 1995;92:9288–9292. doi: 10.1073/pnas.92.20.9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioannini TL, Teghanemt A, Zhang D, Coussens NP, Dockstader W, Ramaswamy S, Weiss JP. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc Natl Acad Sci U S A. 2004;101:4186–4191. doi: 10.1073/pnas.0306906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioannini TL, Weiss JP. Regulation of interactions of Gram-negative bacterial endotoxins with mammalian cells. Immunol Res. 2007;39:249–260. doi: 10.1007/s12026-007-0069-0. [DOI] [PubMed] [Google Scholar]
- Gunn JS, Ernst RK. The structure and function of Francisella lipopolysaccharide. Ann N Y Acad Sci. 2007;1105:202–218. doi: 10.1196/annals.1409.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdeman LV, Cato ED, Moore WEC. Anaerobe Laboratory Manual. Blacksburg, VA: Virginia Polytechnic Institute and State University Anaerobe Laboratory; 1977. [Google Scholar]
- Husebye H, Halaas O, Stenmark H, Tunheim G, Sandanger O, Bogen B, Brech A, Latz E, Espevik T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25:683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Barton GM. Emerging Principles Governing Signal Transduction by Pattern-Recognition Receptors. Cold Spring Harb Perspect Biol. 2014;7 doi: 10.1101/cshperspect.a016253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol. 2014;14:821–826. doi: 10.1038/nri3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki K, Nogawa H, Nishijima M. Identification of mouse MD-2 residues important for forming the cell surface TLR4-MD-2 complex recognized by anti-TLR4-MD-2 antibodies, and for conferring LPS and taxol responsiveness on mouse TLR4 by alanine-scanning mutagenesis. J Immunol. 2003;170:413–420. doi: 10.4049/jimmunol.170.1.413. [DOI] [PubMed] [Google Scholar]
- Kelley SL, Lukk T, Nair SK, Tapping RI. The crystal structure of human soluble CD14 reveals a bent solenoid with a hydrophobic amino-terminal pocket. J Immunol. 2013;190:1304–1311. doi: 10.4049/jimmunol.1202446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Lee CJ, Jin MS, Lee CH, Paik SG, Lee H, Lee JO. Crystal structure of CD14 and its implications for lipopolysaccharide signaling. The Journal of biological chemistry. 2005;280:11347–11351. doi: 10.1074/jbc.M414607200. [DOI] [PubMed] [Google Scholar]
- Lakshminarayan R, Wunder C, Becken U, Howes MT, Benzing C, Arumugam S, Sales S, Ariotti N, Chambon V, Lamaze C, et al. Galectin-3 drives glycosphingolipid-dependent biogenesis of clathrin-independent carriers. Nat Cell Biol. 2014;16:595–606. doi: 10.1038/ncb2970. [DOI] [PubMed] [Google Scholar]
- Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol. 2012;12:168–179. doi: 10.1038/nri3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Mullarkey M, Rose JR, Bristol J, Kawata T, Kimura A, Kobayashi S, Przetak M, Chow J, Gusovsky F, Christ WJ, et al. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther. 2003;304:1093–1102. doi: 10.1124/jpet.102.044487. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- Perera PY, Vogel SN, Detore GR, Haziot A, Goyert SM. CD14-dependent and CD14-independent signaling pathways in murine macrophages from normal and CD14 knockout mice stimulated with lipopolysaccharide or taxol. J Immunol. 1997;158:4422–4429. [PubMed] [Google Scholar]
- Saitoh S, Akashi S, Yamada T, Tanimura N, Matsumoto F, Fukase K, Kusumoto S, Kosugi A, Miyake K. Ligand-dependent Toll-like receptor 4 (TLR4)-oligomerization is directly linked with TLR4-signaling. J Endotoxin Res. 2004;10:257–260. doi: 10.1179/096805104225005904. [DOI] [PubMed] [Google Scholar]
- Schromm AB, Lien E, Henneke P, Chow JC, Yoshimura A, Heine H, Latz E, Monks BG, Schwartz DA, Miyake K, et al. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: a point mutation in a conserved region of MD-2 abolishes endotoxin-induced signaling. J Exp Med. 2001;194:79–88. doi: 10.1084/jem.194.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelter F, Bernheiden M, Menzel R, Jack RS, Witt S, Fan X, Pfister M, Schutt C. Mutation of amino acids 39–44 of human CD14 abrogates binding of lipopolysaccharide and Escherichia coli. Eur J Biochem. 1997;243:100–109. doi: 10.1111/j.1432-1033.1997.00100.x. [DOI] [PubMed] [Google Scholar]
- Strittmatter W, Weckesser J, Salimath PV, Galanos C. Nontoxic lipopolysaccharide from Rhodopseudomonas sphaeroides ATCC 17023. J Bacteriol. 1983;155:153–158. doi: 10.1128/jb.155.1.153-158.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Kagan JC. A cross-disciplinary perspective on the innate immune responses to bacterial lipopolysaccharide. Mol Cell. 2014;54:212–223. doi: 10.1016/j.molcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. 2005;106:2543–2550. doi: 10.1182/blood-2005-03-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov E, Perry MB, Conlan JW. Structural analysis of Francisella tularensis lipopolysaccharide. Eur J Biochem. 2002;269:6112–6118. doi: 10.1046/j.1432-1033.2002.03321.x. [DOI] [PubMed] [Google Scholar]
- Visintin A, Halmen KA, Latz E, Monks BG, Golenbock DT. Pharmacological inhibition of endotoxin responses is achieved by targeting the TLR4 coreceptor, MD-2. J Immunol. 2005;175:6465–6472. doi: 10.4049/jimmunol.175.10.6465. [DOI] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yamin TT, Miller DK. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J Biol Chem. 1997;272:21540–21547. doi: 10.1074/jbc.272.34.21540. [DOI] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.