

Abstract
Ilex latifolia Thunb. (Aquifoliaceae), a Chinese bitter tea called “kudingcha,” has been widely consumed as a health beverage and found to possess antioxidant, antidiabetic, antihypertensive, anti-inflammatory, and anti-ischemic activities. The aim of the present study was to investigate the neuroprotective effects of an ethanol extract of I. latifolia against amyloid β protein (Aβ)-induced memory impairment in mice and neurotoxicity in cultured rat cortical neurons. Memory impairment in mice was induced by intracerebroventricular injection of 15 nmol Aβ (25–35) and measured by the passive avoidance test and Morris water maze test. Chronic administration of I. latifolia (25–100 mg/kg, p.o.) significantly prevented Aβ (25–35)-induced memory loss. I. latifolia also prevented the decrease of glutathione concentrations, increased lipid peroxidation, expression of phosphorylated tau (p-tau), and changes in apoptosis-associated proteins in the memory-impaired mouse brain. Exposure of cultured cortical neurons to 10 μM Aβ (25–35) for 36 h induced neuronal apoptotic death. The neuronal cell death, elevation of intracellular Ca2+ concentration, generation of reactive oxygen species, and expression of proapoptotic proteins caused by Aβ (25–35) in the cultured neurons were inhibited by treatment with I. latifolia (1–50 μg/mL). These results suggest that I. latifolia may have a possible therapeutic role in managing cognitive impairment associated with Alzheimer's disease. The underlying mechanism might involve the antiapoptotic effects mediated by antioxidant activity and inhibition of p-tau formation.
Key Words: : Alzheimer's disease, antioxidant, Ca2+ signaling, cognition, neuroprotection
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder defined by a progressive loss of cognitive ability that correlates with various neuropathological changes in the brain. These changes include the presence of extracellular neuritic senile plaques composed of amyloid β protein (Aβ), intracellular neurofibrillary tangles containing phosphorylated tau protein (p-tau), and the loss of basal forebrain cholinergic neurons.1 Aβ, a 39–43 amino acid proteolytic fragment of the amyloid precursor protein, is the major constituent of senile plaques.2 Aβ inserted into the neuronal membrane bilayer generates reactive oxygen species (ROS) and decreases the glutathione (GSH) level as well as superoxide dismutase activity that leads to lipid and protein peroxidation of neurons.3,4 The oxidative stress leads to cellular dysfunction, such as loss of Ca2+ homeostasis, disruption of signaling pathways, and activation of nuclear transcription factors and apoptotic pathways. These disorders are the ultimate basis for apoptotic neuronal death.5
Previous studies have suggested that misfolded tau is a key pathological agent for AD. Tau is normally a soluble protein that stabilizes microtubules within cells; neurons are particularly rich in tau proteins.6 Neurodegeneration is caused by the formation of insoluble tau aggregates, referred to as neurofibrillary tangles, through hyperphosphorylation of the protein.7,8 There is evidence that Aβ can stimulate tau aggregation, which suggests that tau phosphorylation may play an important role in Aβ-induced neurodegeneration.1,9
Ilex latifolia Thunb. (Aquifoliaceae), a primary component of “kudingcha,” has been used as a tea in China for more than 2000 years. In Chinese folk medicine, it is prescribed for treating headache, hypertension, coronary heart diseases, and inflammatory diseases.10 Thus, the antioxidant properties and phenolic compounds of I. latifolia have been evaluated.11,12 Due to the antioxidant activity of I. latifolia, neuroprotective effects of this plant have also been examined.13 In a previous study, we demonstrated that the antioxidative and anti-inflammatory effects of I. latifolia provided protection against apoptotic neuronal death associated with transient focal ischemia-induced brain damage.14 We isolated antioxidant compounds, caffeoylquinic acid derivatives (CQAs), which are potentially responsible for the neuroprotection conferred by I. latifolia,15 as shown by other groups.16–18 In the present study, we determined whether I. latifolia exerts a protective effect against memory deficits induced by a single intracerebroventricular (icv) injection of Aβ (25–35) in mice, an AD-type amnesia model,19,20 and investigated the underlying mechanisms. In addition, the protective effects of I. latifolia on Aβ (25–35)-induced neuronal damage were studied in cultured rat cortical neurons.
Materials and Methods
Plant material and extract preparation
Dried I. latifolia leaves were purchased from a herbal market in Hong Kong by Han Kook Shin Yak Co., Ltd. (Chungnam, Korea) and identified by Dr. Bang Yeon Hwang (Chungbuk National University, Korea). A voucher specimen (CBNU006) was deposited in the herbarium at the College of Veterinary Medicine, Chungbuk National University. The I. latifolia leaves (1 kg) were subjected to extraction with 5 L of 75% ethanol at room temperature for 2 h. The extract was filtered through Whatman NO. 1 filter paper, and the filtrate was concentrated using a rotary vacuum evaporator to yield an ethanol extract (220 g) that was stored at room temperature until required.
Reagents
Thiobarbituric acid (TBA) was purchased from Tokyo Kasei Kogyo Co., Ltd. (Tokyo, Japan). Hematoxylin and eosin (H&E) stains, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), Dulbecco's modified Eagle's medium (DMEM), Joklik-modified MEM, verapamil, NG-nitro-L-arginine methyl ester (L-NAME), and poly-L-lysine were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Hoechst 33342 dye, Fluo-4 AM, and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) were from Molecular Probes Inc. (Eugene, OR, USA). Fetal bovine serum was purchased from JRS Biosciences (Lenexa, KS, USA). (5S,10R)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine hydrogen maleate (MK-801) was acquired from RBI (Natick, MA, USA), and Aβ (25–35) was purchased from Bachem (Bubendorf, Switzerland). The Pro-Prep buffer was purchased from iNtRONBio. Inc. (Ghunggi, Korea). Donepezil was generously provided by Eisai Co., Ltd. (Tokyo, Japan). Rabbit polyclonal antibodies against Bax, cleaved-caspase-3 (c-caspase-3), and p-tau; mouse polyclonal antibody against β-actin; and horseradish peroxidase-conjugated anti-rabbit secondary antibody were purchased from Millipore Inc. (Bedford, MA, USA). Horseradish peroxidase-conjugated anti-mouse secondary antibody was purchased from Koma Biotech Inc. (Seoul, Korea). All chemicals used were of the highest grade available.
Animals
Male ICR mice (5 weeks old, Sam: Tac:gA[ICR]fBR) from Samtako, Inc. (Gyunggi-do, South Korea) and pregnant Sprague Dawley (SD) rats (Hsd:SD®) from Koatech Co., Ltd. (Gyunggi, Korea) were housed in an environmentally controlled room at 22°C ± 2°C with 55% ± 5% relative humidity, a 12-h light/12-h dark cycle, and food and water provided ad libitum. Procedures involving the experimental animals complied with regulations of the Care and Use of Laboratory Animals of the Animal Ethics Committee of Chungbuk National University (CBNUA-177-1001-01).
Induction of memory impairment and administration of I. latifolia extract
Memory impairment in the mice was induced by icv injection of 15 nmol of the aggregated form of Aβ (25–35), as previously described.21 I. latifolia extract (25, 50, and 100 mg/kg) suspended in distilled water and donepezil (2 mg/kg) were orally administered 30 min before Aβ (25–35) treatment and were further given on a daily basis for 7 days.
Measurement of learning and memory abilities using the passive avoidance test
Memory was evaluated using a step-through passive avoidance apparatus (Gemini II Avoidance System, San Diego instruments, CA, USA), according to a previously described method.21 At 30 min after the administration of I. latifolia and donepezil on day 7 following icv injection of Aβ (25–35), the mice were trained for a step-through passive avoidance task. A retention trial was performed 24 h after the training trial. After the test, locomotor activity and motor coordination were measured using a photobeam monitoring system (AM1051; Benwick Electronics, Benwick, United Kingdom) and rota rod apparatus (775 Rota rod, IITC Life Science Inc., Woodland Hills, CA, USA), respectively. These measurements were taken to eliminate any possible effects of Aβ (25–35) on general motor function. Each mouse was placed in the center of the activity cage, and the total number of mobile counts was registered for 5 min. Next, the animals were placed onto a rod with a 3-cm diameter rotating at 10 rpm. Mice that fell from the rod within 2 min were considered to have a motor coordination deficit.
Measurement of learning and memory abilities with a Morris water maze test
A Morris water maze test was performed to measure spatial learning and memory of the mice according to the Morris method22 with slight modification. The experimental apparatus consisted of a circular water tank (100-cm diameter, height of 40 cm) filled with water (25°C ± 2°C) to a depth of 30 cm. An escape platform with an 8-cm diameter was submerged 1 cm below the surface of the water and placed in the middle of the same quadrant throughout the training phase. Four training trials per day were conducted for five consecutive days after the third day of Aβ (25–35) icv injection. For each trial, the mice were placed into the water from one of the four starting points on the edge of the tank. The time required to escape onto the hidden platform was recorded. If the mouse located the platform, it was allowed to remain there for 10 sec. If the mouse failed to find the platform within 120 sec, it would be gently guided to the platform where it was allowed to stay for 10 sec, and the escape latency was recorded as 120 sec.
Measurement of oxidative stress and histological evaluation of neuronal death in mouse brain
Upon completion of the retention trial of the passive avoidance test, the mice were anesthetized with diethyl ether and the brains were quickly removed. Brain homogenates were prepared freshly in an ice bath with a 50-fold volume of a 10 mM phosphate buffer (pH 7.4). The levels of reduced GSH were estimated spectrophotometrically using the Ellman reagent.23 The extent of lipid peroxidation was assayed by measuring thiobarbituric acid reactive substances (TBARS) at 532 nm as described by Yoshioka et al.24 using 1,1,3,3-tetramethoxypropane as a standard. Concentrations of GSH and TBARS are expressed as nmol/mg protein. Protein concentration was determined according to Lowry's method.25 H&E staining was performed for histological examination of brain tissues from separate groups of animals. The number of pyramidal cells in the CA1 region of the hippocampus was determined as previously described.21
Induction of neurotoxicity, analysis of neuronal viability, and measurement of intracellular Ca2+ concentration ([Ca2+]i) and ROS in primary cultures of rat cerebral cortical neurons
Primary cultured cortical neurons were prepared using embryonic day 15–16 SD rat fetuses as previously described.26 Neurotoxicity experiments were performed on cultured neurons after 3–4 days in vitro, as previously described.21 The cells were treated with 10 μM Aβ (25–35) for 36 h (unless otherwise indicated) to induce neurotoxicity. I. latifolia extract (1, 10, and 50 μg/mL), verapamil (5 μM), L-NAME (1 mM), and MK-801 (10 μM) were applied 15 min before treatment with 10 μM Aβ (25–35). These reagents were also present in the medium during incubation with Aβ (25–35). I. latifolia extract was dissolved in dimethyl sulfoxide (DMSO) to produce 100 mg/mL and diluted with each experimental buffer. The final concentration of DMSO was less than 0.1% and did not affect cell viability. At the end of the incubation period, the viability of the neuronal cell was monitored using a colorimetric MTT assay and Hoechst 33342 staining as previously described.26 Changes in [Ca2+]i and ROS generation were monitored with two fluorescent dyes, Fluo-4 AM and H2DCF-DA, respectively, using a laser scanning confocal microscope (TCS SP2 AOBS; Leica, Heidelberg, Germany).26 Neurons were treated with 10 μM Aβ (25–35) for 24 h to monitor ROS production.
Western blotting
After icv delivery of 15 nmol Aβ (25–35) and treatment with I. latifolia (25–100 mg/kg, p.o.) according to the same schedule used for the memory test, the mice were anesthetized with diethyl ether and the brains were removed. The hippocampus was homogenized with the Pro-Prep buffer, and total proteins were extracted according to the manufacturer's instructions. The cultured neurons were treated with 10 μM Aβ (25–35) for 36 h before being lysed with the RIPA buffer (150 mM NaCl, 1 mM Na-EDTA, protease inhibitor cocktail, and 50 mM Tris-HCl at pH 7.4). The total proteins were extracted, and Western blot analysis of Bax, c-caspase-3, and p-tau expression was performed as previously described.14 The amount of total protein was measured using the Bradford method.27 Band intensities were quantified with image analysis software (a freely available application in the public domain for image analysis developed and maintained by Mr. Wayne Rasband at the Research Services Branch, National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM). Student's t-test was used for comparisons between two groups, and one-way analysis of variance followed by Tukey's test was used for multiple comparisons. P-values < .05 were considered significant.
Results
Inhibitory effect of I. latifolia on Aβ (25–35)-induced memory impairment in mice
During the acquisition trial of the passive avoidance test, step-through latency did not differ among the six groups (sham, 15 nmol Aβ (25–35), 15 nmol Aβ (25–35) + 25 mg/kg I. latifolia, 15 nmol Aβ (25–35) + 50 mg/kg I. latifolia, 15 nmol Aβ (25–35) + 100 mg/kg I. latifolia, and 15 nmol Aβ (25–35) + 2 mg/kg donepezil; data not shown). Step-through latency of the Aβ (25–35)-treated group in the retention trial was significantly shorter, 29.9 ± 9.6 sec compared to 259.5 ± 26.3 sec for the sham control group, indicating that Aβ (25–35) impaired the memory of the mice. Memory impairment caused by Aβ was markedly prevented by chronically administered I. latifolia (25–100 mg/kg), resulting in 237.7 ± 25.2 sec of step-through latency with 100 mg/kg (Fig. 1A). Chronically administered donepezil (2 mg/kg) also prevented Aβ (25–35)-induced memory impairment (latency; 190.7 ± 14.9 sec). To determine whether I. latifolia treatment affects general motor functions, we measured the spontaneous locomotor activity and motor coordination of the mice. Neither I. latifolia nor Aβ (25–35) significantly affected the locomotor or rota rod activity. These findings indicated that the observed memory improvement was not due to immobility that might be caused by I. latifolia administration (Table 1).
FIG. 1.
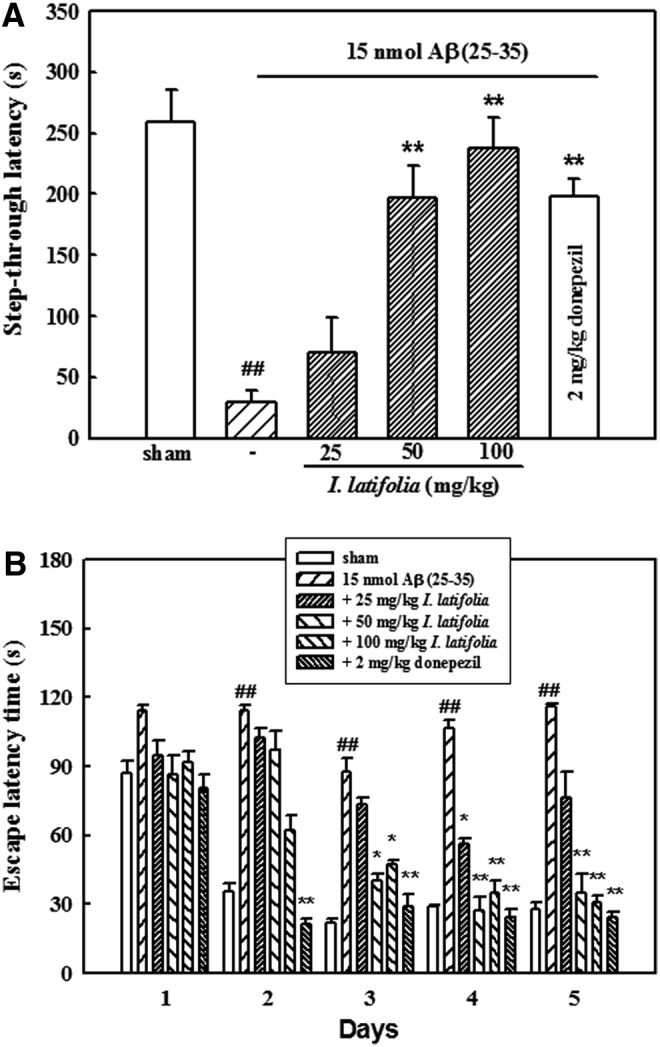
Protective effects of I. latifolia against amyloid β protein (Aβ) (25–35)-induced memory impairment in mice. Learning and memory performance of the mice were assessed by (A) a passive avoidance test (n = 11–13) and (B) a Morris water maze test (n = 8–10). Values are expressed as the mean ± standard error of the mean (SEM) ##P < .01 versus the sham; *P < .05 and **P < .01 versus 15 nmol Aβ (25–35).
Table 1.
Effect of I. Latifolia on Spontaneous Motor Activity and Motor Coordination in Mice
| Group | Dose | Activity (% of prevalue)a | Number of mice which fell downb |
|---|---|---|---|
| Sham | — | 77.0 ± 9.3 | 0 |
| Aβ (25–35) | 15 nmol | 61.1 ± 7.2 | 0 |
| +I. latifolia | 25 mg/kg | 78.6 ± 10.0 | 0 |
| +I. latifolia | 50 mg/kg | 67.0 ± 10.6 | 0 |
| +I. latifolia | 100 mg/kg | 72.8 ± 12.6 | 0 |
After the retention trial for the passive avoidance test, a spontaneous activity was measured using a locomotor activity monitor.
Motor coordination was measured with a rota rod treadmill. Mice that fell off the rod within 2 min were counted. Values are expressed as the mean ± SEM (n = 12–14 mice/group).
SEM, standard error of the mean.
During the first trials using the Morris water maze test, latency time required to reach the hidden platform did not significantly differ among all the groups. The sham mice rapidly learned the location of the hidden platform, reaching it within 30 sec on day 5 compared to the first trials (87.1 ± 10.1 sec). Mice that received Aβ (25–35) had a significantly delayed escape latency time from days 2 to 5 compared to that of the sham control group, indicating that Aβ (25–35) caused a remarkable cognitive deficit. In contrast, chronic administration of I. latifolia (50 and 100 mg/kg) significantly decreased the Aβ (25–35)-induced prolonged latency time (days 3–5; Fig. 1B). Treatment with donepezil also resulted in a significant reduction of Aβ (25–35)-induced memory impairment during the trial.
Inhibitory effect of I. latifolia on Aβ (25–35)-induced GSH depletion and lipid peroxidation
GSH contents of the brains from Aβ (25–35)-treated mice were significantly lower compared to that of the sham group. I. latifolia inhibited the decrease of GSH levels in Aβ (25–35)-treated brains in a dose-dependent manner. TBARS concentration in the brain of Aβ (25–35)-treated mice significantly increased compared to that of the sham group. I. latifolia suppressed the elevation of TBARS concentration induced by Aβ (25–35) in a dose-dependent manner (Table 2).
Table 2.
Effect of I. Latifolia on Aβ (25–35)-Induced Decrease of GSH Concentrations and Increase of TBARS Levels in Mouse Brains
| Group | Dose | GSH (nmol/mg protein) | TBARS (nmol/mg protein) |
|---|---|---|---|
| Sham | — | 14.4 ± 0.5 | 3.7 ± 0.2 |
| Aβ (25–35) | 15 nmol | 7.6 ± 1.2a | 7.2 ± 0.4a |
| +I. latifolia | 25 mg/kg | 9.1 ± 0.2 | 5.3 ± 0.5b |
| +I. latifolia | 50 mg/kg | 11.4 ± 0.1c | 3.9 ± 0.3c |
| +I. latifolia | 100 mg/kg | 12.9 ± 0.6c | 3.4 ± 0.4c |
GSH and TBARS levels were measured after the retention trial for the passive avoidance test. Values are expressed as the mean ± SEM (n = 5–6/group).
P < .01 versus the sham.
P < .05.
P < .01 versus Aβ (25–35) alone.
Aβ, amyloid β protein; GSH, glutathione; TBARS, thiobarbituric acid reactive substances.
Inhibitory effect of I. latifolia on Aβ (25–35)-induced hippocampal neuronal death
H&E staining of the hippocampal CA1 regions in Aβ (25–35)-injected mice revealed neuronal death characterized by condensed nuclei, perinuclear vacuole, and pyknotic nuclei, while no histological changes were observed in the sham mice (Fig. 2A). The number of CA1 pyramidal cells decreased in Aβ (25–35)-treated mice (148.0 ± 8.0) compared to the sham animals (241.5 ± 5.8). Administration of 50 and 100 mg/kg I. latifolia as well as 2 mg/kg donepezil for 7 days protected the hippocampus against Aβ (25–35)-induced neuronal death (178.0 ± 7.1, 201.2 ± 3.2, and 182.0 ± 3.2 cells, respectively; Fig. 2B).
FIG. 2.

Representative photomicrographs of (A) the hippocampus (upper) and CA1 region (lower), and (B) histogram of CA1 pyramidal cell numbers illustrating the inhibitory effect of I. latifolia on Aβ (25–35)-induced hippocampal damage in mice. Brain sections were stained with hematoxylin and eosin. Pathological features of the brain sections 7 days after intracerebroventricular injection of 15 nmol Aβ (25–35) were observed using a brightfield microscope. ##P < .01 versus sham; *P < .05 and **P < .01 versus 15 nmol Aβ (25–35). Color images available online at www.liebertpub.com/jmf
Inhibitory effect of I. latifolia on Aβ (25–35)-induced expression of apoptosis-associated proteins and p-tau in mice
Expression of two proapoptotic proteins, Bax and c-caspase-3, increased upon injection of Aβ (25–35) into the hippocampus. Levels of these proteins were significantly reduced by I. latifolia (Fig. 3A, B). Chronic administration of I. latifolia inhibited the expression of p-tau protein in a dose-dependent manner (Fig. 4A, B) that had been increased by treatment with Aβ (25–35).
FIG. 3.
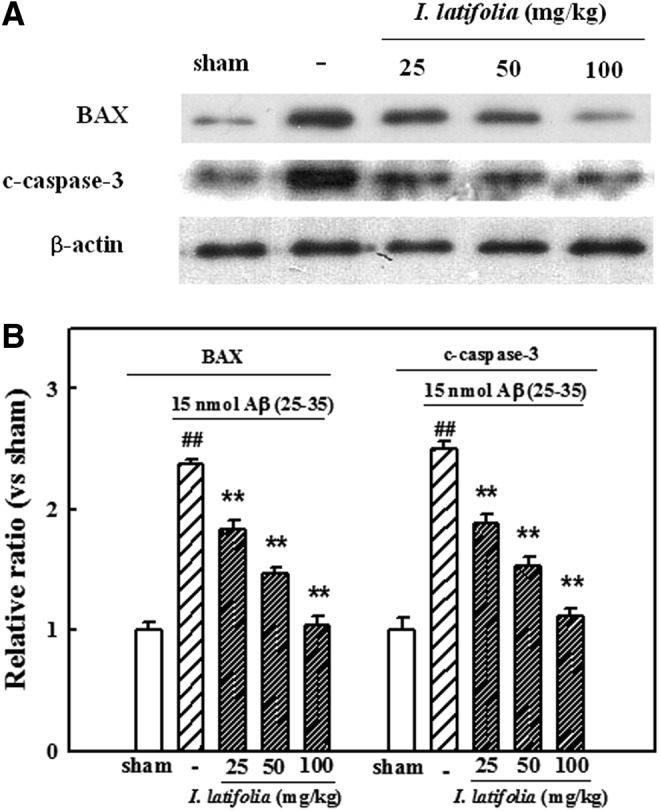
Inhibitory effect of I. latifolia on Aβ (25–35)-induced expression of Bax and c-caspase-3 in mice brains. (A) Representative Western blot of proteins in the brains. (B) Bar graphs showing the relative ratio of Bax and c-caspase-3 proteins compared to β-actin. ##P < .01 versus the sham; **P < .01 versus 15 nmol Aβ (25–35).
FIG. 4.
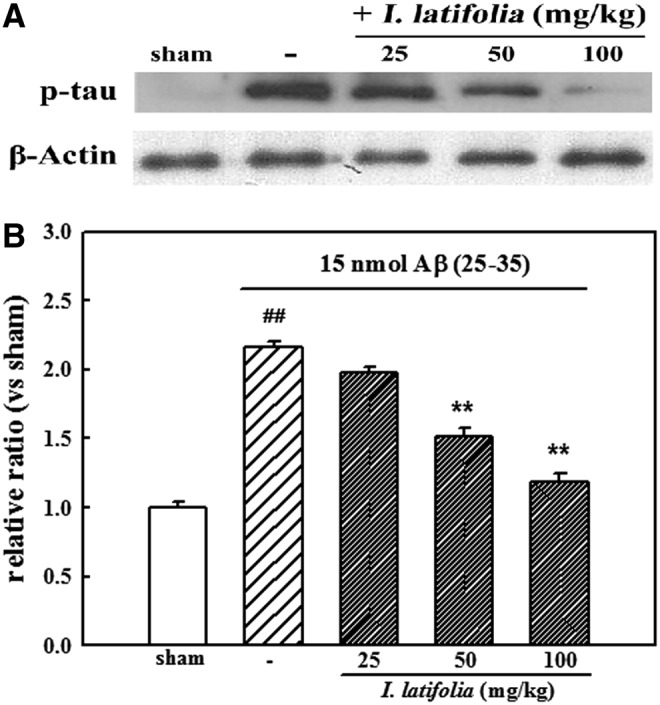
Inhibitory effect of I. latifolia on Aβ (25–35)-induced expression of phosphorylated tau (p-tau) in mice brains. (A) Representative Western blot of proteins in the brains. (B) Bar graphs showing the relative ratio of p-tau protein compared to β-actin. ##P < .01 versus the sham; **P < .01 versus 15 nmol Aβ (25–35).
Inhibitory effect of I. latifolia on Aβ (25–35)-induced death of cultured cortical neurons
Aβ (25–35) at a concentration of 10 μM was used to produce neuronal cell toxicity based on a previous study.28 Treatment of cultured neurons with Aβ (25–35) for 36 h caused neuronal cell death. Viability of Aβ (25–35)-treated cells was 69.3% ± 1.4% of that for the untreated control cells as measured by an MTT assay. I. latifolia (1, 10, and 50 μg/mL) significantly reduced Aβ (25–35)-promoted neuronal cell death. Absorbance with 50 μg/mL I. latifolia was 93.4% ± 3.1% of that observed for the control. Pretreatment with verapamil (5 μM), L-NAME (1 mM), and MK-801 (10 μM) inhibited Aβ (25–35)-induced neuronal cell death (Fig. 5A).
FIG. 5.
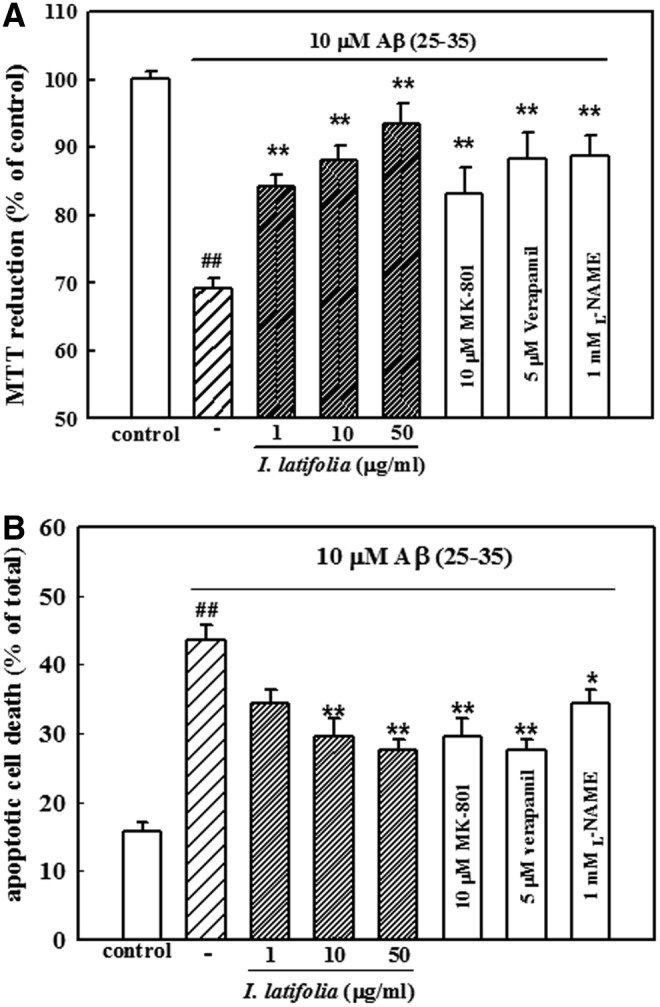
Inhibitory effect of I. latifolia on Aβ (25–35)-induced death of cultured cortical neurons measured by (A) an MTT assay and (B) Hoechst 33342 staining. Values are expressed as the mean ± SEM of data obtained from at least six independent experiments. ##P < .01 versus the control; *P < .05 and **P < .01 versus 10 μM Aβ (25–35).
Condensed or fragmented DNA, indicating apoptotic cell death promoted by Aβ (25–35), was detected with Hoechst 33342 staining. Treatment of neurons with 10 μM Aβ (25–35) caused 43.6% ± 2.2% of the cultured cortical neurons to undergo apoptosis compared to 15.7% ± 1.3% of the control cultures. In contrast, addition of I. latifolia (10 and 50 μg/mL) significantly decreased Aβ (25–35)-induced apoptosis (27.6% ± 1.6% for 50 μg/mL). Verapamil (5 μM), L-NAME (1 mM), and MK-801 (10 μM) also inhibited Aβ (25–35)-induced neuronal cell apoptosis (Fig. 5B).
I. latifolia inhibition of Aβ (25–35)-induced expression of apoptosis-related proteins in cultured cortical neurons
Treatment of cultured neurons with 10 μM Aβ (25–35) increased the expression of Bax and c-caspase-3. In contrast, pretreatment with I. latifolia (1, 10, and 50 μg/mL) inhibited the expression of Bax and c-caspase-3 in a dose-dependent manner (Fig. 6A, B).
FIG. 6.
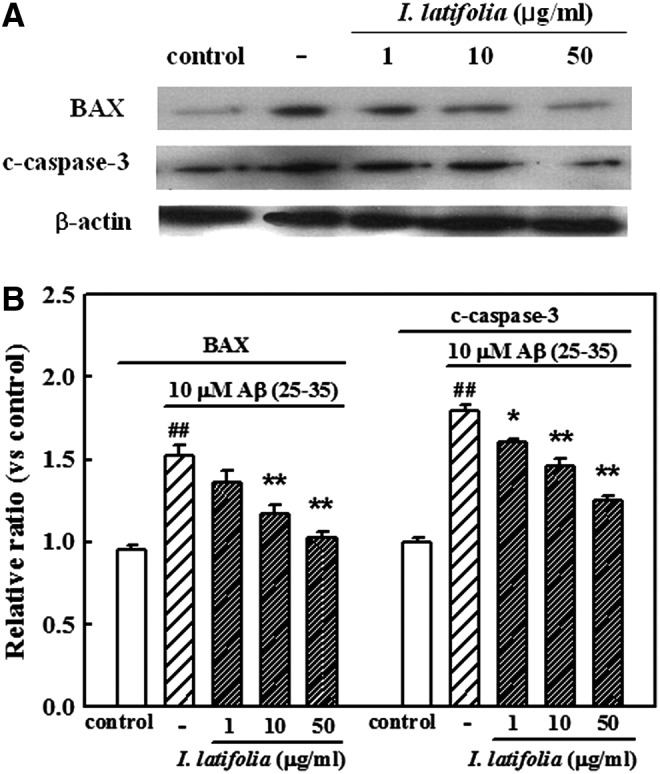
Inhibitory effect of I. latifolia on Aβ (25–35)-induced expression of Bax and c-caspase-3 in cultured cortical neurons. (A) Representative Western blot of proteins in the cultured neurons. (B) Bar graphs showing the relative ratio of Bax and c-caspase-3 proteins compared to β-actin. ##P < .01 versus the control;*P < .05 and **P < .01 versus 10 μM Aβ (25–35).
I. latifolia inhibition of Aβ (25–35)-induced intracellular biochemical changes in neuronal cell cultures
As shown in Figure 7, [Ca2+]i slowly and gradually increased in response to treatment with 10 μM Aβ (25–35) that was maintained for approximately 10 min. However, pretreatment with I. latifolia (1 and 50 μg/mL) significantly inhibited Aβ (25–35)-induced increase of [Ca2+]i throughout the experimental period. Verapamil (5 μM), L-NAME (1 mM), and MK-801 (10 μM) also suppressed the increase of [Ca2+]i resulting from Aβ (25–35) treatment (Fig. 7). I. latifolia, verapamil, L-NAME, and MK-801 did not affect basal [Ca2+]i.
FIG. 7.
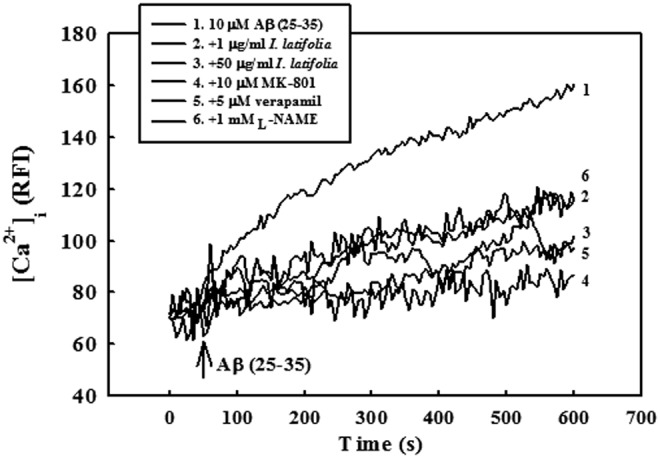
Inhibitory effect of I. latifolia on Aβ (25–35)-induced elevation of [Ca2+]i in cultured cortical neurons. [Ca2+]i was monitored using a laser scanning confocal microscope. All images were processed to analyze changes in [Ca2+]i at the single-cell level. Values are expressed as the relative fluorescence intensity (RFI). Each trace is a single-cell representative of at least four independent experiments.
In the H2DCF-DA-loaded cortical neurons, 10 μM Aβ (25–35) increased the fluorescence intensity, indicating the generation of ROS. In neurons treated with 10 μM Aβ (25–35) for 24 h, the fluorescence intensity increased approximately 4.5-fold to 217.1 ± 10.6 compared to that of the control neurons (47.6 ± 4.2). As presented in Figure 8, Aβ (25–35)-induced increases in ROS production were significantly inhibited by I. latifolia (60.0 ± 1.9 with 50 μg/mL). Verapamil (5 μM), L-NAME (1 mM), and MK-801 (10 μM) prevented Aβ (25–35)-induced ROS generation (Fig. 8). I. latifolia, verapamil, L-NAME, and MK-801 did not have any direct reaction with H2DCF-DA that could have generated fluorescence.
FIG. 8.
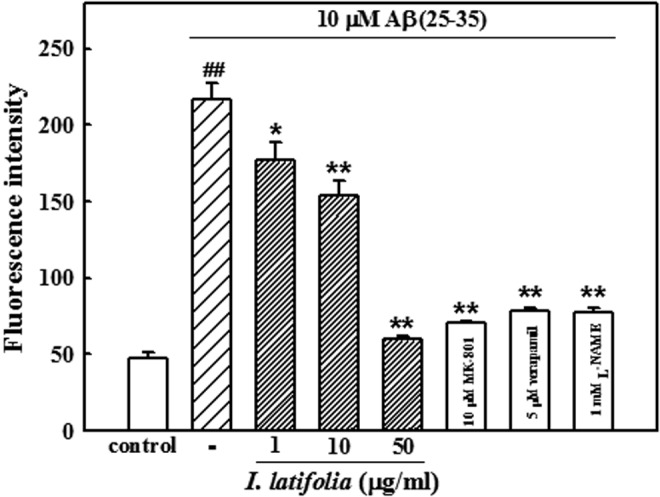
Inhibitory effect of I. latifolia on Aβ (25–35)-induced reactive oxygen species generation in cultured cortical neurons. RFI was monitored using a laser scanning confocal microscope. Values are expressed as the mean ± SEM of data obtained from at least four independent experiments. ##P < .01 versus the control; *P < .05 and **P < .01 versus 10 μM Aβ (25–35).
Discussion
Aβ (25–35), containing a 11-amino acid sequence, is an active peptide fragment with neurotoxic properties of the full-length sequence (1–42).29 A number of studies have reported that Aβ (25–35) acts as a potent neurotoxin both in vitro and in vivo.29,30 Intracerebroventricular injection of Aβ-related peptide into rodents impairs learning and memory based on different behavioral paradigms, including spontaneous alternation along with spatial and nonspatial short-term memory.19,20,30 In the present study, memory impairment was confirmed in mice following icv injection of Aβ (25–35). Chronic oral administration of I. latifolia (50 and 100 mg/kg) ameliorated memory deficits in the Aβ (25–35)-treated mice that showed an increase in avoidance retention time measured by the passive avoidance test and a decrease in escape latency time in the Morris water maze test. General motor functions were not affected. Moreover, histological examination revealed that orally administered I. latifolia prevented Aβ (25–35)-induced neuronal death in the hippocampus. Treatment with Aβ (25–35) led to significant cell loss in the CA1 pyramidal cell layer, and I. latifolia conferred protection against this effect.
One common factor that underlies AD pathogenesis is neuronal Ca2+ dysregulation.31 Increased Ca2+ levels through Aβ-mediated mechanisms can lead to mitochondrial Ca2+ overload, ROS generation, and activation of proapoptotic mitochondrial proteins such as caspases and cytochrome c.31 These activities are linked to cell death and neurodegeneration in several AD models.32 Mitochondrial dysfunction is observed in the AD brain, which exacerbates the release of ROS.33,34 Mitochondrial ROS generation is associated with reduced mitochondrial membrane potential that triggers cytochrome c release into the cytosol.35
Bax, which is a Bcl-2 family member that resides mainly in the cytosol of healthy cells, translocates to the mitochondria following exposure to Aβ and increases cytochrome c release from the mitochondria.36 Cytosolic cytochrome c forms a functional apoptosome that activates caspase-9 and caspase-3. Numerous proteins are cleaved by activated caspase-3, which initiates biochemical cascades that lead to cell death.2,37 In the present study, using cultured cortical neurons, Aβ (25–35) increased [Ca2+]i, ROS generation, and apoptotic neuronal cell death. These effects were inhibited by verapamil, an L-type Ca2+ channel blocker; L-NAME, a nitric oxide synthase (NOS) inhibitor; and MK-801, an NMDA antagonist. Aβ (25–35) also increased the expression of two proapoptotic proteins, Bax and c-caspase-3, in cultured neurons. The Aβ (25–35)-induced increases of [Ca2+]i and ROS levels as well as apoptotic changes were prevented by treatment with I. latifolia (1–50 μg/mL) in cultured neurons. Furthermore, the increased expression of Bax and c-caspase-3 in the memory-impaired mouse brain by treatment with Aβ (25–35) was inhibited by chronic administration of I. latifolia (25–100 mg/kg). We previously isolated antioxidant molecules, QAs, from I. latifolia, which showed neuroprotective activity against excitotoxicity through the inhibition of [Ca2+]i increase and ROS generation in cultured neurons.15 Moreover, we have demonstrated that the suppression of intracellular [Ca2+]i elevation by numerous antioxidants such as oxyresveratrol, gallic acid, catechin, and epicatechin is primarily responsible for the ability of these compounds to prevent Aβ (25–35)-induced neuronal death.26,38–40 Therefore, results from the present study suggested that the antidementia effect of I. latifolia might be attributable to the prevention of apoptosis by suppressing intracellular [Ca2+]i increases and ROS generation due to the antioxidant components of this plant.
GSH is a major intracellular antioxidant that plays a key role in protecting cells from oxidative stress. Thus, GSH levels are an important indicator of oxidative stress in the brain.41 Infusion of Aβ into the brain causes depletion of GSH that results in DNA damage, lipid and protein peroxidation, and subsequent apoptosis of neuronal cells.34 Oxidative stress increases the formation of TBARS, a commonly used maker of lipid peroxidation, in Aβ-infused brain tissue.4,42 In the present study, GSH levels significantly decreased, while TBARS concentrations increased in mouse brains treated with Aβ (25–35). Administration of I. latifolia prevented the decrease in GSH contents and the increase of TBARS in Aβ (25–35)-treated mouse brains. This result further demonstrated that the antioxidant activity of I. latifolia could play an important role in protection from apoptotic neuronal death followed by memory deficits in mice.
Hyperphosphorylated tau protein, as the major component of neurofibrillary tangles, is another hallmark of AD pathology that correlates with neurodegeneration and cognitive deficits.43 In the AD brain, senile plaques consist of extracellular deposits of fibrillar Aβ, while neurofibrillary tangles are formed by intracellular bundles of self-assembled p-tau.44,45 Studies have shown that the deposition of fibrillar Aβ induces the phosphorylation of tau followed by the progressive degeneration of neuronal processes.6–9 Increased intracellular [Ca2+]i through the NMDA receptor leads to the activation of tau.46 Furthermore, oxidative stress facilitates the phosphorylation and polymerization of tau during neurodegeneration in the AD brain.47 In the present study, the level of p-tau was increased in the memory-impaired mouse brain following the injection of Aβ (25–35). This effect was inhibited by treatment with I. latifolia. These results suggest that I. latifolia could also inhibit Aβ (25–35)-induced p-tau formation through the inhibition of intracellular [Ca2+]i increase, thereby preventing memory impairment.
Various kinds of antioxidant molecules, including triterpenoid saponins and CQAs, have been isolated from I. latifolia.12,16–18 Since oxidative stress plays a key role in Aβ-induced neurotoxicity,4,42 antioxidants have been studied to exploit their neuroprotective effects in AD models. CQAs have been reported to protect neurons against Aβ-promoted neuronal cell damage.48 We also previously isolated seven kinds of CQAs from I. latifolia and found that three CQAs showed neuroprotective activity against glutamate-induced and oxygen glucose-deprivation neurotoxicity in cultured neurons.15 These results indicate that these CQAs could contribute to the neuroprotection provided by I. latifolia, although we could not find the inhibitory effects of seven CQAs on Aβ (25–35)-induced neurotoxicity. Further studies will be performed to verify the active compounds from I. latifolia that can help protect against Aβ (25–35)-induced memory impairment.
In the present study, the protective effects of I. latifolia against Aβ (25–35)-induced neurotoxicity were evaluated in vitro and in vivo. The neuroprotection provided by I. latifolia against Aβ-induced memory impairment might be due to inhibition of apoptotic neuronal cell death triggered by oxidative stress and p-tau formation. These results suggest that I. latifolia might have therapeutic efficacy for preventing the progression of AD.
Acknowledgment
This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (2012-0014760).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S: Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 2002;115:201–211 [DOI] [PubMed] [Google Scholar]
- 2.Estus S, Tucker HM, van Rooyen C, et al. : Aggregated amyloid-beta protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J Neurosci 1997;17:7736–7745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim HC, Yamada K, Nitta A, et al. : Immunocytochemical evidence that amyloid β(1–42) impairs endogenous antioxidant systems in vivo. Neuroscience 2003;119:399–419 [DOI] [PubMed] [Google Scholar]
- 4.Butterfield DA, Lauderback CM: Lipid peroxidation and protein oxidation in Alzheimer's disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic Biol Med 2002;32:1050–1060 [DOI] [PubMed] [Google Scholar]
- 5.Yatin SM, Varadarajan S, Link CD, Butterfield DA: In vitro and in vivo oxidative stress associated with Alzheimer's amyloid β-peptide (1–42). Neurobiol aging 1999;20:325–330 [DOI] [PubMed] [Google Scholar]
- 6.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI: Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 1986;83:4913–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee VM, Goedert M, Trojanowski JQ: Neurodegenerative tauopathies. Annu Rev Neurosci 2001;24:1121–1159 [DOI] [PubMed] [Google Scholar]
- 8.Brunden KR, Ballatore C, Crowe A, Smith AB, 3rd, Lee VM, Trojanowski JQ: Tau-directed drug discovery for Alzheimer's disease and related tauopathies: A focus on tau assembly inhibitors. Exp Neurol 2010;223:304–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A: Tau is essential to β-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 2002;99:6364–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang KC: The Pharmacology of Chinese Herbs, 2nd ed. CRC Press, Florida, USA: 1999 [Google Scholar]
- 11.Woo AY, Jiang JM, Chau CF, et al. : Inotropic and chronotropic actions of Ilex latifolia inhibition of adenosine-5′-triphosphatases as a possible mechanism. Life Sci 2001;68:1259–1270 [DOI] [PubMed] [Google Scholar]
- 12.Zhu F, Cai YZ, Sun M, Ke J, Lu D, Corke H: Comparison of major phenolic constituents and in vitro antioxidant activity of diverse Kudingcha genotypes from Ilex kudingcha, Ilex cornuta, and Ligustrum robustum. J Agric Food Chem 2009;57:6082–6089 [DOI] [PubMed] [Google Scholar]
- 13.Colpo G, Trevisol F, Teixeira AM, et al. : Ilex paraguariensis has antioxidant potential and attenuates haloperidol-induced orofacial dyskinesia and memory dysfunction in rats. Neurotox Res 2007;12:171–180 [DOI] [PubMed] [Google Scholar]
- 14.Kim JY, Jeong HY, Lee HK, Yoo JK, Bae K, Seong YH: Protective effect of Ilex latifolia, a major component of “kudingcha”, against transient focal ischemia-induced neuronal damage in rats. J Ethnopharmacol 2011;133:558–564 [DOI] [PubMed] [Google Scholar]
- 15.Kim JY, Lee HK, Hwang BY, Kim S, Yoo JK, Seong YH: Neuroprotection of Ilex latifolia and caffeoylquinic acid derivatives against excitotoxic and hypoxic damage of cultured rat cortical neurons. Arch Pharm Res 2012;35:1115–1122 [DOI] [PubMed] [Google Scholar]
- 16.Negishi O, Negishi Y, Yamaguchi F, Sugahara T: Deodorization with ku-ding-cha containing a large amount of caffeoyl quinic acid derivatives. J Agric Food Chem 2004;52:5513–5518 [DOI] [PubMed] [Google Scholar]
- 17.Huang J, Wang X, Ogihara Y, Shimizu N, Akiyama T, Takeda T: Latifolosides K and L, two new triterpenoid saponins from the bark of Ilex latifolia. Chem Pharm Bull (Tokyo) 2001;49:765–767 [DOI] [PubMed] [Google Scholar]
- 18.Huang J, Wang X, Ogihara Y, Shimizu N, Takeda T, Akiyama T: Latifolosides I and J, two new triterpenoid saponins from the bark of Ilex latifolia. Chem Pharm Bull (Tokyo) 2001;49:239–241 [DOI] [PubMed] [Google Scholar]
- 19.Stepanichev MY, Moiseeva YV, Lazareva NA, Onufriev MV, Gulyaeva NV: Single intracerebroventricular administration of amyloid β(25–35) peptide induces impairment in short-term rather than long-term memory in rats. Brain Res Bull 2003;61:197–205 [DOI] [PubMed] [Google Scholar]
- 20.Maurice T, Lockhart BP, Privat A: Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res 1996;706:181–193 [DOI] [PubMed] [Google Scholar]
- 21.Cho SO, Ban JY, Kim JY, et al. : Aralia cordata protects against amyloid β protein (25–35)-induced neurotoxicity in cultured neurons and has antidementia activities in mice. J Pharmacol Sci 2009;111:22–32 [DOI] [PubMed] [Google Scholar]
- 22.Morris R: Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Meth 1984;11:47–60 [DOI] [PubMed] [Google Scholar]
- 23.Ellman GL, Burkhalter A, Ladou J: A fluorometric method for the determination of hippuric acid. J Lab Clinic Med 1961;57:813–818 [PubMed] [Google Scholar]
- 24.Yoshioka T, Kawada K, Shimada T, Mori M: Lipid peroxidation in maternal and cord blood and protective mechanism against activated-oxygen toxicity in the blood. Am J Obstet Gynecol 1979;135:372–376 [DOI] [PubMed] [Google Scholar]
- 25.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265–275 [PubMed] [Google Scholar]
- 26.Ban JY, Jeon SY, Bae K, Song KS, Seong YH: Catechin and epicatechin from Smilacis chinae rhizome protect cultured rat cortical neurons against amyloid β protein (25–35)-induced neurotoxicity through inhibition of cytosolic calcium elevation. Life Sci 2006;79:2251–2259 [DOI] [PubMed] [Google Scholar]
- 27.Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254 [DOI] [PubMed] [Google Scholar]
- 28.Lee BY, Ban JY, Seong YH: Chronic stimulation of GABAA receptor with muscimol reduces amyloid β protein (25–35)-induced neurotoxicity in cultured rat cortical cells. Neurosci Res 2005;52:347–356 [DOI] [PubMed] [Google Scholar]
- 29.Meunier J, Ieni J, Maurice T: The anti-amnesic and neuroprotective effects of donepezil against amyloid β 25–35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br J Pharmacol 2006;149:998–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruan CJ, Si JY, Zhang L, Chen DH, Du GH, Su L: Protective effect of stilbenes containing extract-fraction from Cajanus cajan L. On A β (25–35)-induced cognitive deficits in mice. Neurosci Lett 2009;467:159–163 [DOI] [PubMed] [Google Scholar]
- 31.Demuro A, Parker I, Stutzmann GE: Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem 2010;285:12463–12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stutzmann GE: The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist 2007;13:546–559 [DOI] [PubMed] [Google Scholar]
- 33.Muller WE, Eckert A, Kurz C, Eckert GP, Leuner K: Mitochondrial dysfunction: Common final pathway in brain aging and Alzheimer's disease-therapeutic aspects. Molecul Neurobiol 2010;41:159–171 [DOI] [PubMed] [Google Scholar]
- 34.Butterfield DA: Amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res 2002;36:1307–1313 [DOI] [PubMed] [Google Scholar]
- 35.Kroemer G: The mitochondrion as an integrator/coordinator of cell death pathways. Cell Death Differ 1998;5:547. [DOI] [PubMed] [Google Scholar]
- 36.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB: BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 2001;15:1481–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicholson DW, Thornberry NA: Caspases: Killer proteases. Trends Biochem Sci 1997;22:299–306 [DOI] [PubMed] [Google Scholar]
- 38.Ban JY, Cho SO, Jeon SY, Bae K, Song KS, Seong YH: 3,4-dihydroxybenzoic acid from Smilacis chinae rhizome protects amyloid β protein (25–35)-induced neurotoxicity in cultured rat cortical neurons. Neurosci Lett 2007;420:184–188 [DOI] [PubMed] [Google Scholar]
- 39.Ban JY, Jeon SY, Nguyen TT, Bae K, Song KS, Seong YH: Neuroprotective effect of oxyresveratrol from smilacis chinae rhizome on amyloid β protein (25–35)-induced neurotoxicity in cultured rat cortical neurons. Biol Pharm Bull 2006;29:2419–2424 [DOI] [PubMed] [Google Scholar]
- 40.Ban JY, Nguyen HT, Lee HJ, et al. : Neuroprotective properties of gallic acid from Sanguisorbae radix on amyloid β protein (25–35)-induced toxicity in cultured rat cortical neurons. Biol Pharm Bull 2008;31:149–153 [DOI] [PubMed] [Google Scholar]
- 41.Woltjer RL, Nghiem W, Maezawa I, et al. : Role of glutathione in intracellular amyloid-alpha precursor protein/carboxy-terminal fragment aggregation and associated cytotoxicity. J Neurochem 2005;93:1047–1056 [DOI] [PubMed] [Google Scholar]
- 42.Tayler H, Fraser T, Miners JS, Kehoe PG, Love S: Oxidative balance in Alzheimer's disease: relationship to APOE, Braak tangle stage, and the concentrations of soluble and insoluble amyloid-beta. J Alzheimers Dis 2010;22:1363–1373 [DOI] [PubMed] [Google Scholar]
- 43.Goedert M, Spillantini MG: A century of Alzheimer's disease. Science 2006;314:777–781 [DOI] [PubMed] [Google Scholar]
- 44.Kosik KS, Joachim CL, Selkoe DJ: Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 1986;83:4044–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selkoe DJ: Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer's disease. Annu Rev Cell Dev Biol 1994;10:373–403 [DOI] [PubMed] [Google Scholar]
- 46.Mondragon-Rodriguez S, Perry G, Zhu X, Moreira PI, Acevedo-Aquino MC, Williams S: Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: Implications for Alzheimer's disease. Oxid Med Cell Longev (Online) 2013;2013:940603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Zhao B: Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev (Online) 2013;2013:316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hur JY, Soh Y, Kim BH, et al. : Neuroprotective and neurotrophic effects of quinic acids from Aster scaber in PC12 cells. Biol Pharm Bull 2001;24:921–924 [DOI] [PubMed] [Google Scholar]