

Abstract
We have previously shown that a DNA-prime followed by an adenovirus-5 boost vaccine containing CSP and AMA1 (DNA/Ad) successfully protected 4 of 15 subjects to controlled human malaria infection (CHMI). However, the adenovirus-5 vaccine alone (AdCA) failed to induce protection despite eliciting cellular responses that were often higher than those induced by DNA/Ad. Here we determined the effect of CHMI on pre-CHMI cellular and antibody responses against CSP and AMA1 expressed as fold-changes in activities. Generally, in the DNA/Ad trial, CHMI caused pre-CHMI ELISpot IFN-γ and CD8+ T cell IFN-γ responses of the protected subjects to fall but among non-protected subjects, CHMI caused rises of pre-CHMI ELISpot IFN-γ but falls of CD8+ T cell IFN-γ responses. In contrast in the AdCA trial, CHMI caused both pre-CHMI ELISpot IFN-γ and CD8+ T cell IFN-γ responses of the AdCA subjects to fall. We suggest that the falls in activities are due to migration of peripheral CD8+ T cells to the liver in response to developing liver stage parasites, and this fall, in the DNA/Ad trial, is masked in ELISpot responses of the non-protected subjects by rises in other immune cell types. In addition, CHMI caused falls in antibody activities of protected subjects, but rises in non-protected subjects in both trials to CSP, and dramatically in the AdCA trial to AMA1, reaching 380 μg/ml that is probably due to boosting by transient blood stage infection before chloroquine treatment. Taken together, these results further define differences in cellular responses between DNA/Ad and AdCA trials, and suggest that natural transmission may boost responses induced by these malaria vaccines especially when protection is not achieved.
Keywords: antibody, adenovirus-boost, AMA1, CSP, CHMI, DNA -prime, efficacy, malaria, T cells, vaccine
Introduction
A heterologous DNA-prime/adenovirus-5 (Ad5) boost (DNA/Ad) malaria vaccine, using the circumsporozoite protein (CSP) and the apical membrane antigen-1 (AMA1) protected 4 of 15 subjects (27%) against controlled human malaria infection (CHMI).1 A single dose of Ad-5 vaccine (AdCA) administered alone did not induce protection, demonstrating that DNA priming was required for protection.2 In the DNA/Ad trial, 2 protected subjects had higher ELISpot and CD8+ T cell IFN-γ responses to CSP, and 3 protected subjects had higher ELISpot and CD8+ T cell IFN-γ responses to AMA1, than those of non-protected subjects.3 CD8+ T cell IFN-γ effector memory (EM) responses to CSP or AMA1 only occurred in the protected subjects, suggesting that this activity was a correlate of protection in the DNA/Ad trial.3 In contrast, CD4+ T cell and antibody responses were much lower, and not associated with protection.1,2 Unexpectedly, non-protected subjects in the AdCA trial developed ELISpot and CD8+ T cell IFN-γ responses, including EM responses, to CSP and AMA1 that were equal to or higher than the protected subjects in the DNA/Ad trial. However, responses of protected subjects were focused on fewer CSP and AMA1 epitopes than those of non-protected subjects in the DNA/Ad and AdCA trial.3 Only peripheral responses can be measured in human subjects and there is considerable evidence that liver-resident cellular responses play a key role in mediating protection.4-6
CHMI routinely consists of the bite of 5 P. falciparum-infected mosquitoes and subjects are treated with chloroquine when blood stage infection is detected in blood films.1,3,7,8 In the DNA/Ad trial, all 6 infectivity controls developed parasitemia detected by qPCR at mean 7.1 days and blood films at mean day 12.3, indicating that these subjects experienced at least 2 cycles of asexual P. falciparum development.1 Non-protected subjects in the DNA/Ad trial and all subjects in the AdCA trial (infectivity controls and immunized subjects) became positive by qPCR at mean day 7.2 and blood film at mean day 12.1, also suggesting they experienced 2 cycles of asexual P. falciparum development.1,9 CHMI itself induced low levels of recall IFN-γ responses to whole sporozoites, but cellular responses to known antigens including CSP and AMA1 were not detected in these studies.10,11 Although CHMI has been reported to boost vaccine induced responses, especially humoral activities,12-14 particularly following live vector immunization,15,16 the effects on cellular responses are less clear. We sought to determine the effect of CHMI on DNA/Ad and AdCA-induced cellular and humoral responses.
We framed 2 hypotheses of the effects of CHMI on peripheral pre-CHMI cellular and antibody activities: (1) did cellular responses fall in protected subjects suggesting migration of antigen-specific immune cells from the periphery to the liver in response to liver stage infections, as hypothesized by others following immunization with radiation-attenuated sporozoites,17,18 and (2) did antibody and cellular responses of non-protected volunteers rise, based on the findings that CHMI induced higher responses in volunteers previously immunized with sporozoites under chloroquine prophylaxis.10 We performed 2 statistical comparisons: The first compared the geometric mean activities of all volunteers before and after CHMI. To determine whether activities of individual subjects rose or fell after CHMI, and we measured changes as fold-differences and whether these were significantly related to pre-CHMI activities. We used a similar analysis to demonstrate that fold-changes in antibody responses to the Ad5 vaccine backbone (NAb) after a second immunization with an Ad5-CSP vaccine were associated with decreases in antibody and cellular responses to the CSP transgene.8 Although ten of the 15 subjects in the DNA/Ad trial were NAb positive, there was no association between NAb and antibody and cellular responses to the malaria antigens after immunization,1 and all subjects in the AdCA trial were NAb negative.2 We also determined whether NAb activities after immunization and pre-CHMI affected CHMI-induced changes in activities against the vaccine antigens. These analyses may provide insights into whether cellular and humoral activities induced by these vaccines might be boosted by natural malaria transmission. We are cautious in our interpretations since we could only measure peripheral activities, as functional activities of liver-resident T cells may differ from those in the periphery,17 and because of the small number of subjects, especially those with activities to CSP. We are using these analyses to develop new hypotheses that may then be tested in pre-clinical or further clinical trials.
Results
We report summed responses against all CSP and AMA peptide pools tested in each assay. Since the 4 protected subjects in the DNA/Ad trial recognized a restricted number of CSP (v11/Cp9; v18/Cp6) and AMA1 (v10/Ap8; v11/Ap10; v18/Ap8) peptide pools associated with individual AMA1 Class I-restricted epitopes,19 we also examined whether responses to these immunodominant peptide pools were affected by CHMI. We first determined if CHMI itself induced responses in the infectivity controls in the DNA/Ad and AdCA trials, using ex vivo ELISpot IFN-γ assays.
Infectivity controls: ex vivo ELISpot IFN-γ
After CHMI, summed ELISpot IFN-γ activities to CSP and AMA1 of the infectivity controls changed by less than 1.5-fold (Fig. 1); all remained negative to CSP as shown by others;10 however, 3 subjects each developed low positive responses to one of the 12 tested AMA1 peptide pools that included Ap1, Ap2, and Ap3 (data not shown). Previous studies using flow cytometry did not measure positive responses to CSP or AMA1,10 and our findings may reflect the greater sensitivity of ELISpot IFN-γ assays. We therefore defined any change in DNA/Ad or AdCA-immunized subjects as positive if fold-changes exceeded 1.5.
Figure 1.

Ex vivo ELISpot IFN-γ activities of infectivity controls. Five infectivity controls from the DNA/Ad trial and 4 infectivity controls from the AdCA trial were used. The associations of fold-changes of pre-CHMI and post-CHMI activities with pre-CHMI activities are shown as log-transformed values, and the dotted line represents no-change. The shaded box shows ±1.5 range (log ±0.18)-. (A) CSP: the fold change was ≤1.5, except one outlier, and all 9 subjects remained negative after CHMI. (B) AMA1: the fold change of all subjects was <1.5, and 6/9 subjects remained negative after CHMI, but 3/9 subjects developed positive activities after CHMI each to a single AMA1 peptide pool (not shown).
We next examined the effect of CHMI on cellular IFN-γ activities of DNA/Ad-immunized subjects, and then compared these to activities of AdCA-immunized subjects.
DNA/Ad trial
Ex vivo ELISpot IFN-γ: activities of protected subjects fell and non-protected subjects rose
We could not find any correlation between the effects of CHMI and pre-existing NAb. We calculated the fold-changes between pre- and post-CHMI (log-transformed) of all subjects that were positive pre-CHMI and/or post-CHMI and the correlations and significance were calculated using Pearson product moment calculations.8 Fold-changes of individual subjects are shown in Table 1 and the relationship between fold-changes with pre-CHMI activities are shown in Figures 2–6. As previously reported 2,3 we found large ranges of activities among all subjects that we interpret as reflecting the expression of different HLA alleles of individual subjects.
Table 1.
Effects of CHMI on activities of individual subjects in the DNA/Ad and AdCA trials
| ELISpot | CD8+ T cell IFN-γ | CD8+ T cell IFN-γ EM | Antibody |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vaccine | Status | Vol. | CSP | AMA1 | CSP | AMA1 | CSP | AMA1 | CSP | AMA1 |
| DNA-Ad | P | v06 | 0 | +1.2 | 0 | 0 | 0 | 0 | −1.8 | −1.3 |
| v10 | 0 | −2.01 | 0 | −4.2 | 0 | −11.51 | −2.3 | −1.4 | ||
| v11 | −3.21 | −2.61 | −9.91 | −4.51 | −28.91 | −5.21 | −4.0 | −4.4 | ||
| v18 | −1.61 | −1.81 | −2.41 | −2.51 | −2.41 | −3.01 | −1.4 | −2.1 | ||
| Non-P | v01 | +4.9* | +1.8 | 0 | +1.4 | 0 | 0 | −1.7 | +1.1 | |
| v02 | 0 | +3.0 | 0 | 0 | 0 | 0 | −1.2 | +2.3 | ||
| v03 | +1.6 | +2.5 | −3.5 | −2.5 | 0 | 0 | −2.2 | +1.6 | ||
| v05 | 0 | +3.3 | 1.0* | +1.1 | 0 | 0 | +2.7 | +2.5 | ||
| v09 | 0 | +4.8* | +2.2* | 1.0 | 0 | 0 | +1.4 | +2.0 | ||
| v12 | +1.8 | +1.2 | +1.1* | 1.0 | 0 | 0 | 1.0 | −2.8 | ||
| v13 | 0 | +1.4 | 0 | 0 | 0 | 0 | 1.0 | +4.2 | ||
| v15 | 0 | +2.8 | −4.4 | −1.2 | 0 | 0 | 1.0 | +1.1 | ||
| v16 | 0 | +4.7 | 0 | −3.3 | 0 | 0 | 1.0 | +3.0 | ||
| v17 | +1.1* | +2.0 | +1.45* | −4.0 | 0 | 0 | +6.7 | +3.7 | ||
| v19 | 0 | +5.7* | −2.2 | −4.2 | 0 | 0 | −3.6 | 1.0 | ||
| Ad-CA | Non-P | v118 | −2.1 | −1.4 | NT | NT | NT | NT | +4.6 | +133.3 |
| v119 | −2.1 | −3.8 | −1.1 | −1.7 | −1.2 | −1.7 | +3.9 | +2.0 | ||
| v125 | −1.7 | −1.6 | 1.0 | −2.1 | 0 | −1.9 | −1.5 | +7.1 | ||
| v126 | −2.4 | −1.4 | −8.5 | −2.2 | 0 | −1.5 | +8.5 | +74.5 | ||
| v127 | −1.9 | −2.4 | −1.2 | −3.2 | −1.2 | −3.2 | −1.7 | +7.3 | ||
| v128 | −4.1 | −1.9 | +1.4 | +1.3 | NT | +1.4 | −1.7 | +10.0 | ||
| v147 | −1.3 | −1.2 | −2.7 | +1.3 | −2.1 | 0 | 1.0 | +146.0 | ||
| v149 | −1.1 | −1.2 | NT | NT | NT | NT | +5.8 | +9.7 | ||
| v156 | −2.5 | −2.1 | −1.2 | −2.4 | −1.6 | −1.0 | +1.5 | +9.2 | ||
| v160 | −1.5 | −1.3 | −3.6 | −3.2 | −6.5 | −23.8 | +1.8 | +2.1 | ||
| v172 | −2.0 | −1.6 | −1.1 | −2.7 | 0 | −24.9 | +1.1 | +7.7 | ||
| v173 | 1.0 | −1.3 | 0 | −1.2 | 0 | 0 | 0 | +15.3 | ||
| v179 | −2.4 | −1.1 | −2.1 | −1.9 | 0 | −1.4 | −1.4 | +28.2 | ||
| v184 | −4.9 | −3.1 | −1.7 | −1.1 | 0 | 0 | 0 | +475.0 | ||
| v194 | −3.5 | −1.9 | 0 | −1.2 | 0 | 0 | −2.4 | +5.1 | ||
| v195 | 0 | −4.2 | 1.0* | −1.5 | 0 | −1.2* | −1.4 | +10.1 | ||
| v196 | −2.9 | −1.8 | −1.2 | −2.6 | 0 | −2.4 | +2.0 | +16.2 | ||
The effects of CHMI on pre-CHMI cellular and antibody activities are shown for each volunteer in the DNA/Ad and AdCA trials. The fold-change of pre- and post-CHMI activities is shown as a rise (+) or fall (−). The threshold for significance was fold-changes >±1.5, which was the range of changes in non-immunized infectivity controls: rises are shown in blue boxes, and falls are shown in green boxes.
Fold-changes of responses to immunodominant peptide pools.
Activity was negative pre-CHMI but was positive post-CHMI. 0: Pre-CHMI and post-CHMI activities were negative.
Figure 2.
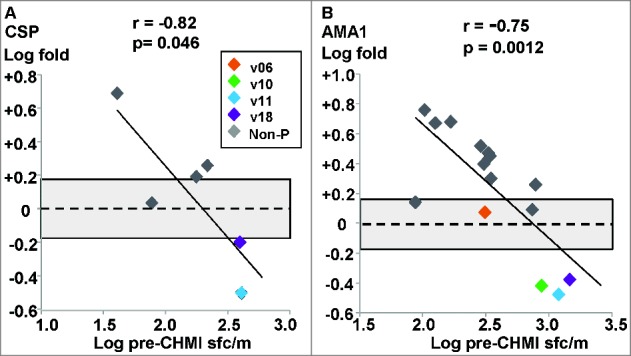
DNA/Ad trial: ex vivo ELISpot IFN-γ activities to CSP and AMA1. The associations of fold-changes of pre-CHMI and post-CHMI activities with pre-CHMI activities are shown as log-transformed values, and the dotted line represents no-change. The shaded box shows ±1.5 range. (A) CSP: there was a significant relationship between fold-change and pre-CHMI activities; the fold changes of 2 protected (v11, v18) and 3 non-protected subjects were greater than ±1.5 (shaded box). (B) AMA1: there was a significant relationship between fold-change and pre-CHMI activities; the fold changes of theAMA1 immunodominant pools of v10 (Ap8), v11 (Ap10) and v18 (Ap8) were used as they represented most of the total summed activities. The fold changes of 3 protected subjects (v10, v11, v18) and 9 non-protected subjects were greater than ±1.5 (shaded box).
Figure 3.
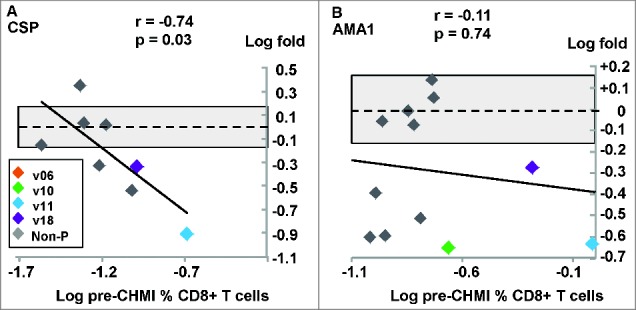
DNA/Ad trial: CD8+ T cell IFN-γ activities to CSP and AMA1. The associations of fold-changes of pre-CHMI and post-CHMI activities with pre-CHMI activities are shown as log-transformed values, and the dotted line represents no-change. The shaded box shows ±1.5 range. (A) CSP: there was a significant relationship between fold-change and pre-CHMI activities; the fold changes of 2 protected (v11, v18) and 3 non-protected subjects were greater than ±1.5 (shaded box). (B) AMA1: there was no significant relationship between fold-change and pre-CHMI activities; the fold changes of 3 protected (v10, v11, v18) and 4 non-protected subjects were greater than −1.5 (shaded box).
Figure 4.

AdCA trial: ex vivo ELISpot IFN-γ activities to CSP and AMA1. The associations of fold-changes of pre-CHMI and post-CHMI activities with pre-CHMI activities are shown as log-transformed values, and the dotted line represents no-change. The shaded box shows ±1.5 range. (A) CSP: there was a significant negative association between fold-change and pre-CHMI activities; activities of all 13/17 positive before CHMI fell after CHMI. (B) AMA1: although activities of all 17/17 subjects that were positive pre-CHMI all fell after CHMI, there was no relationship between fold-change and pre-CHMI activities as only 10/17 were greater than −1.5.
Figure 5.

AdCA trial: CD8+ T cell IFN-γ activities to CSP and AMA1. The associations of fold-changes of pre-CHMI and post-CHMI activities with pre-CHMI activities are shown as log-transformed values, and the dotted line represents no-change. The shaded box shows ±1.5 range. (A) CSP: there was no significant relationship between fold-change and pre-CHMI activities, although activities fell in 5/15 subjects. (B) AMA1: There was a significant relationship between fold-change and pre-CHMI and activities fell in 9/15 subjects.
Figure 6.

DNA/Ad and AdCA trials: association of fold changes of antibody responses to CSP and AMA1 with pre-CHMI activities. The associations of fold-changes of pre-CHMI and post-CHMI activities with pre-CHMI activities are shown as log-transformed values, and the dotted line represents no-change. (A) DNA-Ad trial: AMA1: there was no relationship between fold change after CHMI and pre-CHMI activities. (B) AdCA trial: CSP: there was a significant relationship between fold change after CHMI and pre-CHMI activities. (C): AdCA trial: AMA1: there was a stronger significant relationship than CSP between fold change and pre-CHMI activities. In the AdCA trial, CHMI had a greater effect on lower than higher pre-CHMI activities, and for AMA1, CHMI greatly increased pre-CHMI activities.
CSP: The geometric mean of all subjects pre-CHMI (86 sfc/m, range 13–408 sfc/m) did not significantly change post-CHMI (119 sfc/m, range 19–402 sfc/m). Pre-CHMI activities were positive in 4 subjects and 6 subjects were positive post-CHMI. Post-CHMI activities fell in 2 of 4 protected subjects (v11 and v18) who had the highest pre-CHMI activities including activities to the immunodominant peptide pools (v11 to Cp9: −3.2-fold; v18 to Cp9:-1.6-fold), but rose in 3 non-protected subjects including one that was negative pre-CHMI (Table 1). There was a weakly significant negative association of fold-change with pre-CHMI activities of the 6 subjects that were positive pre-CHMI or post-CHMI (Fig. 2A), but given the small number of positive subjects we include this only for information. AMA1: The geometric mean of all subjects pre-CHMI (348 sfc/m, range 88–1270 sfc/m) significantly (p = 0.012) rose post-CHMI (672 sfc/m, range 121–1426 sfc/m). Twelve/15 subjects were positive pre-CHMI and 14/15 subjects were positive post-CHMI including 2 that were negative pre-CHMI. Post-CHMI activities fell in 3 of 4 protected subjects (Table 1), including activities to the immunodominant peptide pools (v10 to Ap8:-2.0-fold; v11 to Ap10:-2.6-fold; v18 to Ap8:-1.8-fold) and rose in 9/11 non-protected subjects (Table 1). There was a significant negative association between fold-change and pre-CHMI activities of the 14 subjects that were positive pre-CHMI or post-CHMI and CHMI increased activities in subjects with lowest pre-CHMI activities who were all non-protected (Fig. 2B). Activity of the fourth protected subject (v06) was unchanged after CHMI and no single AMA1 peptide pool was immunodominant.
CD8+ T cell IFN-γ activities: activities of protected subjects fell and non-protected subjects fell or were unchanged
CSP: The geometric mean of all subjects pre-CHMI (0.045%, range 0–0.29%) did not significantly change post-CHMI (0.03%, range 0–0.11%). Four/15 subjects were positive pre-CHMI, and 6 subjects were positive post-CHMI including 4 non-protected subjects that were negative pre-CHMI. Post-CHMI activities fell in 2 of 4 protected subjects (Table 1) including activities to the immunodominant peptide pools (v11 to Cp9:-9.9-fold; v18 to Cp9: −2.4-fold). There was a weakly significant negative association between fold-changes of these 8 positive subjects and pre-CHMI activities (Fig. 3A) showing that CHMI caused falls in 4 subjects with the highest pre-CHMI activities (Fig. 3A). AMA1: The geometric mean activity of all subjects pre-CHMI (0.14%, range 0.05–0.98%) did not significantly change post-CHMI (0.08%, range 0–0.25%). 12/15 subjects were positive pre-CHMI and this fell to 8/15 positive subjects post-CHMI. Post-CHMI activities of 3 of 4 protected subjects fell post-CHMI (Table 1) including activities to the immunodominant peptide pools (v10 to Ap8:-4.2-fold; v11 to Ap10:-4.5-fold; v18 to Ap8:-2.5-fold), but also fell in 4 non-protected subjects (Table 1). Although there was no association between fold-changes of the 12 positive subjects and pre-CHMI activities, activities of 3 protected and 4 non-protected subjects fell post-CHMI (Fig. 3B).
CD8+ T cell IFN-γ effector memory (EM) activities by ICS/flow cytometry: activities of protected subjects fell
DNA/Ad induced CD8+ T cell IFN-γ EM activities in 2 protected subjects (v11, v18) to CSP, and in 3 protected subjects (v10, v11, v18) to AMA1, but not in non-protected subjects.
CSP: At pre-CHMI, 2 protected subjects (v11 and v18) were positive and summed activities of each fell post-CHMI (Table 1) and became negative, and post-CHMI activities to the immunodominant peptide pools also fell (v11 to Cp9:-28.9-fold; v18 to Cp6:-2.3-fold). AMA1: at pre-CHMI, 3 protected subjects (v10, v11 and v18) were positive and summed activities of each fell post-CHMI (Table 1), and v10 became negative; post-CHMI activities to the immunodominant peptide pools also fell: v10 to Ap8 (−11.5-fold); v11 to Ap10 (−5.2-fold); v18 to Ap8 (−3.0-fold).
We next analyzed the effect of CHMI on subjects immunized with AdCA alone,2 to compare the outcomes with the DNA/Ad trial.
AdCA trial
Ex vivo ELISpot IFN-γ: activities fell or were unchanged
CSP: The geometric mean of all subjects pre-CHMI (243 sfc/m, range 14–1333 sfc/m) fell but not significantly post-CHMI (135 sfc/m, range 24–1327 sfc/m). Thirteen/17 subjects were positive pre-CHMI and this fell to 7/17 positive subjects post-CHMI. There was a significant negative association between fold-changes in activities and pre-CHMI activities and CHMI caused a fall in activities of all these non-protected subjects (Fig. 4A). AMA1: The geometric mean of all subjects pre-CHMI (1304 sfc/m, range 435–4593 sfc/m) significantly (p = 0.02) fell after CHMI (731 sfc/m, range 165–1335 sfc/m). There was no association between fold-changes in activities and pre-CHMI activities although activities of 10/17 subjects fell and 7/17 were unchanged (Table 1; Fig. 4B).
CD8+ T cell IFN-γ activities by ICS/flow cytometry: activities fell or were unchanged
CSP: The geometric mean of all subjects pre-CHMI (0.11%, range 0.05–0.57%) did not significantly change post-CHMI (0.08%, range 0–0.49%). 12/15 subjects were positive pre-CHMI, and fell to 8/15 subjects post-CHMI including one subject that was negative pre-CHMI. There was no association between fold-changes and pre-CHMI activities, but activities of 5/15 subjects fell and 10/15 subjects were unchanged (Table 1; Fig. 5A). AMA1: The geometric mean of all subjects pre-CHMI (0.47%, range 0.10–2.44%) significantly (p = 0.04) fell post-CHMI (0.28%, range 0.07–1.00%) and 15/15 subjects that were positive pre-CHMI remained positive post-CHMI (Table 2). In contrast to CSP, there was a highly significant negative association between the fold-changes in activities compared to pre-CHMI activities, suggesting that the falls in activities due to CHMI was greatest in 9 subjects with the highest pre-CHMI activities (Table 1; Fig. 5B).
Table 2.
Summary of effects of CHMI
| Antigen |
|||
|---|---|---|---|
| Vaccine | Assay | CSP | AMA1 |
| DNA-Ad | ELISpot | Fell in protected; rose in non-protected | Fell in protected; rose in non-protected |
| CD8+ T IFN-γ | Fell in protected; fell/unchanged in non-protected | Fell in protected; fell/unchanged in non-protected | |
| CD8+ T IFN-γ EM | Fell in protected; non-protected all negative | Fell in protected; non-protected all negative | |
| Antibody | Unchanged | Fell in protected/rose in non-protected | |
| AdCA | ELISpot | Fell/unchanged | Fell/unchanged |
| CD8+ T IFN-γ | Fell/unchanged | Fell/unchanged | |
| CD8+ T IFN-γ EM | Fell/unchanged | Fell | |
| Antibody | Rose | Rose | |
Fall, rises, no changes (none) derived from Figures 2–6.
CD8+ T cell IFN-γ EM activities by ICS/flow cytometry: activities fell or were unchanged
CSP: The geometric mean activity of all subjects pre-CHMI (<0.03%, range 0–0.38%) was unchanged post-CHMI (<0.03%, range 0–0.23%). At pre-CHMI only 5/15 subjects had positive activities and post-CHMI activities of 3/15 subjects fell and one became negative (Table 1). AMA1: The geometric mean of all subjects pre-CHMI (0.08%, range 0–1.54%) did not significantly change post-CHMI (0.06%, range 0–0.0.40%). At pre-CHMI, 11/15 subjects had positive activities and post-CHMI activities fell in 7/15 subjects (Table 1), and were dramatic in 2 subjects (−23.8, −24.9) who became negative.
We have previously reported that DNA/Ad induced low antibody activities to CSP and AMA1,1 whereas AdCA induced higher levels of antibodies, especially to AMA1.9 We expected, based on our second hypothesis, that antibody activities would rise after CHMI. CHMI itself did not induce antibody responses to CSP or AMA1 in the infectivity controls (data not shown).
DNA/Ad trial: ELISA antibody: activities rose in non-protected and fell in protected subjects (AMA1)
CSP: pre-CHMI activities of all subjects were low (data not shown), as seen with the infectivity controls, but post-CHMI activities of 3 of 4 protected subjects fell, and also fell in 3 and rose in 2 non-protected subjects (Table 1). AMA1: the geometric mean activities pre-CHMI (11.9 μg/mL, range 1.5–102 μg/mL) did not significantly change post-CHMI (14.6 μg/mL, range 1.8–70.8 μg/mL). The association between fold-changes and pre-CHMI activities was not significant although post-CHMI activities of 2 of 4 protected subjects fell and rose in 7 non-protected subjects (Table 1; Figure 6A).
AdCA trial: ELISA antibody: activities rose in some to CSP and all to AMA1
CSP: the geometric mean activities pre-CHMI (2.2 μg/mL, range 0.5.1–7.8 μg/mL) did not significantly change after CHMI (3.2 μg/mL, 1.1–6.9 μg/mL μg/mL). However, there was a highly significant negative association between fold-change and pre-CHMI activities, and activities rose in 7 subjects with the lowest pre-CHMI activities (Table 1; Fig. 6B). AMA1: the geometric mean activities pre-CHMI (5.6 μg/mL, range 2.1–38.0 μg/mL) rose considerably (16.2-fold) after CHMI (90.7 μg/mL, range 19–380 μg/mL) and the change was highly significant (p = < 0.001). Activities of all subjects rose after CHMI and there was a highly significant negative association between fold-changes in activities compared to pre-CHMI activities (Fig. 6B), suggesting that CHMI caused the largest increases (+475-fold, +146-fold) in activities of subjects with the lowest pre-CHMI activities.
CHMI affected pre-CHMI activities differently in the DNA/Ad and AdCA trials
The effect of CHMI on pre-CHMI activities differed when the AdCA vaccine was primed with DNA (Table 2). In the DNA/Ad trial, CHMI caused falls in ELISpot activities of protected and rises in activities of non-protected subjects, whereas CHMI predominantly caused falls in ELISpot activities in the AdCA trial. However, CHMI caused falls in CD8+ T cell IFN-γ activities in both protected and non-protected subjects in the DNA-Ad and AdCA trials. It thus appears that ELISpot measured other cell types in addition to CD8+ T cell activities in the non-protected subjects in the DNA/Ad trial.. However, in the AdCA trials, the effects of CHMI on ELISpot and CD8+ T cell activities were more similar suggesting that CD8+ T cells predominated in ELISpot activities. In contrast CHMI caused falls in antibody activities of protected subjects and rises in non-protected subjects in the DNA/Ad trial but rises in the AdCA trial to CSP and often dramatically to AMA1.
Discussion
The aim of this study was to examine the effect of CHMI on cellular and antibody responses in the DNA/Ad and AdCA trials according to 2 pre-analysis hypotheses. CHMI itself did not induce significant ELISpot IFN-γ responses in the infectivity controls, except very low activities to a single AMA1 peptide pool in 3 subjects, nor did CHMI induce significant antibody responses to CSP and AMA1. In some assays the numbers of subjects with positive ELISpot IFN-γ (Fig. 2A) and CD8+ T cell IFN-γ EM responses (Table 1) to CSP were low and the effects of CHMI are shown for information only.
In the DNA/Ad trial, the first finding was that CHMI caused falls in cellular responses of protected subjects measured by ELISpot and CD8+ T cell IFN-γ assays to CSP and AMA1 as hypothesized. This was especially true of CD8+ T cell IFN-γ EM activities. ELISpot activities of these protected volunteers were abolished by CD8+ T cell depletion,1 suggesting that in these subjects ELISpot is predominantly measuring CD8+ T cell IFN-γ, probably explaining the consistency of these assays, ELISpot IFN-γ and ICS. One interpretation of this effect is that, in response to liver stage infections that develop after CHMI, CD8+ IFN-γ cells might migrate from the periphery to the liver, causing a fall in peripheral activities. In agreement with this hypothesis, after immunization with radiation-attenuated sporozoites, the frequencies of antigen specific CD8+ IFN-γ secreting cells to a few antigens were low in the peripheral circulation of some subjects but not detected in other subjects, and are consistent with the finding that antigen-specific CD8+ T cells were found in the liver in non-human primates similarly immunized.17 Rodent models have suggested that hepatic-resident CD8+ T memory responses are associated with protection5 and may persist in the liver.20-22 It is possible, but difficult to prove, that antigen-specific T cells detected in the peripheral circulation of human subjects reflect T cells resident in the liver. Liver-resident memory T cells in mice display a unique transcriptional profile,22 and identification of such a profile in humans could aid vaccine development.
However, CHMI affected ELISpot and CD8+ T cell IFN- γ activities of non-protected subjects in the DNA/Ad and AdCA trials differently; ELISpot activities rose and CD8+ T cell IFN- γ activities were either unchanged or fell. The most straightforward interpretation is that DNA/Ad immunization elicited CD8+ T cell IFN- γ activities and other cell types at post CHMI in the non-protected subjects, such as natural killer (NK) and γδ cells that contributed to ELISpot activities. Indeed, NK cells have been shown to control P. falciparum blood stage infection23 and may contribute to RTS,S induced protection,24 and NK and γδ cells may contribute to protection induced by live P. falciparum sporozoite/chloroquine immunization.10,25 In this case, CHMI increased activities of these other cells masking the fall of activities of CD8+ T cells that were insufficient to induce protection, even though they apparently migrated to the liver.
In contrast, in the AdCA trial, CHMI predominantly caused falls in ELISpot and CD8+ T cell IFN- γ activities of these subjects who were all non-protected. This is consistent with the interpretation that AdCA induced predominantly CD8+ T cells, not other types of immune cells, and these possibly also migrated to the liver, but did not induce protection. We have previously shown that CD8+ T cells of the protected volunteers recognize small regions of CSP and AMA associated with class 1-restricted epitopes in AMA1 [20], whereas CD8+ T cells of non-protected subjects in the DNA/A and AdCA trials recognize multiple regions of CSP and AMA1. We suggest therefore that CD8+ T cells may migrate to the liver following CHMI in protected and non-protected subjects, but only CD8+ T cells that recognized a small number of CSP and AMA1 epitopes were protective, whereas those that recognized larger numbers of CSP and AMA1 regions were not protective.
An alternative interpretation of the drops in CD8+ T cell IFN-γ activities in the AdCA trial may be that AdCA immunization drove CD8+ T cell EM differentiation to exhaustion,3 as their cytokine profile lacked TNF and IL2 secretion,26-28 and differs after DNA/Ad immunization when memory T cells secreted both TNF and IL-2.3 In that case, it is possible that the CHMI boost further drove CD8+ T EM cells to exhaustion leading to apoptosis and an apparent reduction in peripheral activities. In future vaccine trials, we will measure markers of T cell exhaustion including programmed cell death-1,29-31 that has been implicated in malaria infection and T cell exhaustion.32,33
Perhaps the most surprising outcome was the dramatic boost of anti-AMA1 antibodies of non-protected subjects in the DNA/Ad trial, and the AdCA trial, where there was a significant relationship between fold-change and pre-CHMI activities. CHMI itself did not induce antibodies to CSP or AMA1; CHMI also failed to induce anti-AMA1 antibodies in a separate trial. These outcomes suggest that B-cell responses, in contrast to T cell responses, were readily boosted by live sporozoites and probably by the transient blood stage infection before chloroquine treatment. However, the effect of CHMI was greater on antibody responses to AMA1 than CSP, and this might be due to the small numbers of sporozoites delivered by CHMI compared to the much larger numbers of red blood stages derived from each sporozoite, especially as 2 cycles of asexual development occurred before chloroquine treatment. Anti-AMA1 antibody activities rose to a geometric mean of 91 µg/mL, and ranged up to 380 µg/mL. These activities are comparable to those achieved in a field trial with a recombinant protein AMA1 vaccine FMP2.1/AS02A, measured in the same laboratory as this study, and induced efficacy against clinical malaria identical to the vaccine strain.34 Although the AdCA vaccine failed to induce protection9 it is interesting to speculate that natural transmission might be expected to boost anti-AMA1 antibody activities to levels previously only achieved with AMA1 recombinant protein vaccines.
Other studies have shown that CHMI also boosts antibody responses in mice and non-human primates, particularly following live vector immunization. CHMI boosted antibody but not CD8+ T cell responses in mice primed with a Salmonella typhimurium and boosted with recombinant protein containing a CD8+ T cell epitope from P. berghei and appeared to enhance protection.16 Similarly, CHMI boosted antibody responses in a DNA-prime pox virus boost containing 2 malaria blood stage antigens MSP142 and AMA1 induced low antibody responses in rhesus monkeys immunized with DNA-pox virus vaccines containing MSP1 and AMA1.15 In humans CHMI has also been shown to increase antibody activities in a human study using chimpanzee adenovirus ChAd63 followed by pox virus boost encoding AMA1, or a pox-based vector (NYVAC) encoding MSP119 and AMA1.14
These outcomes suggest that in future trials we should consider more detailed analyses of T cell responses, especially measuring markers of T cell exhaustion. They also suggest that natural transmission may boost vaccine-induced responses, especially antibody responses; we are planning further development of the DNA/Ad vaccine that if successful would be transferred to a malaria-endemic trial site. While presentation of antigens derived from sporozoites and blood stages may differ from protein vaccines, these result also suggest that a further boost of the DNA/Ad vaccine with recombinant CSP or AMA1 may improve immunogenicity, and such studies are underway in mice.
Generalizability
This analysis extends our understanding of the effect of DNA priming on the immunogenicity of adenovirus vectors and identified that live sporozoite challenge particularly boosted antibody responses. These outcomes may be applicable to live whole sporozoite vaccines where priming with live vectors may beneficially affect responses induced by live sporozoites.
Limitations
Our findings, that CHMI boosted antibody activities, have not been tested for improved efficacy; this could be addressed, at least preliminarily for AMA1, using functional antibody growth inhibition assays.35
Methods
Human ethics statement
The study protocols for these clinical trials were approved by the Institutional Review Boards at the Walter Reed Army Institute of Research (WRAIR) and the National Naval Medical Center (NNMC). The study was conducted at the WRAIR Clinical Trials Center in accordance with the principles described in the Nuremberg Code and the Belmont Report; all federal regulations regarding the protection of human participants as described in 32 CFR 219 (The Common Rule) and instructions from the Department of Defense, the Department of the Army, the Department of the Navy and the Bureau of Medicine and Surgery of the United States Navy; and the internal policies for human subject protections and the standards for the responsible conduct of research of the US Army Medical Research and Materiel Command (USAMRMC) and the Naval Medical Research Center (NMRC). NMRC and WRAIR each hold a Federalwide Assurance from the Office of Human Research Protections (OHRP) under the Department of Health and Human Services. NMRC also holds a Department of Defense/Department of the Navy Addendum to the Federalwide Assurance for human subject protections. All key personnel were certified as having completed mandatory human research ethics education curricula and training. All potential study subjects provided written, informed consent before screening and enrollment and had to pass an assessment of understanding.
Human subjects
The full details of the clinical findings of these trials, including patient recruitment and flow, safety and tolerability have been previously reported.1,9 Fifteen subjects were immunized with DNA/Ad and 4 were fully protected against CHMI, and none of the 11 non-protected subjects showed a significant delay to parasitemia.1 In a separate trial, 18 subjects were immunized with AdCA, and one showed a significant delay to onset of parasitemia after CHMI but none were fully protected.9
Immunological endpoints
DNA/Ad samples used in this study were collected 22/23 days post Ad administration, 5 or 6 days before CHMI (pre-CHMI), and 4 weeks after CHMI (post-CHMI). AdCA samples were collected 22/23 days post Ad administration that 5 or 6 days before CHMI (pre-CHMI), and 4 weeks after CHMI (post-CHMI). A rise or fall in activity is calculated as the fold-change post-CHMI compared to pre-CHMI activity: for example a 2-fold rise is expressed as +2.0, and a 2-fold fall is expressed as −2.0.
Ex vivo Enzyme Linked Immunospot Interferon-gamma Assays (ELISpot IFN-γ)
IFN-γ responses were measured by IFN-γ ELISpot assay7 using fresh peripheral blood mononuclear cells (PBMC). The full length P. falciparum 3D7 CSP sequence was covered by 15 amino acid (aa) peptides overlapping by 11 aa and combined into 9 pools (Cp1-Cp9) each containing 3 to 12 peptides.7 Full length AMA1 was covered by 15mers that were combined into 12 pools (Ap1-Ap12) each containing 10–13 peptides.7 Results, expressed as spot forming cells/million PBMC (sfc/m), are shown as: (1) the magnitude of responses of each subject to individual CSP or AMA1 peptide pools, (2) summed responses of each subject, or (3) numbers of positive subjects defined as a subject with a positive response to at least one CSP or one AMA1 peptide pool.7,8 A positive response to a given CSP or AMA1 peptide pool was defined as positive after showing (1) a statistically significant difference between the number of spot forming cells in triplicate or quadruplicate test wells and triplicate or quadruplicate negative control wells (Student's 2 tailed t-test), plus (2) at least a doubling of spot forming cells in test wells relative to negative control wells, plus (3) a difference of at least 10 spots between test and negative control wells.
Flow cytometry with intracellular cytokine staining (ICS)
Frozen PBMC were stimulated by each CSP (Cp1, Cp2, Cp6 and Cp9) or AMA1 (Ap1, Ap3, Ap4, Ap8, Ap9 and Ap10) peptide pools as previously described.7 Control stimulants were medium alone and the CEF peptide pool (Anaspec, San Jose, CA). Cells were phenotyped as CD4+ and CD8+ T cells and stained for IFN-γ. Data for peptide pools were corrected for media response at each time point. A positive response was defined as a frequency of CD4+ or CD8+ IFN-γ cells exceeding the geometric mean + 3 standard deviations of the medium-stimulated controls, 0.030%.9 A subject was considered positive if activity to one or more peptide pools was at least 0.03%; some subjects who had summed activities >0.03% were considered negative if activities to individual peptide pools did not reach 0.03%. Activities are shown as (1) each subject's responses individual CSP or AMA1 peptide pools, (2) summed responses of all subjects, or (3) numbers of positive subjects defined as a subject with a positive response to at least one CSP or one AMA1 peptide pool.7,8
CD8+ T cell memory responses
Frozen PBMC taken at the same time points were stimulated with the immunodominant CSP or AMA1 peptide pools,7 sorted as CD8+ T cells, and phenotyped by CD45RA and CD27 staining as effector memory (EM) cells that were CD45RA−CD27−.36 Each pool was then stained for IFN-γ, and activities of EM T cells were expressed as per cent of CD8+ T cells. A positive response was defined as exceeding the geometric mean + 3 standard deviations of the medium stimulated controls (0.03%). Responses are shown as activities to individual peptide pools, the sum of responses to individual peptide pools, the geometric mean of summed responses, and the number of positive subjects.
Statistical analyses
The Mann-Whitney U test was used to compare the summed cellular responses of protected and non-protected subjects from the 2 trials. Rank correlations were employed to examine the relationships between the fold-change between pre- and post-CHMI activities.37,38 All values were transformed to base 10 logarithms for analysis. Activities of subjects that had positive activities pre- and post-CHMI were included. Pearson product moment calculations were calculated for the log-transformed activities and correlation was calculated as r. Two-sided tests were used, with p = < 0.05 considered significant. This was previously used to determine the association of neutralizing antibodies to the Ad-5 component that had no effect on immunogenicity in the DNA/Ad trial, where 10/15 subjects were Ad-5 seronegative, and 5 subjects were seropositive.1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank all the study volunteers who participated in the trial. We thank Dr. Thomas L. Richie, former Director of the NMRC Malaria Department, and Dr. Carter Diggs and Dr. Lorraine Soisson of the US Agency for International Development (USAID) for their involvement in the design of the clinical trials used in this study. Active duty military personnel at the time they contributed to this work: CT, IC, and JEE. MS and EV were US Government employees. The work of these individuals was prepared as part of official government duties. Title 17 USC. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 USC. §101 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person's official duties. The study protocol for the clinical trial presented in this manuscript was approved by the WRAIR and NMRC Institutional Review Boards, in compliance with all applicable Federal Regulations governing protection of human subjects. All volunteers gave written informed consent. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, the Department of the Army, the Department of Defense, or the US Government.
Funding
This work was supported by USAID “Development of Adenovirus-Vectored Malaria Vaccines” Grant # GHA-P-00-03-00006-01, Project Number 936–3118; and the Congressionally Directed Medical Research Program “Development of Recombinant Adenoviral-based Vaccines against Malaria” Grant # W81XWH- 05-2-0041. Website https://cdmrp.org; Military Infectious Research Program “Phase 1/2a clinical trials assessing the safety, tolerability, immunogenicity and protective efficacy of Ad5-CA, a 2-antigen, adenovirus-vectored Plasmodium falciparum malaria vaccine, in healthy, malaria-naive adults work unit number 62787A 870 F 1432. The funders reviewed and approved the study design. The funders had no role in the data collection and analysis, or decision to prepare and publish this manuscript.
Trial Registrations
DNA/Ad: ClinicalTrials.gov NCT00870987; AdCA: NCT00392015
References
- 1.Chuang I, Sedegah M, Cicatelli S, Spring M, Polhemus M, Tamminga C, Patterson N, Guerrero M, Bennett JW, McGrath MG, et al.. DNA prime/adenovirus boost malaria vaccine encoding P. falciparum CSP and AMA1 induces sterile protection associated with cell-mediated immunity. PLoS One 2013; 8:1371; PMID:23457473; http://dx.doi.org/ 10.1371/journal.pone.0055571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tamminga C, Sedegah M, Maiolatesi S, Fedders C, Reyes S, Reyes A, Vasquez C, Alcorta Y, Chuang I, Spring M, et al.. Human adenovirus 5-vectored Plasmodium falciparum NMRC-M3V-Ad-PfCA vaccine encoding CSP and AMA1 is safe, well-tolerated and immunogenic but does not protect against controlled human malaria infection. Hum Vaccin Immunother 2013; 9:2165-2177; PMID:23899517; http://dx.doi.org/ 10.4161/hv.24941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sedegah M, Hollingdale MR, Farooq F, Ganeshan H, Belmonte M, Kim Y, Peters B, Sette A, Huang J, McGrath S, et al.. Sterile Immunity to Malaria after DNA Prime/Adenovirus Boost Immunization Is Associated with Effector Memory CD8+T Cells Targeting AMA1 Class I Epitopes. PLoS One 2014; 9:e106241; PMID:25211344; http://dx.doi.org/ 10.1371/journal.pone.0106241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zarling S, Berenzon D, Dalai S, Liepinsh D, Steers N, Krzych U. The survival of memory CD8 T cells that is mediated by IL-15 correlates with sustained protection against malaria. J Immunol 2013; 190:5128-5141; PMID:23589611; http://dx.doi.org/ 10.4049/jimmunol.1203396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nganou-Makamdop K, van Gemert GJ, Arens T, Hermsen CC, Sauerwein RW. Long term protection after immunization with P. berghei sporozoites correlates with sustained IFNgamma responses of hepatic CD8+ memory T cells. PLoS One 2012; 7:e36508; PMID:22563506; http://dx.doi.org/ 10.1371/journal.pone.0036508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epstein JE, Tewari K, Lyke KE, Sim BK, Billingsley PF, Laurens MB, Gunasekera A, Chakravarty S, James ER, Sedegah M, et al.. Live attenuated malaria vaccine designed to protect through hepatic CD8(+) T cell immunity. Science 2011; 334:475-480; PMID:21903775; http://dx.doi.org/ 10.1126/science.1211548 [DOI] [PubMed] [Google Scholar]
- 7.Sedegah M, Tamminga C, McGrath S, House B, Ganeshan H, Lejano J, Abot E, Banania G, Sayo R, Farooq F, et al.. Adenovirus 5-vectored P. falciparum Vaccine Expressing CSP and AMA1. Part A: Safety and Immunogenicity in Seronegative Adults. PLoS One 2011; 6:e24586; PMID:22003383; http://dx.doi.org/ 10.1371/journal.pone.0024586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamminga C, Sedegah M, Regis D, Chuang I, Epstein JE, Spring M, Mendoza-Silveiras J, McGrath S, Maiolatesi S, Reyes S, et al.. Adenovirus-5-vectored P. falciparum vaccine expressing CSP and AMA1. Part B: safety, immunogenicity and protective efficacy of the CSP component. PLoS One 2011; 6:e25868; PMID:22003411; http://dx.doi.org/ 10.1371/journal.pone.0025868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamminga C, Sedegah M, Maiolatesi S, Fedders C, Reyes S, Reyes A, Vasquez C, Alcorta Y, Chuang I, Spring M, et al.. Human Adenovirus 5-Vectored Plasmodium falciparum NMRC-M3V-Ad-PfCA Vaccine encoding CSP and AMA1 is safe, well tolerated and immunogenic but does not protect against controlled human malaria infection. Hum Vaccin Immunother 2013; 9:2165-2177; PMID:23899517; http://dx.doi.org/ 10.4161/hv.24941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teirlinck AC, McCall MB, Roestenberg M, Scholzen A, Woestenenk R, de Mast Q, van der Ven AJ, Hermsen CC, Luty AJ, Sauerwein RW. Longevity and composition of cellular immune responses following experimental Plasmodium falciparum malaria infection in humans. PLoS Pathog 2011; 7:e1002389; PMID:22144890; http://dx.doi.org/ 10.1371/journal.ppat.1002389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elias SC, Choudhary P, de Cassan SC, Biswas S, Collins KA, Halstead FD, Bliss CM, Ewer KJ, Hodgson SH, Duncan CJ, et al.. Analysis of human B-cell responses following ChAd63-MVA MSP1 and AMA1 immunization and controlled malaria infection. Immunology 2014; 141:628-644; PMID:24303947; http://dx.doi.org/ 10.1111/imm.12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoyanov CT, Boscardin SB, Deroubaix S, Barba-Spaeth G, Franco D, Nussenzweig RS, Nussenzweig M, Rice CM. Immunogenicity and protective efficacy of a recombinant yellow fever vaccine against the murine malarial parasite Plasmodium yoelii. Vaccine 2010; 28:4644-4652; PMID:20451637; http://dx.doi.org/ 10.1016/j.vaccine.2010.04.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biswas S, Choudhary P, Elias SC, Miura K, Milne KH, de Cassan SC, Collins KA, Halstead FD, Bliss CM, Ewer KJ, et al.. Assessment of Humoral Immune Responses to Blood-Stage Malaria Antigens following ChAd63-MVA Immunization, Controlled Human Malaria Infection and Natural Exposure. PLoS One 2014; 9:e107903; PMID:25254500; http://dx.doi.org/ 10.1371/journal.pone.0107903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ockenhouse CF, Sun PF, Lanar DE, Wellde BT, Hall BT, Kester K, Stoute JA, Magill A, Krzych U, Farley L, et al.. Phase I/IIa safety, immunogenicity, and efficacy trial of NYVAC-Pf7, a pox-vectored, multiantigen, multistage vaccine candidate for Plasmodium falciparum malaria. J Infect Dis 1998; 177:1664-1673; PMID:9607847; http://dx.doi.org/ 10.1086/515331 [DOI] [PubMed] [Google Scholar]
- 15.Hamid MM, Remarque EJ, El Hassan IM, Hussain AA, Narum DL, Thomas AW, Kocken CH, Weiss WR, Faber BW. Malaria infection by sporozoite challenge induces high functional antibody titres against blood stage antigens after a DNA prime, poxvirus boost vaccination strategy in Rhesus macaques. Malar J 2011; 10:29; PMID:21303498; http://dx.doi.org/ 10.1186/1475-2875-10-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tartz S, Deschermeier C, Retzlaff S, Heussler V, Sebo P, Fleischer B, Jacobs T. Plasmodium berghei sporozoite challenge of vaccinated BALB/c mice leads to the induction of humoral immunity and improved function of CD8(+) memory T cells. Eur J Immunol 2012; 43:693-704; PMID:23229763; http://dx.doi.org/ 10.1002/eji.201142262 [DOI] [PubMed] [Google Scholar]
- 17.Epstein JE, Tewari K, Lyke KE, Sim BK, Billingsley PF, Laurens MB, Gunasekera A, Chakravarty S, James ER, Sedegah M, et al.. Live Attenuated Malaria Vaccine Designed to Protect through Hepatic CD8+ T Cell Immunity. Science 2011; 334(6055):475-480; PMID:21903775; http://dx.doi.org/ 10.1126/science.1211548 [DOI] [PubMed] [Google Scholar]
- 18.Seder RA, Chang LJ, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, Holman LA, James ER, Billingsley PF, Gunasekera A, et al.. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 2013; 341:1359-1365; PMID:23929949; http://dx.doi.org/ 10.1126/science.1241800 [DOI] [PubMed] [Google Scholar]
- 19.Sedegah M, Hollingdale MR, Farooq F, Ganeshan H, Belmonte M, Kim Y, Peters B, Sette A, Huang J, McGrath MG, et al.. Sterile Immunity to Hepatic Stage Malaria after DNA Prime/Adenovirus Boost Immunization is Associated with Effector Memory CD8+T Cells Targeting AMA1 Class I Epitopes. PLoS One 2014; 9(9):e106241; PMID:25211344; http://dx.doi.org/ 10.1371/journal.pone.0106241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krzych U, Dalai S, Zarling S, Pichugin A. Memory CD8 T cells specific for plasmodia liver-stage antigens maintain protracted protection against malaria. Front Immunol 2012; 3:370; PMID:23233854; http://dx.doi.org/ 10.3389/fimmu.2012.00370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tse SW, Radtke AJ, Zavala F. Induction and maintenance of protective CD8+ T cells against malaria liver stages: implications for vaccine development. Mem Inst Oswaldo Cruz 2011; 106(Suppl 1):172-178; PMID:21881772; http://dx.doi.org/ 10.1590/S0074-02762011000900022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tse SW, Cockburn IA, Zhang H, Scott AL, Zavala F. Unique transcriptional profile of liver-resident memory CD8 T cells induced by immunization with malaria sporozoites. Genes Immun 2013; 14(5):302-9; PMID:23594961; http://dx.doi.org/ 10.1038/gene.2013.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Q, Amaladoss A, Ye W, Liu M, Dummler S, Kong F, Wong LH, Loo HL, Loh E, Tan SQ, et al.. Human natural killer cells control Plasmodium falciparum infection by eliminating infected red blood cells. Proc Natl Acad Sci U S A 2014; 111:1479-1484; PMID:24474774; http://dx.doi.org/ 10.1073/pnas.1323318111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horowitz A, Hafalla JC, King E, Lusingu J, Dekker D, Leach A, Moris P, Cohen J, Vekemans J, Villafana T, et al.. Antigen-specific IL-2 secretion correlates with NK cell responses after immunization of Tanzanian children with the RTS,S/AS01 malaria vaccine. J Immunol 2012; 188:5054-5062; PMID:22504653; http://dx.doi.org/ 10.4049/jimmunol.1102710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCall MB, Roestenberg M, Ploemen I, Teirlinck A, Hopman J, de Mast Q, Dolo A, Doumbo OK, Luty A, van der Ven AJ, et al.. Memory-like IFN-gamma response by NK cells following malaria infection reveals the crucial role of T cells in NK cell activation by P. falciparum. Eur J Immunol 2010; 40:3472-3477; PMID:21072880; http://dx.doi.org/ 10.1002/eji.201040587 [DOI] [PubMed] [Google Scholar]
- 26.Quinn KM, Da Costa A, Yamamoto A, Berry D, Lindsay RW, Darrah PA, Wang L, Cheng C, Kong WP, Gall JG, et al.. Comparative analysis of the magnitude, quality, phenotype, and protective capacity of simian immunodeficiency virus gag-specific CD8+ T cells following human-, simian-, and chimpanzee-derived recombinant adenoviral vector immunization. J Immunol 2013; 190:2720-2735; PMID:23390298; http://dx.doi.org/ 10.4049/jimmunol.1202861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang SH, Lee CG, Park SH, Im SJ, Kim YM, Son JM, Wang JS, Yoon SK, Song MK, Ambrozaitis A, et al.. Correlation of antiviral T-cell responses with suppression of viral rebound in chronic hepatitis B carriers: a proof-of-concept study. Gene Ther 2006; 13:1110-1117; PMID:16525482; http://dx.doi.org/ 10.1038/sj.gt.3302751 [DOI] [PubMed] [Google Scholar]
- 28.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 2003; 77:4911-4927; PMID:12663797; http://dx.doi.org/ 10.1128/JVI.77.8.4911-4927.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hafalla JC, Claser C, Couper KN, Grau GE, Renia L, de Souza JB, Riley EM. The CTLA-4 and PD-1/PD-L1 inhibitory pathways independently regulate host resistance to Plasmodium-induced acute immune pathology. PLoS Pathog 2012; 8:e1002504; PMID:22319445; http://dx.doi.org/ 10.1371/journal.ppat.1002504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26:677-704; PMID:18173375; http://dx.doi.org/ 10.1146/annurev.immunol.26.021607.090331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 2007; 8:239-245; PMID:17304234; http://dx.doi.org/ 10.1038/ni1443 [DOI] [PubMed] [Google Scholar]
- 32.Wykes MN, Horne-Debets JM, Leow CY, Karunarathne DS. Malaria drives T cells to exhaustion. Front Microbiol 2014; 5:249; PMID:24904561; http://dx.doi.org/ 10.3389/fmicb.2014.00249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Illingworth J, Butler NS, Roetynck S, Mwacharo J, Pierce SK, Bejon P, Crompton PD, Marsh K, Ndungu FM. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J Immunol 2013; 190:1038-1047; PMID:23264654; http://dx.doi.org/ 10.4049/jimmunol.1202438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thera MA, Doumbo OK, Coulibaly D, Laurens MB, Ouattara A, Kone AK, Guindo AB, Traore K, Traore I, Kouriba B, et al.. A field trial to assess a blood-stage malaria vaccine. N Engl J Med 2011; 365:1004-1013; PMID:21916638; http://dx.doi.org/ 10.1056/NEJMoa1008115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergmann-Leitner ES, Duncan EH, Burge JR, Spring M, Angov E. Miniaturization of a high-throughput pLDH-based Plasmodium falciparum growth inhibition assay for small volume samples from preclinical and clinical vaccine trials. Am J Trop Med Hyg 2008; 78:468-471; PMID:18337345 [PubMed] [Google Scholar]
- 36.Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med 1997; 186:1407-1418; PMID:9348298; http://dx.doi.org/ 10.1084/jem.186.9.1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamminga C. Adenovirus-5-Vectored P. falciparumVaccine Expressing CSP and AMA1. Part B: Safety, immunogenicity and Protective Efficacy of the CSP Component. PLoS One 2011; 6:e25868; PMID:22003411; http://dx.doi.org/ 10.1371/journal.pone.0025868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wessa P. Free Statistics Software. Offic Res Dev Edu 2014; 1(1):23-r7 [Google Scholar]