

Abstract
Autocrine, paracrine, endocrine, and neuroendocrine hormonal systems help regulate cardiovascular and renal function. Any change in the balance among these systems may result in hypertension and target organ damage, whether the cause is genetic, environmental or a combination of the two. Endocrine and neuroendocrine vasopressor hormones such as the renin-angiotensin system (RAS), aldosterone, and catecholamines are important for regulation of blood pressure and pathogenesis of hypertension and target organ damage. While the role of vasodepressor autacoids such as kinins is not as well defined, there is increasing evidence that they are not only critical to blood pressure and renal function but may also oppose remodeling of the cardiovascular system.
Here we will primarily be concerned with kinins, which are oligopeptides containing the aminoacid sequence of bradykinin. They are generated from precursors known as kininogens by enzymes such as tissue (glandular) and plasma kallikrein. Some of the effects of kinins are mediated via autacoids such as eicosanoids, nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), and/or tissue plasminogen activator (†PA). Kinins help protect against cardiac ischemia and play an important part in preconditioning as well as the cardiovascular and renal protective effects of angiotensin-converting enzyme (ACE) and angiotensin type 1 receptor blockers (ARB). But the role of kinins in the pathogenesis of hypertension remains controversial. A study of Utah families revealed that a dominant kallikrein gene expressed as high urinary kallikrein excretion was associated with a decreased risk of essential hypertension. Moreover, researchers have identified a restriction fragment length polymorphism (RFLP) that distinguishes the kallikrein gene family found in one strain of spontaneously hypertensive rats (SHR) from a homologous gene in normotensive Brown Norway rats, and in recombinant inbred substrains derived from these SHR and Brown Norway rats this RFLP cosegregated with an increase in blood pressure. However, humans, rats and mice with a deficiency in one or more components of the kallikrein-kinin-system (KKS) or chronic KKS blockade do not have hypertension. In the kidney, kinins are essential for proper regulation of papillary blood flow and water and sodium excretion. B2-KO mice appear to be more sensitive to the hypertensinogenic effect of salt. Kinins are involved in the acute antihypertensive effects of ACE inhibitors but not their chronic effects (save for mineralocorticoidsalt-induced hypertension).
Kinins appear to play a role in the pathogenesis of inflammatory diseases such as arthritis and skin inflammation; they act on innate immunity as mediators of inflammation by promoting maturation of dendritic cells, which activate the body’s adaptive immune system and thereby stimulate mechanisms that promote inflammation. On the other hand, kinins acting via NO contribute to the vascular protective effect of ACE inhibitors during neointima formation. In myocardial infarction produced by ischemia/reperfusion, kinins help reduce infarct size following preconditioning or treatment with ACE inhibitors. In heart failure secondary to infarction, the therapeutic effects of ACE inhibitors are partially mediated by kinins via release of NO, while drugs that activate the angiotensin type 2 receptor act in part via kinins and NO. Thus kinins play an important role in regulation of cardiovascular and renal function as well as many of the beneficial effects of ACE inhibitors and ARBs on target organ damage in hypertension.
Introduction
The kinin-generating system
Kininogenases such as tissue (glandular) and plasma kallikreins are enzymes that generate kinins by hydrolyzing substrates known as kininogens, which circulate at high concentrations in plasma. Kinins are rapidly destroyed by a group of peptidases known as kininases (Fig. 1). Plasma and tissue kallikrein (TK) arc both potent kininogenases as well as serine proteases. A single gene encodes for plasma kallikrein, and there is a large family of glandular kallikrein genes; however, KLK1 is the only TK known to generate kinins (hereafter referred to as TK, or simply kallikrein).
Figure 1.
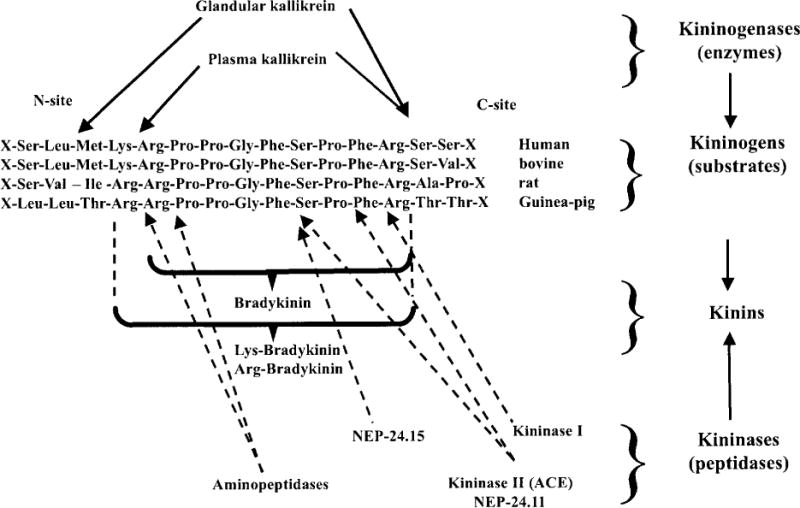
Site of kininogen cleavage (solid arrows) by the main kininogenases (glandular and plasma kallikrein). The broken arrows indicate sites of kinin cleavage by kininases (kininase I, kininase II, neutral endopeptidases 24.11 and 24.1 5, and aminopeptidases). [Modified after Carretero and Scicli (49).]
Plasma kallikrein-kinin system
Plasma kallikrein, also known as Fletcher factor, is expressed mainly in the liver; in plasma, it is found in the zymogen form (prekallikrein) and differs from glandular kallikrein not only biochemically but also immunologically and functionally. It preferentially releases bradykinin from high-molecular-weight kininogen (HMWK), also known as Fitzgerald factor. Together with HMWK and Hageman factor (factor XII), plasma kallikrein is involved in coagulation, fibrinolysis, and possibly activation of the complement system. Prolylcarboxypeptidase (PRCP, also called angiotensinase C) is a membrane protein that activates plasma prekallikrein in endothelial cells (177, 191, 261, 262) and accounts for sustained inflammatory responses to stimuli such as lipopolysaccharide (185). PRCP is constitutively expressed on the surface of the endothelial cell membrane, although it was originally purified from lysosomes (126, 245). When HMWK and plasma prekallikrein bind to the endothelial cell membrane, PRCP rapidly converts plasma prekallikrein to kallikrein, releasing kinins (262, 317). This pathway does not require factor XII. Taken together, the plasma kallikrein-HMWK system, acting through the release of bradykinin, could be involved in local regulation of blood flow as well as some of the effects of ACE inhibitors. On the other hand, patients with a congenital deficiency of plasma HMWK (Fitzgerald trait) have normal amounts of kinins in their blood (253). [For a review of the plasma kallikrein-HMWK system, see (69, 119, 277).]
Tissue (glandular) kallikrein-kinin system
Kallikrein belongs to a family of serine proteases with very high homology expressed by genes that are tightly clustered and arranged in tandem on the same chromosome. The kallikrein family is estimated to contain at least 3 genes in humans, 20 in rats, and 23 to 30 in mice, many of them pseudogenes (65). However, despite their highly homologous amino acid composition, most of these proteases are not kininogenases and act on different substrates. For example, in rats tonin hydrolyzes angiotensinogen and generates angiotensin II; while in humans prostate-specific antigen hydrolyzes semenogelin, a HMW seminal vesicle protein (29, 134). We have isolated a submandibular gland kallikrein that contracts isolated aortic rings and (like tonin) generates angiotensin II (309, 310), suggesting that localized regions of varying gene expression are important in determining kallikrein substrate specificity and possibly function as well. [For a review of the molecular biology of the glandular kallikrein-kininogen system, see (38, 44, 250).]
True TK or KLK1 is encoded by a single gene having five exons and four introns. Other members of the kallikrein gene family have a similar exonic and intronic structure with conserved splice junctions. However, even though the 5′ and 3′ flanking regions are highly homologous among the various genes, their regulation and site of expression are different, suggesting that small variations in the nucleotide sequence of the 5′ region can have a significant influence on gene expression. While the kallikrein gene is expressed mainly in the submandibular gland, pancreas and kidney, we detected small amounts of kallikrein mRNA in the heart, vascular tissue, and adrenal glands on polymerase chain reaction (PCR) (189, 237). Kallikrein and similar enzymes have been found in the arteries and veins (190), heart (188), brain (58), spleen (57), adrenal glands (255), and blood cells (189); they have also been observed in the pituitary gland (66, 216), pancreas (84), large and small intestines (242, 320), and salivary and sweat glands (106) along with their exocrine secretions. Some of these enzymes were probably true kallikrein, while others may represent separate members of the kallikrein family.
Immunoreactive TK can be found in plasma, primarily the inactive form; only a small portion remains active (88, 131, 219, 220, 254). In humans (210) and rabbits (195), (50% of urinary kallikrein is inactive (zymogen), while in rats most of it is active (187). TK can release kinins from low-molecular-weight kininogen (LMWK) and HMWK. In humans, TK releases lys-bradykinin (kallidin), whereas in rodents it releases bradykinin (11, 172).
Kininogens (kallikrein substrates) are the precursors of kinins. In plasma there are two main forms, LMWK and HMWK (114, 115). Both strongly inhibit cysteine proteinases such as calpain and cathepsins H, L, and B (192, 274). In the rat there is a third kininogen known as t-kininogen that releases kinins when incubated with trypsin but not tissue or plasma kallikrein and is one of the main acute reactants of inflammation. All kininogens, including LMWK, HMWK, and t-kininogen, also inhibit thiol proteases such as cathepsin M, H, and calpains (16, 86, 193, 194). HMWK is involved in the early stages of surface-activated coagulation (intrinsic coagulation pathway) (69, 120, 277).
Kinins are oligopeptides that contain the sequence of bradykinin and act mainly as local hormones, since they circulate at very low concentrations (1–50 fmol/ml) and are rapidly hydrolyzed by kininases. Kinin concentrations are higher in the kidney, heart and aorta (100–350 fmol/g) (37), further supporting the hypothesis that there they act mainly as local hormones. Eicosanoids, nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), tissue plasminogen activator (tPA), and cytokines mediate at least some of the effects of exogenously administered kinins (62, 268, 280, 291, 292) (Fig. 2).
Figure 2.
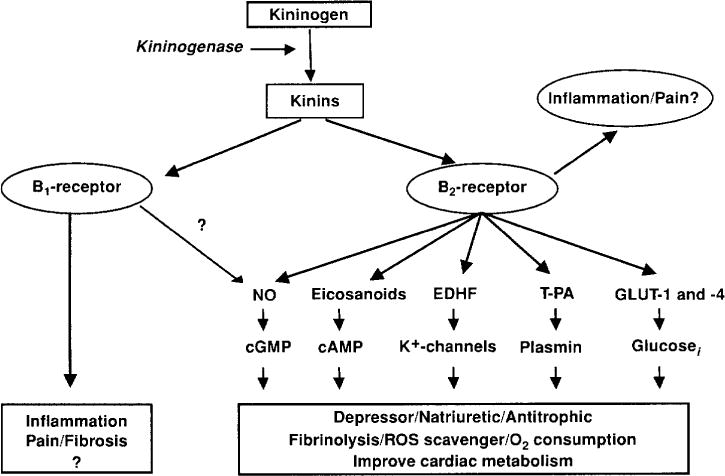
Kinins act via the B2 and B1 receptors. Most of the known effects of kinins are mediated by the B2 receptor that in terms act by stimulating the release of various intermediaries: eicosanoids, endothelial derive hyperpolarizing factor (EDRF), nitric oxide (NO), tissue plasminogen activator (†PA), glucose transporter (GLU-1 and -2 [modified from (42)].
Kininases are peptidases found in blood and other tissues that hydrolyze kinins and other peptides (75). The best known is angiotensin-converting enzyme (ACE) or kininase II, which converts angiotensin I to II and inactivates kinin, N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP), substance P, and other peptides (74, 75). Another important kininase is neutral endopeptidase 24.11 (NEP-24.11), also known as enkephalinase or neprilysin, which not only hydrolyzes kinins and enkephalins but also destroys atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), and endothelin (267, 294). Our research suggests that it may be an important renal kininase, at least in rats (290). When NEP-24.15, aminopeptidases, and carboxypeptidases are suppressed in vivo, endogenous plasma kinins do not increase significantly and their half-life remains less than 20 s, suggesting that other peptidases are also important for kinin metabolism (113). Thus clearance of kinins from plasma is more complex than initially thought.
Receptors
At least two subtypes of kinin receptors have been well characterized using analogues of bradykinin: B1 and B2 (221, 225). Both have been cloned and belong to the family of 7-transmembrane receptors linked to G-proteins (165). B1 receptors are present at very low density (or not at all) in normal tissue but are expressed and synthesized de novo during tissue injury, inflammation, and administration of lipopolysaccharides such as endotoxin. In some species, including rabbits [see Marceau et al. for review (155)], they mediate contraction of the isolated aorta and mesenteric arteries. Their main agonists are des-Arg9-bradykinin and des-Arg10-kallidin. B2 receptors mediate most of the effects of bradykinin and are the main receptors for bradykinin and kallidin (lys-bradykinin).
Kinin Antagonists
Studies using kinin analogues with agonistic and antagonistic properties suggest the existence of other kinin receptors (33, 223, 224, 238, 272). Replacing proline with D-phenylalanine at position 7 of bradykinin reportedly converted it to a B2-specific antagonist, while replacing Phe8 in des-Arg9-BK with a residue containing an aliphatic (Ala, Ile, Leu, D-Leu, norleucine, etc.) or saturated cyclic hydrocarbon chain (cyclohexyalanine) resulted into potent B1 antagonists (155, 222). Further modifications created a highly potent and long-lasting B2 receptor antagonist, DArg0-[Hyp3-Thi5-DTic7-Oic8]-bradykinin or icatibant (Hoe-140) (301), which has become an important tool for studying kinins. More recently, novel kinin antagonists, including nonpeptide agents, have been developed for possible oral treatment of inflammation, hyperalgesia, and even cancer (27, 34, 270, 271, 300).
In humans, the B2 receptor is reportedly activated directly by kallikreins and other serine proteases and this effect can be blocked by icatibant (103). Some ACE inhibitors potentiate the effect of bradykinin not only by inhibiting its hydrolysis but also by cross-talk between ACE and the B2 receptor (158). B2 forms heterodimers with the angiotensin type 1 receptor (AT1), increasing its activation (5). The stability of this heterodimer is not affected by bradykinin or angiotensin or their antagonists (7). B2 also forms a complex with endothelial nitric oxide synthase (eNOS), suppressing NO, and this effect is reversed by bradykinin (116). Moreover, B2 has been found to interact directly with AT2 (3), B1 (15), ACE (236), and even other B2 receptors (forming homodimers) (8). Since preeclampsia has been linked to an increase in AT1 and B2 heterodimers that could mediate the enhanced response to angiotensin II (6), it seems reasonable to posit that interaction between AT1 and B2 could play a pathophysiological role in some conditions. However, Hansen et al. recently reported that heterodimerization does not occur as a natural consequence, nor does B2 influence AT1 signaling (98). Hence whether or not these inter-receptor interactions (either homoor heterodimerization) have any physiological or pathological value remains unclear.
Kinin Agonists
Numerous studies have shown that blockade of B2 kinin receptor or deletion of its gene diminished the beneficial effects of ACE inhibitors (21, 35, 40, 70, 142, 146, 163, 311) (308). These observations have led to the design of new studies focusing on the potential cardiovascular protective effects of chronically infusing bradykinin (BK) or its selective B2 receptor agonists that are highly resistant to degradation by peptidases such as neutral endopeptidase, ACE, or carboxypeptidases. Two of these studies investigated the effect of chronic BK infusion in hypertensive animals, specifically (i) Ang II-induced hypertension (204) and (ii) Dahl salt-sensitive rats (59). In the first study, exogenous BK (low, medium and high doses) opposed the effect of Ang II on vascular tone but was unable to prevent end-organ damage; in the second study, infusion of BK protected the kidneys against salt-induced injury by inhibiting fibrosis and inflammation. In both studies BK was unable to lower blood pressure. Manolis et al. (153) showed that chronic infusion of a selective B2 receptor agonist diminished the extent of myocardial damage following infarction in mice. However, none of these studies distinguished cardiovascular protection from proinflammatory effects (adaptive and innate immune system) resulting from activation of B2 receptors. Thus it becomes imperative to carefully define the role of B2 activation in dendritic cells and different subsets of T lymphocytes to establish the safe therapeutic window between cardioprotective and proinflammatory effects following treatment with agonists of BK or its B2 analogues or therapy involving ACE inhibitors or angiotensin receptor blockers (ARBs) (105).
Because B1 receptors have historically been linked to inflammation and algesia, researchers were understandably surprised to find that activation of B1 protected against inflammation of the central nervous system (CNS) by limiting infiltration by encephalitogenic T lymphocytes, providing evidence that B1 agonists could improve treatment of chronic inflammatory diseases such as multiple sclerosis (MS) (249). This hypothesis may have originated from early studies of expression of kinin receptors on T lymphocytes, showing that B1 receptors are upregulated during the course of MS and a B1 agonist negatively regulated T-cell migration in vitro (217).
The Physiological Role of the Kallikrein-Kinin System
The accumulated literature on (1) antibodies to kinins and kallikrein, (2) kinin antagonists and kininase inhibitors, (3) gene knockout (KO) models of the kallikrein-kinin system, and (4) kininogen-deficient rats as well as spontaneous genetic mutations in humans has allowed us to study the role of kinins in specific physiological and pathological conditions.
Homozygous TK-deficient mice exhibited renal hypercalciuria and became hypocalcemic when placed on a low calcium (Ca) diet or as the result of a defect in tubular Ca reabsorption (208). However, B2−/− mice treated with a B1 antagonist showed no change in urinary Ca excretion and adapted normally to the low Ca diet, suggesting that TK may help regulate renal tubular Ca transport via a non-kinin-mediated mechanism. In humans, a loss-of-function polymorphism at exon 3 of the TK gene caused an active-site arginine at position 53 to be converted to histidine (R53H), resulting in lower kallikrein activity (by 50–60%) compared to R53R subjects (26). In these patients, increased basal Ca reabsorption by the thick ascending limb counteracted the distal tubule defect when they were treated with furosemide, which blocks the Na-K-Cl type 2 co-transporter (NKCC2) and inhibits Na-dependent Ca and Mg reabsorption, thereby increasing Ca delivery to distal segments. However, more pharmacogenetic studies are needed before we can state categorically that abnormal Ca regulation by the kidney in such cases is linked causally to the kallikrein mutation (26).
The kallikrein-kinin system in inflammation
Recent evidence implicates inflammation in the pathogenesis of cardiovascular diseases such as hypertension, target organ damage, atherosclerosis, and cardiac remodeling postmyocardial infarction (MI) (9, 18, 85, 96, 100, 107, 133, 149, 157, 182, 201, 232, 233, 241, 316). Moreover, we already know obesity is a risk factor for hypertension, atherosclerosis, and rheumatoid arthritis, and now we have evidence that it is associated with inflammation as well (56, 259, 305). Thus, we need to know exactly how the KKS is involved in the body’s innate and adaptive immune system before we can understand its role in cardiovascular disease.
Over the last 60 years, accumulated evidence has established that the KKS contributes to the pathogenesis of inflammation. The seminal finding was the demonstration by Holton & Holton, Kellet, and Lewis (109, 125, 132) that subcutaneous injections of bradykinin reproduce some of the cardinal signs of acute inflammation: vasodilatation (rubor and calor), increased vascular permeability (as evidenced by a protein-rich exudate, tumor, or infiltration of injured tissue by neutrophils) and pain. Furthermore, components of the KKS are also found in inflammatory exudates [for a review, see (97)]. Thus, it has been proposed that various components of the KKS are mediators and/or modulators of inflammation in arthritis (54, 63, 263) [for a review, see (166)], pancreatitis (322), skin inflammation (209), asthma (78), rhinitis (71), allergic diseases (71), hereditary angioedema (71), sepsis (211), colitis (99, 156), and many other diseases [for a review, see (1,176)].
The development of B1 and B2 receptor antagonists, genetic deletion of specific components of the KKS in mice, and the discovery of kininogen-deficient rats have allowed researchers to study the role of the KKS in inflammation. Blockade or deletion of B1 or B2 has been found to have a protective effect in experimental models of inflammation, including (1) bacterial infections (118), (2) parasites (246), (3) lipopolysaccharide-induced acute renal inflammation (17), (4) capsaicin-induced skin inflammation, which required deletion of both B1 and B2 (209), (5) streptozotocin-induced diabetes, wherein insulitis was reportedly mediated by B1 and proteinuria by B2 (322), (6) periodontitis induced by Gram-negative bacteria, mediated via cross-talk between Toll-like receptor type 2 (TLR2) and B2 which resulted in the development of either IFN-γ- or IL-17-producing T cells (175), (7) 2,4,6-trinitrobenzene sulfonic acid-induced inflammatory bowel disease, mediated via B1 (99), (8) experimental arthritis (63, 263), and human rheumatoid and osteoid arthritis (54, 166) (Fig. 3).
Figure 3.
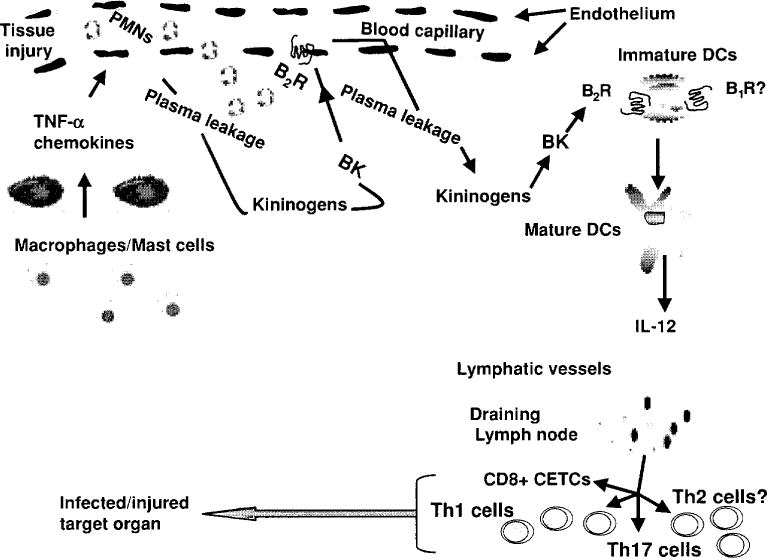
Kinins participate in the activation of the innate/adaptive immunity in a variety of acute and/or chronic inflammatory diseases. Upon tissue injury, kinins via activation of dendritic cells (DCs) B2 receptors triggered mechanism related to maturation of DCs, including migration to sites of inflammation and release of proinflammatory cytokines and chemokines such as IL-12, which promote differentiation of naive CD4+ to Th1 and CD8+ cytotoxic effector T cells (CETCs) producing IFN-γ and other proinflammatory cytokines, reaching infected or injured peripheral tissues such as lung, kidneys, heart or brain (244). [Modified from Monteiro et al. (175).] Copyright 2009. The American Association of Immunologists, Inc.
The acute inflammatory effects of kinins are mediated mainly by the B2 receptor (63, 263), whereas chronic effects are also mediated by B1 induced by the inflammation (17, 99, 154, 209). Kinins regulate expression of kininogen binding sites on endothelial cells, further increasing kininogen concentration at the site of inflammation (245). In addition, some known effects of kinins on inflammation are probably mediated by release of NO, prostaglandins, histamine, and cytokines by fibroblasts, macrophages and mast cells (104, 176). A kinin-induced increase in capillary permeability may be due to dilatation of arterioles, constriction of post-capillary venules, or fenestrations of the endothelium. While many of these effects are mediated by activation of B2, there is evidence that B1 may also participate in the pathogenesis of inflammation. B1 receptors may form as the result of noxious stimuli, tissue damage, cytokines or inflammation itself and become activated by des-arginine originating from the C-terminal (97). Kinins are capable of inducing severe pain, which as noted above is one of the four cardinal signs of inflammation. Recently Liu et al. (139) suggested one possible cause of pain in inflammation, demonstrating that bradykinin acts via the B2 receptors, phospholipase C (PLC) and release of Ca from intracellular stores to close M-type K+ channels and open Ca2+-activated CI channels (CaCCs), thereby acting directly on the neurons that generate the signals the patient experiences as pain.
Bradykinin acting via the B2 receptor reportedly induces maturation of dendritic cells (which stimulate both innate and adaptive immune responses) and release interleukin-12 (IL-12), which drives T-cell polarization (Fig. 3). Thus, bradykinin could send a signal that activates both innate and adaptive immune responses (12, 175), which appears to be important for development of acquired resistance to some infections such as Trypanosoma cruzi and Listeria (118, 174). In this way, kinins might actually play a dual role: they could both mediate inflammation in arthritis and other conditions by activating the body’s adaptive immune responses and at the same time contribute to acquired resistance to some infections.
There is also evidence that the KKS has a protective role both in the cardiovascular system and in the kidney [for a review, see (42, 48)], since kinins reportedly participate in the cardiovascular and renal protective effects of ACE inhibitors, ARBs and ischemia/reperfusion preconditioning. Yet at the same time they act as mediators of inflammation in atherosclerosis (201), hypertension (96), and target organ damage (68, 178, 276, 284, 297). This functional duality has yet to be satisfactorily explained. It could be that kinins act as autocrine and/or paracrine hormones at low concentrations, exerting a protective effect either directly or via the release of NO and prostaglandins which improve blood flow; whereas when kinins are released into tissue at high concentrations, they could activate both innate and adaptive immune systems (243, 244, 246). Recently, Duka et al. (72) reported that in MI both B1 and B2 are needed in order for ACE inhibitors to have a cardioprotective effect. Their data were strengthened by the finding that this cardioprotective effect was not seen in B1−/− mice; but what was particularly intriguing was their discovery that these mice not only exhibited the same lack of cardioprotection following treatment with ACE inhibitors (ACEi) but also greater deterioration of cardiac function compared to untreated B2−/− mice, leading the researchers to postulate that upregulation of B1 may have caused the detrimental proinflammatory effects of BK potentiation by ACE inhibitors to outweigh the slight vasodilator action of B1 (which is already minimal compared to B2) (72). Duka’s data contradict our findings obtained either by blocking the B2 receptors with icatibant or using B2−/− mice (143, 311), perhaps related to differences in genetic background: their mice had a background of C57BL6 × 129/Sv (72) compared to the 129SvEv mice (311), C57BL/6J mice (307, 308), and Lewis rats (143) used in our laboratory. Nevertheless, Duka’s findings certainly fit the proinflammatory profile of B1 receptors demonstrated in experimental colitis (99, 156). Clearly more work is warranted to determine what factors and conditions favor the development of either inflammation or cardiovascular protection associated with activation of the KKS.
The kallikrein-kinin system in the vasculature and regulation of local blood flow
Arteries and veins contain a kallikrein-like enzyme, and both vascular tissue and smooth muscle cells in culture contain kallikrein mRNA (190, 237). Vascular smooth muscle cells in culture release both kallikrein and kininogen (200). Thus the components of the KKS are present in vascular tissue, where they could play an important role in regulation of vascular resistance. Arteries isolated from mice lacking the kallikrein gene reportedly exhibited significantly reduced flow-induced dilatation compared to controls, suggesting that the KKS in the arterial wall participates in this process (25, 168). Moreover, in humans a partial genetic deficiency of TK (R53H) was associated with inward remodeling of the brachial artery that renders it incapable of adapting to a chronic increase in wall shear stress, a form of arterial dysfunction that affects 5% to 7% of Caucasians (13). The effect of ACE inhibitors on local potentiation of kinin-induced vasodilatation appears to be attributable in part to their prevention of bradykinin degradation and increased production of endothelium-derived relaxing factors (EDRFs) such as NO and prostaglandins. In spontaneously hypertensive rats (SHR) and canine coronary arteries, ACE inhibitors potentiate the endothelium-dependent relaxation evoked by bradykinin (67, 173), which appears to primarily involve increased release of NO and/or EDHF. When the arterial endothelium is removed, smooth muscle cells proliferate in the media and then migrate across the internal elastic lamina into the intima where they cause neointimal hyperplasia, mimicking some of the vascular changes seen in atherosclerosis. ACE inhibitors suppress neointima formation (198, 215), and blocking kinins or inhibiting NO synthesis lessens their protective effect, suggesting that it may be mediated by a local increase in kinins that stimulates the release of NO (76, 77).
Kinins play an important role in local regulation of blood flow in organs rich in TK such as the submandibular gland, uteroplacental complex, and kidney (23, 234, 256, 258). In rats nephrectomized 48 h earlier to exclude the renal RAS, inhibition of angiotensin I-converting enzyme (kininase II) significantly increased blood flow in the submandibular gland but did not affect blood pressure. In contrast, 10 min after sympathetic stimulation of the gland to increase vascular kallikrein secretion, the ACE inhibitor markedly decreased blood pressure and increased kinin concentrations in arterial blood (254, 324). Changes in blood flow and blood pressure were blocked by antibodies to kinins and TK. In a separate study, the effect of the same ACE inhibitor on basal glandular blood flow was blocked by a kinin antagonist; at low doses, the antagonist caused no significant change in blood flow when the ACE inhibitor was not administered, whereas at high doses basal blood flow decreased significantly (23). A study using kinin antibodies and antagonists clearly indicated that kinins can act as paracrine hormones, regulating blood flow within the gland, and that the effect of ACE inhibitors on blood flow is mediated by kinins (23). In nephrectomized pregnant rabbits infused with an angiotensin antagonist to block the uterine RAS, ACE inhibitors increased both uterine and placental blood flow and also raised levels of immunore-active PGE2, whereas all of these effects were blocked by a kinin antibody (258). In addition, endogenous bradykinin and its B2 receptors have been shown to play an important role in traumatic brain injury in mice by mediating its cerebrovascular effects (edema, cell death, vasodilatation, and/or changes in blood flow) (286), (323). Moreover, increased muscular activity has been associated with significantly increased interstitial concentrations of bradykinin in both skeletal muscle and the connective tissue around the adjacent tendon, supporting the concept that both bradykinin and adenosine contribute to exercise-induced hyperemia in skeletal muscle and suggesting that bradykinin may help regulate blood flow in peritendinous tissue (130). Taken together, these findings suggest that endogenously generated kinins help regulate skeletal muscle, uterine, and cerebral blood flow, either directly or through the release of prostaglandins.
Kinins in regulation of renal blood flow
Kinins may also play an important role in regulation of renal blood flow. In sodium-depleted dogs, blocking renal kinins by infusing low doses of a kinin antagonist into the renal artery decreased renal blood flow and autoregulation of the glomerular filtration rate (GFR) without altering blood pressure (19). The changes in blood flow could be neutralized by first administering an ACE inhibitor, suggesting that either (i) they were mediated by renin release due to the partial agonistic effect of the kinin antagonist, or else (ii) renal kinins may have increased when ACE was inhibited, thereby competing more effectively with the antagonist. The changes in GFR autoregulation were not altered by the ACE inhibitor and may have been due to modification of either the relationship between afferent and efferent glomerular arteriolar resistance or the coefficient of filtration (19). We recently showed that the dilator effect of bradykinin in the renal efferent arteriole is mediated by cytochrome P450 metabolites of arachidonic acid, called epoxyeicosatrienoic acids (EETs), and that bradykinin stimulates the glomeruli to release another cytochrome P450 metabolite, the constrictor eicosanoid 20-hydroxyeicosatetraenoic acid (20-HETE), and together with an as yet unidentified dilator prostaglandin they help regulate the downstream glomerular circulation and perhaps even the GFR (226, 298).
We examined the role of kinins in regulation of renal blood flow distribution using a laser-Doppler flowmeter (234). A kinin antagonist lowered papillary blood flow without altering outer cortical blood flow, suggesting that intrarenally formed kinins are an important component of blood flow regulation in the inner medulla. This study also showed that the RAS plays an important role in regulation of papillary blood flow; after kinins were blocked, enalaprilat increased flow significantly. We also found that when both ACE and NEP-24.11 were inhibited, flow increased by 50% compared to 25% when they were inhibited separately. These increases were blocked by the kinin antagonist, suggesting that the rise in blood flow induced by both ACE and NEP-24.11 inhibitors was mediated by increased kinins in the interstitial space. We failed to observe any consistent effect on water or sodium excretion; still, water excretion does tend to decrease in animals treated with a kinin antagonist. Moreover, there are other studies that support this hypothesis. In anesthetized rats, blocking kinins slowed renal blood flow (256). In dogs, when kallikrein excretion was stimulated by sodium deprivation, an intrarenally administered kinin antagonist partially blocked the effect of enalaprilat on renal blood flow (319), suggesting that even though blockade of the RAS accounted for a significant portion of the increased blood flow caused by the ACE inhibitor, a substantial component was contributed by endogenous kinins. Similar results were reported in rats when the KKS was stimulated by deoxycorticosterone (181).
In the unchallenged kidney, kinins play a minor role in regulation of blood flow; however, when the KKS is stimulated by low sodium intake or mineralocorticoids, or when endogenous kinin degradation is inhibited, kinins appear to help regulate blood flow (197, 282). Another study suggested that kinins play an important role in regulation of papillary blood flow and may help regulate the GFR when renal perfusion pressure is reduced (283).
Kinins in regulation of water and electrolyte excretion
Renal kallikrein is located in the connecting tubule; it is released in significant amounts and excreted in the urine (195, 196, 251) (Fig. 4). Kallikrein releases kinins into the lumen of the distal nephron, either from filtered kininogen or kininogen produced in the principal cells of this nephron segment (82, 252). Kinin receptors are also present in the collecting duct (281). In addition, kallikrein is released on the basolateral side of the nephron, where it may liberate kinins from plasma kininogens (296). The interstitial renal fluid contains a high concentration of kinins (266). The role of kinins in regulation of water and sodium excretion has been studied by increasing intrarenal kinins, blocking kinins, or deleting the genes that express kinin receptors or TK (121, 160, 170, 208, 239). Infusion of kinins into the late proximal nephron doubled excretion of simultaneously administered 22Na (121), an effect mediated by prostaglandins (122), while infusion of a kinin antagonist into this same nephron segment reduced 22Na recovery significantly (123). Systemic administration of phosphoramidon, which inhibits NEP-24.11 (a major kininase in the nephron), caused urinary kinin excretion to double; diuresis increased by 15% and natriuresis by 37% (290). Although these data support the hypothesis that increased kinins in the nephron heighten intrarenal control of water and electrolyte excretion, it is also possible that the effect of phosphoramidon is mediated by blocking hydrolysis of other peptides such as ANF (279).
Figure 4.
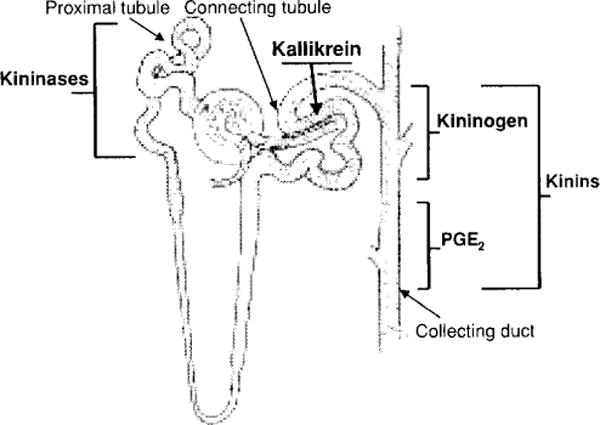
Localization of the kallikrein-kinin system along the nephron. Kallikrein is produced in the convoluted connecting tubule, kininogens in the cortical portion of the collecting duct and kinins along the collecting duct. Filtered blood kinins are mainly destroyed by kininases located along the proximal tubule. Activation of kinin receptors in the nephron leads to production of prostaglandin E2 (PGE2) along the medullary portion of the collecting duct. Drawing of the nephron was kindly provided by Dr. William H. Beierwaltes.
Infusion of aprotinin inhibited the enzymatic activity of urinary kallikrein but did not affect acute water or electrolyte excretion in euvolemic and sodium- or water-expanded rats (212). A transient decrease in sodium excretion has been observed during administration of aprotinin in mineralocorticoid-treated rats (184) or kinin antibodies in saline-expanded rats (160); however, these data should be interpreted with caution, since antibodies may stimulate release of histamine, cause an anaphylactoid reaction, or form a HMW complex with kininogen which is then deposited in the nephron, any of which might alter water and sodium excretion. To avoid these problems, we use Fab fragments of kinin antibodies, which are rapidly distributed in the extracellular fluid and excreted by the kidney; moreover, they do not form HMW complexes or activate complement and other proteolytic systems in plasma, thus reducing the risk of anaphylactoid reactions. In nonanesthetized rats, Fab fragments blocked 70% of the effect of 100 ng bradykinin on blood pressure and appeared rapidly in the urine, suggesting that they neutralize kinins not only in the vascular and interstitial spaces but also in the lumen of the distal nephron. We used these Fab fragments and a kinin antagonist to study rats treated with deoxycorticosterone and salt (DOCA-salt), which stimulates the renal KKS. Both the Fab fragments and the kinin antagonist significantly lowered urine volume and raised urinary osmolarity; however, only the Fab fragments significantly lessened urinary sodium excretion without affecting blood pressure, renal blood flow, or GFR (282). The resulting antidiuretic effect may have been due to blockade of kinins in the renovascular interstitial space, since the kinin antagonist was most likely hydrolyzed in the proximal tubule and never reached the lumen of the distal nephron. On the other hand, the antinatriuretic effect of the Fab fragments may have been due to blockade of kinins in both the vascular/interstitial and urinary compartments, since the antibody appeared in the urine and the kinin antagonist did not have an antidiuretic effect. From this, we conclude that kinins may aid in regulation of water and sodium excretion when the KKS is stimulated. In normal nonanesthetized rats, blocking kinins in the lumen with Fab fragments of monoclonal antibodies to kallikrein decreased urinary PGE2, UV, and UNaV. The changes in UV and UNaV mimicked those of PGE2, suggesting that the natriuretic and diuretic effects of kinins are mediated in part by PGE2 (239).
Mice deficient in TK reportedly lack the 70-kDa form of γ-ENaC (epithelial sodium channel), consistent with reduced renal ENaC activity in tissues that normally express TK such as the cortical collecting duct; however, in mice lacking B2 receptors, the 70-kDa form of γ-ENaC was more abundant, suggesting that its absence in TK-deficient mice is not kinin mediated (207). In vitro, stimulation of EDRF release by endothelial cells using bradykinin or acetylcholine increased cGMP and inhibited Na+ transport by cortical collecting duct cells (273). In vivo, stimulation of EDRF release by bradykinin caused natriuresis and diuresis without affecting the GFR (128). Thus it would appear that kinins acting as local hormones contribute to regulation of renal hemodynamic and excretory function, either directly or via release of PGE2 and EDRF.
Kinins in blood pressure regulation and the pathogenesis of hypertension
Both genetic and environmental factors acting via intermediary phenotypes participate in regulation of blood pressure, the etiology of hypertension and development of target organ damage. Vasoactive systems are an important component of these intermediary phenotypes. They can act as local hormones (intracrine, autocrine, and paracrine) or as endocrine and neuroendocrine systems. We use the term intracrine to indicate hormones that act within the cells that synthesize them, such as reactive oxygen species (O2−) and products of protooncogenes. The word autocrine denotes hormones that act on the cell membrane receptors where they are produced, such as growth factors. Paracrine hormones act near the site where they are produced; these include kinins, eicosanoids, NO, and EDHF. Endocrine hormones such as aldosterone are released into the extracellular fluid and act on distant tissues, though they can also have both autocrine and paracrine effects. Finally, neuroendocrine hormones such as catecholamines are released by neurons and act near or distant from the site of release.
Blood pressure is the result of a balance between vasopressor and vasodepressor systems. Alteration of this equilibrium may result in (i) hypertension, (ii) target organ damage, (iii) ineffective antihypertensive treatment, or (iv) hypotension and shock. Changes in this balance could be due to (i) genetic factors such as a mutation in one or more genes of the vasoactive system and/or (ii) environmental factors that alter the activity of vasoactive systems. Endocrine and neuroendocrine vasopressor systems, such as the renin-angiotensin-aldosterone system and catecholamines, play a well-established and important role in regulation of blood pressure, the pathogenesis of some forms of hypertension, and target organ damage. The role of vasodepressor systems is less well established; however, evidence suggests that they play an important role in regulation of blood flow, renal function, the pathogenesis of salt-induced hypertension and target organ damage, and the cardio-renal protective effects of ACE inhibitors and ARBs (49, 145, 203, 311, 312). Concentrations of kinins in tissue may well exceed blood levels and could conceivably contribute to the anti-hypertensive and vasodilator effects of ACE inhibitors in humans (130, 203). Vasodepressor hormones such as kinins, eicosanoids, NO, carbon monoxide (CO), and EDHF act as local hormonal systems, opposing the effects of vasopressor systems. Some vasodepressors such as atrial (ANF), brain (BNP), and C-type (CNP) natriuretic peptides may act as both endocrine and local hormones.
The role of the kallikrein-kinin system in the pathogenesis of hypertension has been studied by (i) measuring various components of the system, (ii) bradykinin B2 receptor antagonists, (iii) mice with B1, B2, or both deleted by homologous recombination, (iv) deletion of the TK gene, and (v) rats deficient in kininogen. Decreased activity of the KKS may play a role in hypertension. Low urinary kallikrein excretion in children is one of the major genetic markers associated with a family history of essential hypertension, and children with high urinary kallikrein are less likely to be genetically predisposed to hypertension (264, 289, 304, 321). A restriction fragment length polymorphism (RFLP) for the kallikrein gene family in SHR has been linked to high blood pressure (218), and urinary kallikrein excretion is decreased in several models of genetic hypertension. Urinary and/or arterial TK can also be decreased in renovascular hypertension and genetically hypertensive rats (43, 47, 52, 124). Although these reductions may be secondary to increases in blood pressure, decreased urinary kallikrein in (i) normotensive children of patients with essential hypertension, (ii) genetically hypertensive rats, and (iii) Dahl salt-sensitive rats that have not yet developed hypertension (48, 108, 159, 257, 278) suggests that the cause of the decrease may differ in each model.
Circulating kinin concentrations range from 5 to 50 pg/ml blood (253). These concentrations need to be increased to at least 100 pg in humans (28) and 1000 pg in rats (240) to acutely lower blood pressure. Although blood kinin levels may increase in some conditions, they are rarely high enough to explain changes in blood pressure, save for nonphysiological situations such as stimulation of the sympathetic nerve of the submandibular gland in animals treated with ACE inhibitors (see section on blood flow regulation). Thus, kinins can only act as paracrine hormones, regulating local vascular resistance and organ function.
In early studies, acute administration of a kinin antagonist at high doses increased blood pressure in most rats tested, though some exhibited a vasodepressor effect (22). We used a more potent antagonist (20)—also at high doses—and found that it produced a transient biphasic response: first a small pressor effect, followed by a depressor effect (39). At smaller doses—though still sufficient to block exogenous bradykinin—the same antagonist did not alter normal blood pressure. These studies seem compatible with the hypothesis that kinins help regulate blood pressure; however, to produce a pressor effect, the antagonist has to be administered at much higher doses than those needed to block the vasodepressor effect of exogenous bradykinin if bound kinins are to be displaced from tissue receptors. We must be cautious in interpreting these data, since we cannot rule out the possibility that these antagonists have a vasopressor effect unrelated to kinin-blocking activity. Our studies using kinin antibodies or their Fab fragments showed that although they partially blocked the vasodepressor effect of kinins, they did not acutely increase blood pressure (282), (160), (239).
Blood pressure and cardiovascular function are often normal in HMWK-deficient rats and B1−/− or B2−/− mice (171, 229, 230). However, mice lacking TK have normal blood pressure and yet both the structure and function of the heart are clearly abnormal (168). Chronic blockade of B2 receptors with a potent and selective antagonist, icatibant, did not increase blood pressure under normal conditions or in situations that favor hypertension in rats, such as (i) chronic infusion of a subpressor or pressor dose of angiotensin II (Ang II), (ii) a high salt diet, or (iii) mineralocorticoids and salt (148, 229). However, other researchers have had entirely different results, who found that lack circulating kininogen or blockade of B2 receptors are associated with significant increases in blood pressure under normal conditions or when animals are challenged with a pressor agent or diet (147, 150–152).
Mice with the bradykinin B2 receptor deleted by homologous recombination (gene KO) initially have normal blood pressure (Fig. 5); however, they develop hypertension when fed a high sodium diet (8%) for at least 2 months (10, 55). Thus low kinin activity may be involved in the development and maintenance of salt-sensitive high blood pressure. However, in these mice hypertension induced by mineralocorticoids (renin independent) or aortic coarctation (renin dependent) was not exacerbated (228). Also, others have reported that as these mice grow older, they also develop hypertension and left ventricular hypertrophy even on a normal sodium diet (73). However, we were unable to confirm that B2 ablation renders mice spontaneously hypertensive (230), while others tried but could not confirm that the B2 receptor is a major component in maintenance of normal blood pressure and cardiac structure (55, 170, 228, 285). Mice deficient in B1, B2, or both, as well as mice with low TK, had blood pressure readings similar to wild-type controls, confirming that kinins are not an important determinant of blood pressure (285). Moreover, mice lacking both B1 and B2 did not show any significant change in blood pressure when dietary salt was increased (32). That contrasts with previous observations demonstrating the chronic hypertensive effect of a high salt diet in B2−/− mice (10, 55), although differing duration of high salt intake could account for this disparity. Indeed, Alfie et al. (10) did not observe hypertension until after 8 weeks of a high salt diet, while Cervenka et al. (55) started the high salt diet during gestation and continued it for up to four months whereas the B1/B2−/− mice were only fed high salt for 5 weeks (32). We do not know whether B1−/− mice develop hypertension when given high salt. Also, although lack of both B1 and B2 (as in Akita mice) exacerbates diabetic complications as well as oxidative stress, mitochondrial DNA damage, and overexpression of fibrogenic genes, these mice showed no sign of hypertension (117). Moreover, wild-type mice or mice with the TK gene deleted exhibited a similar increase in blood pressure when renovascular hypertension was induced (129), further supporting the concept that the KKS plays no more than a minor role in blood pressure regulation either under normal conditions or during hypertension. In kininogen-deficient Brown Norway Katholiek rats (BNK), administration of mineralocorticoids and salt or angiotensin II reportedly increased blood pressure to the same degree as rats with a normal KKS (229), contradicting reports by other investigators (150–152). Thus, taken together the published data would suggest that kinins are not critical for blood pressure regulation, nor are they required for the development of hypertension, save perhaps in salt-sensitive animals. Even though the data are inconsistent, absent more convincing evidence, it seems reasonable to conclude that chronic blockade of the KKS does not cause hypertension.
Figure 5.
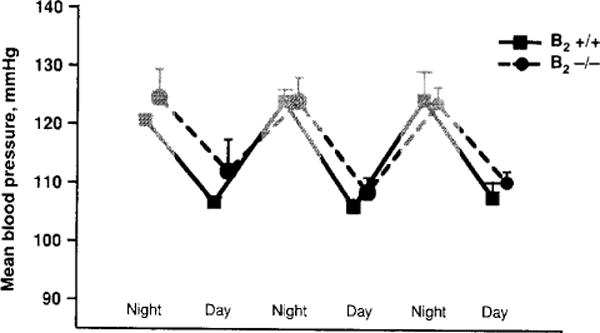
Diurnal and nocturnal mean blood pressure of bradykinin B2 receptor knockout (−/−) and wild type (+/+) mice. Blood pressure was measured 24 h by a telemetric system (Rhaleb N-E and Carretero O.A. unpublished data).
Role of Kinins in the Antihypertensive Effect of ACE Inhibitors
Inhibition of kinin degradation and hydrolysis of other oligopeptides may contribute to the antihypertensive effect of ACE inhibitors. While blockade of angiotensin II formation appears to play an important role in this process, the role of kinins is less well established. Orally active ACE inhibitors are effective antihypertensive agents, not only in high-renin hypertension but also in clinical and experimental models that do not involve the systemic RAS (45, 162). Thus some of their effects may be mediated by a local RAS, kinins, or some other undetermined mechanism, since ACE can hydrolyze numerous other peptides (Fig. 6). ACE inhibitors may also augment the effect of kinins by interacting directly with the B2 receptor (158).
Figure 6.
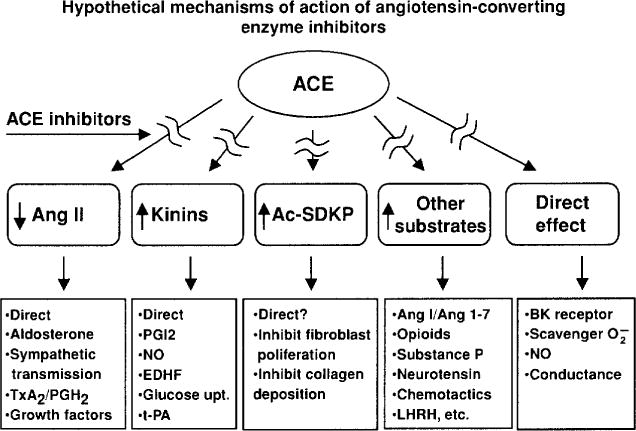
ACE has multiple substrates, and inhibition of their hydrolysis may explain the cardioprotective effect of ACE inhibitors.
Blood kinins are unchanged or moderately increased after treatment with ACE inhibitors (35, 40, 49) [for a review, see (36, 50)]. Kinins in the urine reportedly increase more consistently following ACE inhibiton therapy, which suggests that their renal concentration increases too (64, 164, 183, 290, 295), strengthening the antihypertensive effect of ACE inhibitors by altering renovascular resistance and increasing sodium and water excretion.
Studies involving various experimental models of hypertension have shown that the acute antihypertensive effect of ACE inhibitors is attenuated by blocking kinins with either hightiter kinin antibodies (46, 51, 53) or a B2 antagonist (35, 40, 70). Kinin antagonists also partially reversed their antihypertensive action in rats with renovascular hypertension (21); however, lack of B2 receptors did not influence ACEi treatment in mice with renovascular (2 kidney-1 clip or 2K1C) hypertension (Fig. 7). This is not surprising, since it is well established that the RAS plays a major role in the development of renovascular hypertension. We assessed the influence of kinins on the acute antihypertensive effect of enalaprilat in rats with severe hypertension induced by aortic ligation between the renal arteries (40). In this model, renin is necessary for the pathogenesis of hypertension (45); however, acute and severe hypertension can damage the endothelium enough to activate plasma kallikrein and increase kinin formation. We found that enalaprilat lowered mean blood pressure by 48 ± 6 mmHg in the controls and 21 ± 4 mmHg in the kinin antagonist group (P < 0.01); however, kinins in arterial plasma were not significantly altered by the ACE inhibitor (41 ±10 vs. 68 ± 20 pg/ml) (Fig. 8). As indicated earlier, in order for mean blood pressure in a nonanesthetized rat to decrease, kinins in arterial blood must reach at least 1000 pg/ml (240). Thus the effect of the ACE inhibitor may have been due to an increase in tissue kinins, which could regulate vascular resistance acting as a paracrine hormonal system. Cachofeiro et al. (35) demonstrated that pretreatment with a bradykinin antagonist or NO synthesis inhibitor attenuated the acute antihypertensive effect of both Captopril and ramipril in SHR, whereas a prostaglandin synthesis inhibitor made no difference, suggesting that this effect was due to bradykinin stimulating the release of NO. However, in dogs kinins may strengthen the acute hypotensive effect of ACE inhibitors via prostaglandins (214).
Figure 7.
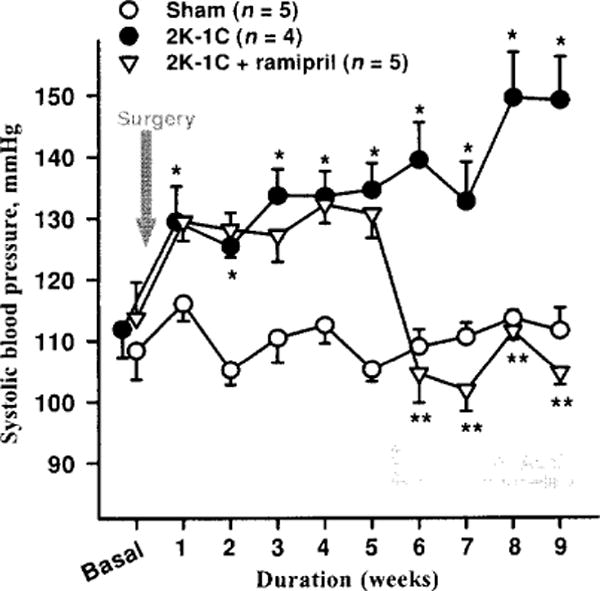
Antihypertensive effect of ACE inhibitor in bradykinin B2 receptor knockout (−/−) mice. Mice with 2K-1C hypertension were given plain water (vehicle) or water mixed with ACE inhibitor, ramipril, (4 mg/kg/day) to drink 5 weeks after blood pressure was increased. ACE inhibitor normalized blood pressure in B2−/− hypertensive mice. *P < 0.001, 2K-1C versus sham; **P < 0.001, 2K-1 C versus 2K-1C + ramipril. (Rhaleb N-E and Carretero O.A. unpublished data.)
Figure 8.
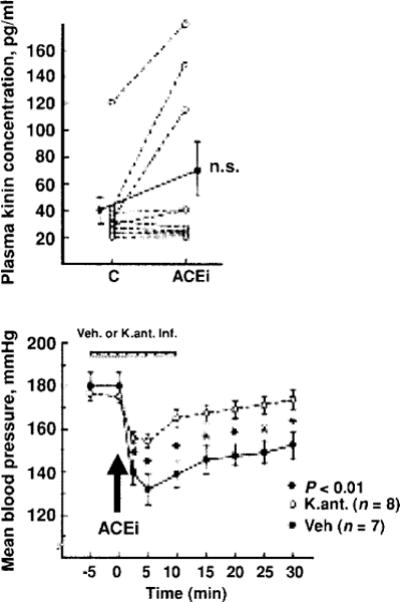
Role of kinins in the acute antihypertensive effects an ACE inhibitor (enalaprilat) in rats with severe hypertension. Top: Blood kinin concentrations before (C) and after administration of the ACE inhibitor. Bottom: Mean blood pressure before and after ACE inhibition, open and closed circles represent rats pretreated with a kinin antagonist or vehicle, respectively. Values are mean ± SEM (bottom). [Reprinted from Carbonell et al. (40).]
In humans, an ACE insertion/deletion polymorphism at intron 16 of the ACE gene could be important for bradykinin metabolism (179); ACE activity is higher in subjects with ACE deletion and correlates with rapid bradykinin degradation. In normotensive subjects and hypertensive patients with low or normal renin, aprotinin (an inhibitor of kallikrein and other proteases) partially blocked the acute antihypertensive effect of Captopril (199). While that could have been due to kinin inhibition, other investigators tested a specific B2 kinin receptor antagonist (icatibant) and found that the short-term blood pressure effects of ACE inhibitors were attenuated in both normotensive and hypertensive subjects (87), suggesting that the acute effect of ACE inhibitors is mediated in part by kinins affecting local and peripheral vascular resistance either directly or through release of prostaglandins and NO.
But the contribution of kinins to the chronic antihypertensive effects of ACE inhibitors remains more controversial. In renovascular hypertension (2K1C), chronic blockade of kinin receptors interferes with ramipril’s ability to lower blood pressure (14). In mineralocorticoid hypertension, where KKS and ACE activity are reportedly increased (180), chronic ACE inhibitors have a small but significant antihypertensive effect that can be blunted by blocking the B2 receptor with icatibant (41, 229), suggesting that kinins may be involved; however, they are ineffective in SHR (14) or hypertension induced by aortic coarctation (35, 91, 231). Therefore, it would seem that the role of kinins in the long-term antihypertensive effect of ACE inhibitors depends on the model. To our knowledge, no studies of chronic KKS blockade have been conducted in humans.
Role of kinins in the cardiac antihypertrophic effect of ACE inhibitors
ACE inhibitors have been shown to reverse LV hypertrophy in essential hypertension and in various experimental models of hypertension, partly due to reduced afterload; however, even apart from their blood-pressure lowering effect they may decrease formation of angiotensin II, which stimulates various proto-oncogenes and growth factors. The cardiac KKS may also strengthen the effect of ACE inhibitors on the heart. At doses that do not lower blood pressure, they may reverse LV hypertrophy in rats with hypertension due to aortic coarctation (248); however, direct 24-h measurements are needed before we can be certain that blood pressure does not decrease. Although Linz et al. reported they were able to reverse the antihypertrophic effects of an ACE inhibitor using a kinin antagonist (136), we have not been able to confirm this (231). Further studies are needed to determine whether ACEi doses that do not lower blood pressure (monitored for 24 h) reverse cardiac hypertrophy, and whether kinins contribute to this effect. Capillary length and density both increase in hearts of SHR treated with an ACE inhibitor at both “antihypertensive” and “nonantihypertensive” doses, and there is strong evidence that angiotensin II also has significant angiogenic effects (79); however, the cardioprotective effect of an ACE inhibitor on capillary growth cannot be attributed solely to inhibition of angiotensin II production, since Gohlke et al. demonstrated that blockade of B2 receptors with a selective antagonist (icatibant) blocked part of the ACE inhibitor’s effects, suggesting that the increased capillary growth may have been partly due to kinins (90) [for review, see (287)].
Role of kinins in myocardial ischemia and the protective effect of ischemic preconditioning and ACE inhibitors
Both human and animal studies have demonstrated that kinins are released from the heart, especially during ischemia. This could have a cardioprotective effect (102, 186, 202); indeed, studies showed that intracoronary bradykinin infusion significantly limited infarct size, reduced ventricular arrhythmias, improved cardiac performance, and normalized myocardial metabolism (135, 137, 163). In angioplasty patients, balloon inflation for 1 minute (simulating ischemic preconditioning) increased coronary sinus kinins 50-fold (203). Preconditioning nearly doubled cardiac interstitial kinins compared to non-preconditioned ischemic hearts (202), while infusing bradykinin prior to coronary artery bypass grafting (CABG) reduced myocardial reperfusion injury as indicated by a drop in creatine kinase myocardial isoenzyme (299). Moreover, we have shown that preconditioning reduced infarct size and is-chemia/reperfusion injury in mice and rats and these effects were abolished or significantly blunted in B2 kinin receptor KO mice (B2−/−) or HMWK-deficient rats (312). TK may also contribute to the cardioprotective effects of preconditioning, as reduction of infarct size was significantly reduced in TK KO mice (TK−/−) (95, 213), whereas transfection of the TK gene into the heart reduced ischemia/reperfusion injury and enhanced myocyte survival (314). Taken together, the accumulated data suggest that ischemic preconditioning activates TK and/or plasma prekallikrein in endothelial cells and causes kinins to be generated from both LMWK and HMWK. These kinins then act on the B2 receptor, triggering and/or mediating intracellular signal transduction pathways that contribute to the cardioprotective effect of preconditioning.
ACE inhibitors reduce ischemia/reperfusion injury, including infarct size and reperfusion arrhythmias. These effects involve blocking not only angiotensin II formation but also kinin degradation (101, 142, 143, 145, 302, 311); moreover, they may conceivably be mediated in part via a kinin-NO-dependent mechanism, since they were suppressed by blocking synthesis of NO or prostaglandins (140, 144) and diminished in eNOS gene KO mice (313). In animal models of ischemia/reperfusion injury, ACE inhibitors reduced infarct size and ventricular arrhythmias and these effects were attenuated or abolished by co-administering a B2 antagonist (163) or deleting the TK gene (95, 169).
The cardioprotective effects of kinins may be mediated in several ways. Release of NO from the endothelium may be stimulated either directly or via prostaglandins (275). Myocardial ischemia enhanced kinin release, accompanied by increased cGMP (an indicator of NO production) and 6-keto-PGF1α (a metabolite of prostacyclin) (138, 235), whereas blocking NO or prostaglandin synthesis diminished or abolished cardioprotection (94, 293). Kinins improve cardiac metabolism by increasing high-energy phosphate production and glycogen content in the heart, which could be mediated by facilitating translocation of intracellular glucose transporters (GLUT1 and GLUT4) and thereby increasing glucose uptake (247) (227). This is important because during ischemia the source of energy production is shifted from oxidation of fatty acids to glycolysis. Activation of protein kinase C (PKC) has also been shown to be involved in the protective mechanism of preconditioning (269, 303, 315). Activation of kinins phos-phorylates a secondary effector, presumably ATP-sensitive potassium channels (KATP). When kinins activate PKC, KATP channels open, promoting cardioprotection in hearts subjected to ischemia/reperfusion injury (30, 167). Such responses may favorably influence functional and metabolic events during ischemic episodes and protect against ischemia/reperfusion injury.
Role of kinins in the cardioprotective effect of ACE inhibitors in heart failure post-MI
There is overwhelming evidence that ACE inhibitors reduce morbidity and mortality, improve cardiac function, regress LV remodeling and prolong life in patients with heart failure (HF), not only improving cardiac function and increasing survival but also lessening myocardial re-infarction (206). Since ACE inhibitors prevent kinin degradation in the coronary circulation, one hypothesis is that kinins stimulate NO and PGI2, which are important inhibitors of platelet aggregation. They also greatly stimulate tPA (89, 268), thereby activating plasmin and fibrinolysis. Although the specific benefit of ACE inhibitors in re-infarction is still not fully understood, these hypotheses certainly open up a promising new area of cardiovascular research. We showed that in a rat model of HF due to surgically induced MI, ACE inhibitors improved cardiac function and attenuated remodeling as evidenced by an increased ejection fraction and decreased LV dilatation, myocyte hypertrophy and interstitial fibrosis, and these beneficial cardiac effects were diminished by blocking kinins (143). Moreover, in B2−/− mice (Fig. 9) and kininogen-deficient rats post-MI, ACE inhibitors had little or no effect (142, 311). Although we do not know exactly how kinins protect the heart, accumulated evidence suggests that kinin-stimulated release of NO and/or prostaglandins is largely responsible. Kinins stimulated release of NO from the mouse myocardium and decreased myocardial oxygen consumption, but these effects were blocked by a B2 kinin antagonist and in B2−/− mice they were absent altogether (146), while in endothelial NO synthase KO mice (eNOS−/−) with HF post-MI ACEi cardioprotection was almost abolished (141). Taken together, these findings suggest that kinins acting on the B2 receptor via release of NO play an important role in the cardioprotective action of ACE inhibitors.
Figure 9.
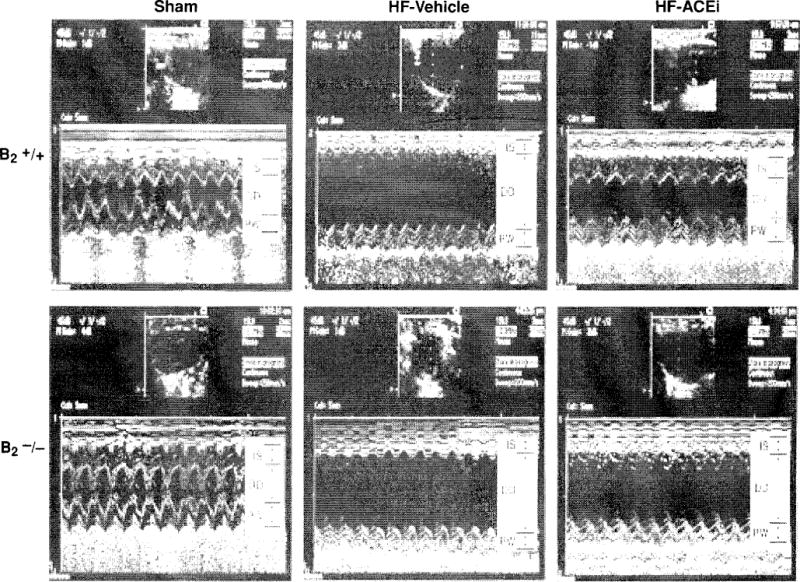
Two-dimensional M-mode echocardiographs of B2−/− mice and B2+/+ mice with sham coronary ligation (sham) or heart failure induced by coronary artery ligation. ACE inhibitor ramipril (2.5 mg/kg/day) reduced LV chamber size and thickeness in B2+/+, but only marginally in B2−/−, indicating an important role of kinin in the cardoprotective effects of ACE inibition in heart failure model. IS indicates interventricular septum; DD, left ventricular (LV) diastolic dimension; and PW, LV posterior wall. (Reprinted from Yang et al. (311).]
Recent reports suggest that the B1 receptors also contribute to the cardiac therapeutic effect of ACE inhibitors (111, 112, 308). We found that while they do not appear to play an essential role in cardiac hemodynamics and function under normal conditions or during the development of HF, they may help maintain morphological integrity, since mice with targeted B1 deletion (B1−/−) had increased basal LV mass and chamber dimensions and ACEi cardioprotection was reduced by induction of MI (308). ACEi can also upregulate and directly activate B1 receptors in cultured cells as well as the heart and vasculature (112, 161). They activate the HEAWH amino acid sequence in the second extracellular loop [residues 195–199 in the human receptor], a conserved sequence in B1 receptors in several species. This sequence matches the consensus pentameric HEXXH sequence of the catalytic domains in ACE and other metalloenzymes required to couple the Zn2+ cofactor. Activation of B1 receptors by ACE inhibitors leads to increased L-arginine uptake and prolonged NO release from cultured endothelial cells (110, 112), contributing to the therapeutic effect of ACE inhibitors (110, 112).
Role of kinins in the cardioprotective effect of angiotensin receptor blockers
Two subtypes of Ang II receptors, AT1, and AT2, have been identified. Most biological actions of Ang II are mediated by AT1, whereas less is known about the function of AT2. In general, activation of AT2 has been considered cardioprotective, partially due to stimulation of kinins and NO/cGMP (92, 127, 260). It has been shown that in mouse carotid arteries, flow-induced dilatation is AT2- and kinin dependent, since this effect is diminished in mice that lack TK (TK−/−) or AT2 receptors (AT2−/−) (24). Both in vitro and in vivo studies have demonstrated that Ang II is able to stimulate NO/cGMP production in the vasculature and this effect is blocked by either an AT2 or kinin B2 antagonist (2, 205, 260), suggesting that Ang II-stimulated NO/cGMP is mediated via activation of AT2 and increased endogenous kinins. However, the mechanism(s) responsible for AT2-stimulated kinin release is not fully understood. Tsutsumi et al. (288) reported that overexpression of AT2 in vascular smooth muscle cells decreased cellular pH associated with increased kininogenase activity, suggesting the existence of an acid-optimal kininogenase in the mouse aorta that is responsible for release of kinins from the vasculature. However, it is questionable whether this enzyme is TK, since the optimum pH for TK is approximately 8.5. In this regard, we questioned whether activation of plasma prekallikrein by PRCP contributes to AT2-stimulated kinin release, since PRCP is a known plasma prekallikrein activator in the endothelium and its enzyme activity appears to occur at acidic pH levels (191). We transfected cultured mouse coronary artery endothelial cells with the AT2 gene carried by an adenovirus and found that overexpression of AT2 increased bradykinin release associated with upregulation of PRCP mRNA and protein expression (318). Bradykinin release was further enhanced by activating AT2 with a specific agonist and this effect was blocked with a PRCP siRNA (Fig. 10). Our study provides evidence that AT2-stimulated bradykinin release is mediated at least in part via a PRCP-dependent mechanism.
Figure 10.
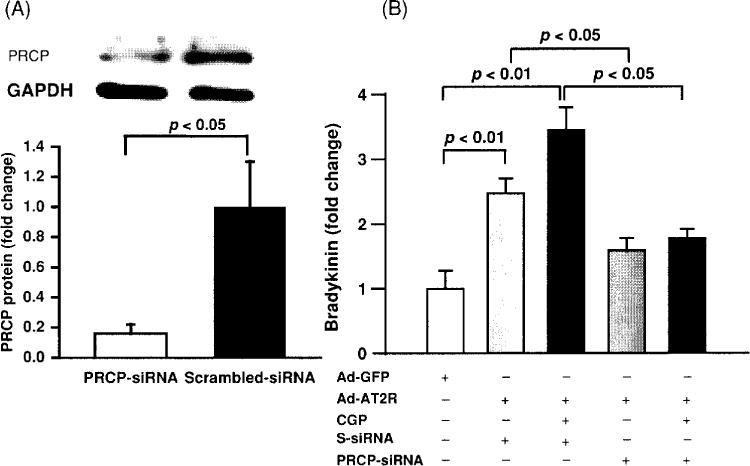
Effect of prolylcarboxypeptidase (PRCP) blockade by a siRNA on bradykinin (BK) release in Ad-AT2R-transfected mouse coronary endothelial cells (ECs). (A) Representative Western blot showing PRCP protein expression (top) and quantitative analysis of PRCP protein (bottom); PRCP SiRNA, but not scarmbled-SiRNA (S-siRNA) significantly blocked PRCP expression. (B) Fold change in bradykinin release relative to Ad-GFP-transfected cells; BK release was further increased when Ad-AT2R-transfected ECs were treated with CGP42112A, a selective AT2 agonist (CGP; 0.1 μmol/l). SiRNA, but not S-siRNA significantly blunted Ad-AT2R-induced bradykinin release, n = 4. [Reprinted from Zhu, Carretero, Yang et al., (318).]
Since blockade of AT1 increases Ang II, which in turn may activate AT2, it seems reasonable that the cardioprotective effect of ARBs is mediated in part by kinins via activation of AT2. In fact, we found that ARBs improved cardiac function and ameliorated remodeling in rats with HF post-MI and these effects were significantly attenuated by an AT2 or B2 antagonist (143) or in mice lacking AT2 receptors (AT2−/−) (306). Using B2−/− or eNOS−/− mice and kininogen-deficient rats, we confirmed that lack of kinins or endothelium-derived NO diminished the cardioprotective effect of ARBs (141, 145, 311), suggesting they are mediated in large part by increased kinins and NO via AT2. We also found that B1 partially mediated the therapeutic effects of ARB in mice with HF post-MI (308).
Because Ang II also plays a critical role in regulation of blood pressure as well as the pathogenesis of hypertension, we should be careful not to underestimate the contribution of both the RAS and the KKS to the effects of ACE inhibitors, both separately and acting together (Fig. 11). AbdAlla et al. reported that both AT1 and B2 self-assemble into stable heterodimers, increasing Gαq and Gαi (the two major signaling proteins triggered by AT1) and altering the endocytotic pathways of both receptors (7). This appears to be the first reported example of signal enhancement triggered by heterodimerization of two different vasoactive hormone receptors. Moreover, interaction of AT1 and B2 potentiates the pressor effect of Ang II (7). On the other hand, interaction of Ang (1–7) with bradykinin has emerged as an endogenous antihypertensive/antitrophic mechanism, opposing many of the effects of Ang II that are mediated by AT1 (80, 81, 83). Ang (1–7) acting via its receptor Mas (different from AT1 or AT2) induced bradykinin-mediated hypotension in SHR and normal rats (93) as well as dilatation of porcine coronary arteries (4, 31). Cleavage of Ang I and II to Ang (1–7) by various endopeptidases (60, 61) is another way kinins could expand the beneficial effects of ACE inhibitors or ARBs. In addition, kinins may be important in mediating the counter-regulatory protective effect of AT2, which opposes AT1 (143, 306), (265). Therefore, there seems to be a close interaction between kinins and angiotensins in the regulation of cardiovascular and renal function.
Figure 11.
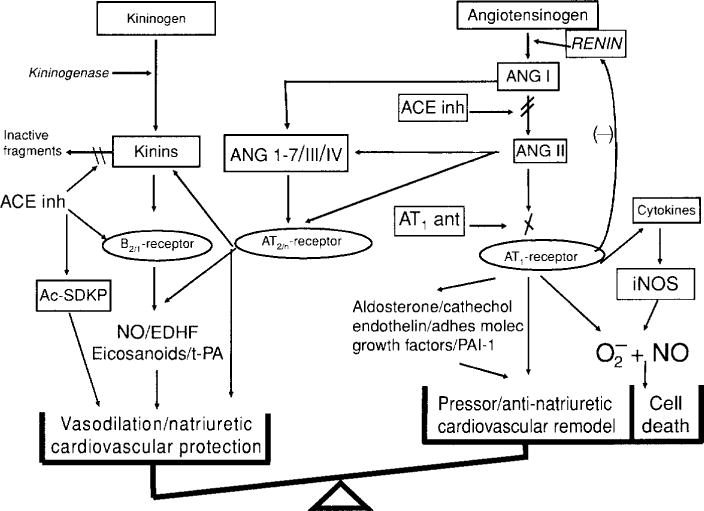
The renin-angiotensin and kallikrein-kinin systems. In both systems, a substrate is cleaved by an enzyme of restricted specificity, releasing a peptide that is either already active (lysbradykinin, bradykinin) or inactive (angiotensin I). Upon further processing by a specific peptidase, angiotensin I is converted to a vasoactive peptide (angiotensin II). In turn, vasoactive peptides are inactivated by peptidases. Angiotensin-Converting enzyme is common to both systems but has different roles: it processes angiotensin I to angiotensin II and is the main kinin-inactivating peptidase. [Reprinted from Liu et al. (143).]
Conclusion
We must conclude that kinins do not play a fundamental role in the pathogenesis of hypertension, since humans, rats, and mice deficient in one or more components of the KKS or found to have chronic KKS blockade do not have hypertension. Renal kinins help regulate papillary blood flow and water and sodium excretion, which explains why B2-KO mice are more salt-sensitive. Kinins are also potent mediators of inflammation by mediating the cardinal signs of inflammation, acting mainly via inducible B1 and in certain diseases B2. While kinins participate in the acute antihypertensive effect of ACE inhibitors, in general they are not involved in their chronic effects except for mineralocorticoid-salt-induced hypertension. Kinins acting via NO enhance the vascular protective effect of ACE inhibitors during neointima formation. In MI produced by ischemia/reperfusion, kinins play an important role in the infarct reduction seen after preconditioning or ACEi treatment. In HF secondary to infarction, the therapeutic effects of ACEi are partially mediated by kinins via NO while that of ARBs is due in part to activation of AT2 via kinins and NO. Thus kinins play an important role in regulation of cardiovascular and renal function as well as many of the beneficial effects of ACEi and ARBs.
Acknowledgments
The authors wish to acknowledge the editorial work by Steve Haller. This work was supported by NIH grants HL-028982 to OAC and HL-071806 to NER.
References
- 1.Farmer SG, editor. The Kinin System (The Handbook of lmmunopharmacology) San Diego, CA: Academic Press Inc; 1997. pp. 1–323. [Google Scholar]
- 2.Abadir PM, Carey RM, Siragy HM. Angiotensin AT2 receptors directly stimulate renal nitric oxide in bradykinin B2-receptor-null mice. Hypertension. 2003;42:600–604. doi: 10.1161/01.HYP.0000090323.58122.5C. [DOI] [PubMed] [Google Scholar]
- 3.Abadir PM, Periasamy A, Carey RM, Siragy HM. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension. 2006;48:1–7. doi: 10.1161/01.HYP.0000228997.88162.a8. [DOI] [PubMed] [Google Scholar]
- 4.Abbas A, Gorelik G, Carbini LA, Scicli AG. Angiotensin-(1-7) induces bradykinin-mediated hypotensive responses in anesthetized rats. Hypertension. 1997;30:217–221. doi: 10.1161/01.hyp.30.2.217. [DOI] [PubMed] [Google Scholar]
- 5.AbdAlla S, Abdel-Baset A, Lother H, el Massiery A, Quitterer U. Mesangial AT1/B2 receptor heterodimers contribute to angiotensin II hyperresponsiveness in experimental hypertension. J Mol Neurosci. 2005;26:185–192. doi: 10.1385/JMN:26:2-3:185. [DOI] [PubMed] [Google Scholar]
- 6.AbdAlla S, Lother H, el Massiery A, Quitterer U. Increased AT1 receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat Med. 2001;7:1003–1009. doi: 10.1038/nm0901-1003. [DOI] [PubMed] [Google Scholar]
- 7.AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature. 2000;407:94–98. doi: 10.1038/35024095. [DOI] [PubMed] [Google Scholar]
- 8.AbdAlla S, Zaki E, Lother H, Quitterer U. Involvement of the amino terminus of the B(2) receptor in agonist-induced receptor dimerization. J Biol Chem. 1999;274:26079–26084. doi: 10.1074/jbc.274.37.26079. [DOI] [PubMed] [Google Scholar]
- 9.Agarwal D, Haque M, Sriramula S, Mariappan N, Pariaut R, Francis J. Role of proinflammatory cytokines and redox homeostasis in exercise-induced delayed progression of hypertension in spontaneously hypertensive rats. Hypertension. 2009;54:1393–1400. doi: 10.1161/HYPERTENSIONAHA.109.135459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alfie ME, Yang X-P, Hess F, Carretero OA. Salt-sensitive hypertension in bradykinin B2 receptor knockout mice. Biochem Biophys Res Commun. 1996;224:625–630. doi: 10.1006/bbrc.1996.1076. [DOI] [PubMed] [Google Scholar]
- 11.Alhenc-Gelas F, Marchetti J, Allegrini J, Corvol P, Menard J. Measurement of urinary kallikrein activity. Species differences in kinin production. Biochim Biophys Acta. 1981;677:477–488. doi: 10.1016/0304-4165(81)90262-2. [DOI] [PubMed] [Google Scholar]
- 12.Aliberti J, Viola JP, Vieira-de-Abreu A, Bozza PT, Sher A, Scharfstein J. Cutting edge: Bradykinin induces IL-12 production by dendritic cells: a danger signal that drives Th1 polarization. J Immunol. 2003;170:5349–5353. doi: 10.4049/jimmunol.170.11.5349. [DOI] [PubMed] [Google Scholar]
- 13.Azizi M, Boutouyrie P, Bissery A, Aghrarazii M, Verbeke F, Stern N, Bura-Rivière A, Laurent S, Alhenc-Gelas F, Jeunemaitre X. Arterial and renal consequences of partial genetic deficiency in tissue kallikrein activity in humans. J Clin Invest. 2005;115:780–787. doi: 10.1172/JCI23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao G, Gohlke P, Qadri F, Unger T. Chronic kinin receptor blockade attenuates the antihypertensive effect of ramipril. Hypertension. 1992;20:74–79. doi: 10.1161/01.hyp.20.1.74. [DOI] [PubMed] [Google Scholar]
- 15.Barki-Harrington L, Bookout AL, Wang G, Lamb ME, Leeb-Lundberg LM, Daaka Y. Requirement for direct cross-talk between B1 and B2 kinin receptors for the proliferation of androgen-insensitive prostate cancer PC3 cells. Biochem J. 2003;371:581–587. doi: 10.1042/BJ20021708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barias A, Okamoto H, Greenbaum LM. T-kininogen — the major plasma kininogen in rat adjuvant arthritis. Biochem Biophys Res Commun. 1985;129:280–286. doi: 10.1016/0006-291x(85)91434-2. [DOI] [PubMed] [Google Scholar]
- 17.Bascands JL, Bachvarova M, Neau E, Schanstra JP, Bachvarov D. Molecular determinants of LPS-induced acute renal inflammation: Implication of the kinin B1 receptor. Biochem Biophys Res Commun. 2009;386:407–412. doi: 10.1016/j.bbrc.2009.06.063. [DOI] [PubMed] [Google Scholar]
- 18.Baumgarten G, Knuefermann P, Kalra D, Gao F, Taffet GE, Michael L, Blackshear PJ, Carballo E, Sivasubramanian N, Mann DL. Load-dependent and -independent regulation of proinflammatory cytokine and cytokine receptor gene expression in the adult mammalian heart. Circulation. 2002;105:2192–2197. doi: 10.1161/01.cir.0000015608.37608.18. [DOI] [PubMed] [Google Scholar]
- 19.Beierwaltes WH, Carretero OA, Scicli AG. Renal hemodynamics in response to a kinin analogue antagonist. Am J Physiol. 1988;255:F408–F414. doi: 10.1152/ajprenal.1988.255.3.F408. [DOI] [PubMed] [Google Scholar]
- 20.Beierwaltes WH, Carretero OA, Scicli AG, Vavrek RJ, Stewart JM. Competitive analog antagonists of bradykinin in the canine hindlimb. Proc Soc Exp Biol Med. 1987;186:79–83. doi: 10.3181/00379727-186-42588. [DOI] [PubMed] [Google Scholar]
- 21.Benetos A, Gavras H, Stewart JM, Vavrek RJ, Hatinoglou S, Gavras I. Vasodepressor role of endogenous bradykinin assessed by a bradykinin antagonist. Hypertension. 1986;8:971–974. doi: 10.1161/01.hyp.8.11.971. [DOI] [PubMed] [Google Scholar]
- 22.Benetos A, Gavras I, Gavras H. Hypertensive effect of a bradykinin antagonist in normotensive rats. Hypertension. 1986;8:1089–1092. doi: 10.1161/01.hyp.8.11.1089. [DOI] [PubMed] [Google Scholar]
- 23.Berg T, Carretero OA, Scicli AG, Tilley B, Stewart JM. Role of kinin in regulation of rat submandibular gland blood flow. Hypertension. 1989;14:73–80. doi: 10.1161/01.hyp.14.1.73. [DOI] [PubMed] [Google Scholar]
- 24.Bergaya S, Hilgers RH, Meneton P, Dong Y, Bloch-Faure M, Inagami T, Alhenc-Gelas F, Lévy BI, Boulanger CM. Flow-dependent dilation mediated by endogenous kinins requires angiotensin AT2 receptors. Circ Res. 2004;94:1623–1629. doi: 10.1161/01.RES.0000131497.73744.1a. [DOI] [PubMed] [Google Scholar]
- 25.Bergaya S, Meneton P, Bloch-Faure M, Mathieu E, henc-Gelas F, Levy BI, Boulanger CM. Decreased flow-dependent dilation in carotid arteries of tissue kallikrein-knockout mice. Circ Res. 2001;88:593–599. doi: 10.1161/01.res.88.6.593. [DOI] [PubMed] [Google Scholar]
- 26.Blanchard A, Azizi M, Peyrard S, Stern N, henc-Gelas F, Houillier P, Jeunemaitre X. Partial human genetic deficiency in tissue kallikrein activity and renal calcium handling. Clin J Am Soc Nephrol. 2007;2:320–325. doi: 10.2215/CJN.02630706. [DOI] [PubMed] [Google Scholar]
- 27.Bock MG, Longmore J. Bradykinin antagonists: new opportunities. Curr Opin Chem Biol. 2000;4:401–406. doi: 10.1016/s1367-5931(00)00107-1. [DOI] [PubMed] [Google Scholar]
- 28.Bönner G, Preis S, Schunk U, Toussaint C, Kaufmann W. Hemodynamic effects of bradykinin on systemic and pulmonary circulation in healthy and hypertensive humans. J Cardiovasc Pharmacol. 1990;15(Suppl 6):S46–S56. [PubMed] [Google Scholar]
- 29.Boucher R, Demassieux S, Garcia R, Genest J. Tonin, angiotensin II system. Circ Res. 1977;41:26–29. doi: 10.1161/01.res.41.4.26. [DOI] [PubMed] [Google Scholar]
- 30.Brew EC, Mitchell MB, Rehring TF, Gamboni-Robertson F, Mclntyre RC, Jr, Harken AH, Banerjee A. Role of bradykinin in cardiac functional protection after global ischemia-reperfusion in rat heart. Am J Physiol. 1995;269:H1370–H1378. doi: 10.1152/ajpheart.1995.269.4.H1370. [DOI] [PubMed] [Google Scholar]
- 31.Brosnihan KB, Li P, Ferrarlo CM. Angiotensin-(1-7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension. 1996;27:523–528. doi: 10.1161/01.hyp.27.3.523. [DOI] [PubMed] [Google Scholar]
- 32.Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776–781. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- 33.Burch RM, Farmer SG, Steranka LR. Bradykinin receptor antagonists. Med Res Rev. 1990;10:237–269. doi: 10.1002/med.2610100204. [DOI] [PubMed] [Google Scholar]
- 34.Burgess GM, Perkins MN, Rang HP, Campbell EA, Brown MC, Mcintyre P, Urban L, Dziadulewicz EK, Ritchie TJ, Hallett A, Snell CR, Wrigglesworth R, Lee W, Davis C, Phagoo SB, Davis AJ, Phillips E, Drake GS, Hughes GA, Dunstan A, Bloomfield GC. Bradyzide, a potent non-peptide B(2) bradykinin receptor antagonist with long-lasting oral activity in animal models of inflammatory hyperalgesia. Br J Pharmacol. 2000;129:77–86. doi: 10.1038/sj.bjp.0703012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cachofeiro V, Sakakibara T, Nasjletti A. Kinins, nitric oxide, and the hypotensive effect of captopril and ramiprilat in hypertension. Hypertension. 1992;19:138–145. doi: 10.1161/01.hyp.19.2.138. [DOI] [PubMed] [Google Scholar]
- 36.Campbell DJ. The kallikrein-kinin system in humans. Clin Exp Pharmacol Physiol. 2001;28:1060–1065. doi: 10.1046/j.1440-1681.2001.03564.x. [DOI] [PubMed] [Google Scholar]
- 37.Campbell DI, Kladis A, Duncan A-M. Bradykinin peptides in kidney, blood, and other tissues of the rat. Hypertension. 1993;21:155–165. doi: 10.1161/01.hyp.21.2.155. [DOI] [PubMed] [Google Scholar]
- 38.Carbini LA, Scicli AG, Carretero OA. The molecular biology of the kallikrein-kinin system: Ill. The human kallikrein gene family and kallikrein substrate. J Hypertens. 1993;11:893–898. doi: 10.1097/00004872-199309000-00002. [DOI] [PubMed] [Google Scholar]
- 39.Carbonell LF, Carretero OA, Madeddu P, Scicli AG. Effects of a kinin antagonist on mean blood pressure. Hypertension. 1988;11(Suppl I):I-84–I-88. doi: 10.1161/01.hyp.11.2_pt_2.i84. [DOI] [PubMed] [Google Scholar]
- 40.Carbonell LF, Carretero OA, Stewart JM, Scicli AG. Effect of a kinin antagonist on the acute antihypertensive activity of enalaprilat in severe hypertension. Hypertension. 1988;11:239–243. doi: 10.1161/01.hyp.11.3.239. [DOI] [PubMed] [Google Scholar]
- 41.Carretero OA. High-mineralocorticoid conditions: Kinins (paracrine hormones) in the regulation of renal function and blood pressure. In: Mornex R, Jaffiol C, Ledère J, editors. Progress in Endocrinology. The Proceedings of the Ninth International Congress of Endocrinology, Nice 1992. Carnforth, Lancastershire, UK: Parthenon Publications Group; 1993a. pp. 536–540. [Google Scholar]
- 42.Carretero OA. Kinins: local hormones in regulation of blood pressure and renal function. Choices Cardiali. 1993b;(Suppl 1):10–14. [Google Scholar]
- 43.Carretero OA, Amin VM, Ocholik T, Scicli AG, Koch J. Urinary kallikrein in rats bred for their susceptibility and resistance to the hypertensive effect of salt. A new radioimmunoassay for its direct determination. Circ Res. 1978;42:727–731. doi: 10.1161/01.res.42.5.727. [DOI] [PubMed] [Google Scholar]
- 44.Carretero OA, Carbini LA, Scicli AG. The molecular biology of the kallikrein-kinin system: I. General description, nomenclature and the mouse gene family. J Hypertens. 1993;11:693–697. doi: 10.1097/00004872-199307000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Carretero OA, Kuk P, Piwonska Ss, Houle JA, Marin-Grez M. Role of the renin-angiotensin system in the pathogenesis of severe hypertension in rats. Circ Res. 1971;29:654–663. doi: 10.1161/01.res.29.6.654. [DOI] [PubMed] [Google Scholar]
- 46.Carretero OA, Miyazaki S, Scicli AG. Role of kinins in the acute antihypertensive effect of the converting enzyme inhibitor, Captopril. Hypertension. 1981;3:18–22. doi: 10.1161/01.hyp.3.1.18. [DOI] [PubMed] [Google Scholar]
- 47.Carretero OA, Polomski C, Hampton A, Scicli AG. Urinary kallikrein, plasma renin and aldosterone in New Zealand genetically hypertensive (GH) rats. Clin Exp Pharmacol Physiol. 1976;3(Suppl):55–59. [Google Scholar]
- 48.Carretero OA, Scicli AG. The renal kallikrein-kinin system in human and in experimental hypertension. Klin Wochenschr. 1978;56(Suppl. I):113–125. doi: 10.1007/BF01477462. [DOI] [PubMed] [Google Scholar]
- 49.Carretero OA, Scicli AG. Kinins paracrine hormone. Kidney Int. 1988;34(Suppl 26):S-52–S-59. [PubMed] [Google Scholar]
- 50.Carretero OA, Scicli AG. The kallikrein-kinin system as a regulator of cardiovascular and renal function. In: Laragh JH, Brenner BM, editors. Hypertension: Physiology, Diagnosis, and Management. New York: Raven Press; 1995. pp. 983–999. [Google Scholar]
- 51.Carretero OA, Scicli AG, Maitra SR. Role of kinins in the pharmacological effects of converting enzyme inhibitors. In: Horovitz ZP, editor. Angiotensin Converting Enzyme Inhibitors Mechanisms of Action and Clinical Implications. Baltimore: Urban & Schwarzenberg; 1981. pp. 105–121. [Google Scholar]
- 52.Carretero OA, Scicli AG, Piwonska A, Koch J. Urinary kallikrein in rats bred for susceptibility and resistance to the hypertensive effect of salt and in New Zealand genetically hypertensive rats. Mayo Clin Proc. 1977;52:465–467. [PubMed] [Google Scholar]
- 53.Carretero OA, Ørstavik TB, Rabito SF, Scicli AG. Interference of converting enzyme inhibitors with the kallikrein-kinin system. Clin Exp Hypertens A. 1983;5:1277–1285. doi: 10.3109/10641968309048857. [DOI] [PubMed] [Google Scholar]
- 54.Cassim B, Shaw OM, Mazur M, Misso NL, Naran A, Langlands DR, Thompson PJ, Bhoola KD. Kallikreins, kininogens and kinin receptors on circulating and synovial fluid neutrophils: role in kinin generation in rheumatoid arthritis. Rheumatology (Oxford) 2009;48:490–496. doi: 10.1093/rheumatology/kep016. [DOI] [PubMed] [Google Scholar]
- 55.Cervenka L, Harrison-Bernard LM, Dipp S, Primrose G, Imig JD, El-Dahr SS. Early onset salt-sensitive hypertension in bradykinin B2 receptor null mice. Hypertension. 1999;34:176–180. doi: 10.1161/01.hyp.34.2.176. [DOI] [PubMed] [Google Scholar]
- 56.Chambers JC, Eda S, Bassett P, Karim Y, Thompson SG, Gallimore JR, Pepys MB, Kooner JS. C-reactive protein, insulin resistance, central obesity, and coronary heart disease risk in Indian Asians from the United Kingdom compared with European whites. Circulation. 2001;104:145–150. doi: 10.1161/01.cir.104.2.145. [DOI] [PubMed] [Google Scholar]
- 57.Chao J, Chao L, Margolius HS. Isolation of tissue kallikrein in rat spleen by monoclonal antibody-affinitv chromatography. Biochim Biophys Acta. 1984;801:244–249. doi: 10.1016/0304-4165(84)90073-4. [DOI] [PubMed] [Google Scholar]
- 58.Chao J, Chao L, Swain CC, Tsai J, Margolius HS. Tissue kallikrein in rat brain and pituitary: regional distribution and estrogen induction in the anterior pituitary. Endocrinology. 1987;120:475–482. doi: 10.1210/endo-120-2-475. [DOI] [PubMed] [Google Scholar]
- 59.Chao J, Li HJ, Yao YY, Shen B, Gao L, Bledsoe G, Chao L. Kinin infusion prevents renal inflammation, apoptosis, and fibrosis via inhibition of oxidative stress and mitogen-activated protein kinase activity. Hypertension. 2007;49:490–497. doi: 10.1161/01.HYP.0000255925.01707.eb. [DOI] [PubMed] [Google Scholar]
- 60.Chappell MC, Allred AJ, Ferrario CM. Pathways of angiotensin-(1-7) metabolism in the kidney. Nephrol Dial Transplant. 2001;16(Suppl 1):22–26. doi: 10.1093/ndt/16.suppl_1.22. [DOI] [PubMed] [Google Scholar]
- 61.Chappell MC, Gomez MN, Pirro NT, Ferrario CM. Release of angiotensin-(1-7) from the rat hindlimb. Influence of angiotensin-converting enzyme inhibition. Hypertension. 2000;35:348–352. doi: 10.1161/01.hyp.35.1.348. [DOI] [PubMed] [Google Scholar]
- 62.Cherry PD, Furchgott RF, Zawadzki JV, Jothianandan D. Role of endothelial cells in relaxation of isolated arteries by bradykinin. Proc Natl Acad Sci USA. 1982;79:2106–2110. doi: 10.1073/pnas.79.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cialdai C, Giuliani S, Valenti C, Tramontana M, Maggi CA. Effect of intra-articular 4-(S)-amino-5-(4-{4-[2,4-dichloro-3-(2,4-dimethyl-8-quinolyloxymethyl)phenylsulfo namido|-tetrahydro-2H-4-pyranylcarbonyl} piperazino)-5-oxopentyl](trimethyl)ammonium chloride hydrochloride (MEN16132), a kinin B2 receptor antagonist, on nociceptive response in monosodium iodoacetate-induced experimental osteoarthritis in rats. J Pharmacol Exp Ther. 2009;331:1025–1032. doi: 10.1124/jpet.109.159657. [DOI] [PubMed] [Google Scholar]
- 64.Clappison BH, Anderson WP, Johnston CI. Role of the kallikrein-kinin system in the renal effects of angiotensin-converting enzyme inhibition in anaesthetized dogs. Clin Exp Pharmacol Physiol. 1981;8:509–513. doi: 10.1111/j.1440-1681.1981.tb00758.x. [DOI] [PubMed] [Google Scholar]
- 65.Clements JA. The glandular kallikrein family of enzymes: Tissue-specific expression and hormonal regulation. Endocr Rev. 1989;10:393–419. doi: 10.1210/edrv-10-4-393. [DOI] [PubMed] [Google Scholar]
- 66.Clements JA, Matheson BA, MacDonald RJ, Funder JW. The expression of the kallikrein gene family in the rat pituitary: oestrogen effects and the expression of an additional family member in the neurointermediate lobe. J Neuroendocrinal. 1989;1:199–203. doi: 10.1111/j.1365-2826.1989.tb00103.x. [DOI] [PubMed] [Google Scholar]
- 67.Clozel M. Mechanism of action of angiotensin converting enzyme inhibitors on endothelial function in hypertension. Hypertension. 1991;18(Suppl 11):II-37–II-42. doi: 10.1161/01.hyp.18.4_suppl.ii37. [DOI] [PubMed] [Google Scholar]
- 68.Cohuet G, Struijker-Boudier H. Mechanisms of target organ damage caused by hypertension: Therapeutic potential. Pharmacol Ther. 2006;111:81–98. doi: 10.1016/j.pharmthera.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 69.Colman RW. Patho-physiology of kallikrein system. Ann Clin Lab Sci. 1980;10:220–226. [PubMed] [Google Scholar]
- 70.Danckwardt L, Shimizu I, Bönner G, Rettig R, Unger T. Converting enzyme inhibition in kinin-deficient Brown Norway rats. Hypertension. 1990;16:429–435. doi: 10.1161/01.hyp.16.4.429. [DOI] [PubMed] [Google Scholar]
- 71.Dear JW, Foreman JC. The nasal airway pharmacology of bradykinin. In: Farmer SG, editor. The Kinin System. San Diego, CA: Academic Press Inc; 1997. pp. 287–300. [Google Scholar]
- 72.Duka A, Kintsurashvili E, Duka I, Ona D, Hopkins TA, Bader M, Gavras I, Gavras H. Angiotensin-Converting enzyme inhibition after experimental myocardial infarct: role of the kinin B1 and B2 receptors. Hypertension. 2008;51:1352–1357. doi: 10.1161/HYPERTENSIONAHA.107.108506. [DOI] [PubMed] [Google Scholar]
- 73.Emanueli C, Maestri R, Corradi D, Marchione R, Minasi A, Tozzi MG, Salis MB, Straino S, Capogrossi MC, Olivetti G, Madeddu P. Dilated and failing cardiomyopathy in bradykinin B2 receptor knockout mice. Circulation. 1999;100:2359–2365. doi: 10.1161/01.cir.100.23.2359. [DOI] [PubMed] [Google Scholar]
- 74.Erdös EG. Angiotensin I converting enzyme. Circ Res. 1975;36:247–255. doi: 10.1161/01.res.36.2.247. [DOI] [PubMed] [Google Scholar]
- 75.Erdös EG. Kininases. In: Erdös EG, editor. Handbook of Experimental Pharmacology, Vol XXV Suppl: Bradykinin, Kallidin and Kallikrein. Berlin: Springer-Verlag; 1979. pp. 427–487. [Google Scholar]
- 76.Farhy R, Ho K-L, Carretero OA, Scicli AG. Kinins mediate the antiproliferative effect of ramipril in rat carotid artery. Biochem Biophys Res Commun. 1992;182:283–288. doi: 10.1016/s0006-291x(05)80142-1. [DOI] [PubMed] [Google Scholar]
- 77.Farhy RD, Carretero OA, Ho K-L, Scicli AG. Role of kinins and nitric oxide in the effects of angiotensin converting enzyme inhibitors on neointima formation. Circ Res. 1993;72:1202–1210. doi: 10.1161/01.res.72.6.1202. [DOI] [PubMed] [Google Scholar]
- 78.Farmer SG. The kallikrein-kinin system in asthma and acute respiratory distress syndrome. In: Farmer SG, editor. The Kinin System. San Diego, CA: Academic Press Inc; 1997. pp. 249–263. [Google Scholar]
- 79.Fernandez LA, Twickler J, Mead A. Neovascularization produced by angiotensin II. J Lab Clin Med. 1985;105:141–145. [PubMed] [Google Scholar]
- 80.Ferrario CM. Angiotensin-(1-7) and antihypertensive mechanisms. J Nephrol. 1998;11:278–283. [PubMed] [Google Scholar]
- 81.Ferrario CM, Averill DB, Brosnihan KB, Chappell MC, Iskandar SS, Dean RH, Diz DI. Vasopeptidase inhibition and Ang-(l-7) in the spontaneously hypertensive rat. Kidney Int. 2002;62:1349–1357. doi: 10.1111/j.1523-1755.2002.kid559.x. [DOI] [PubMed] [Google Scholar]
- 82.Figueroa CD, Maclver AG, Mackenzie JC, Bhoola KD. Localisation of immunoreactive kininogen and tissue kallikrein in the human nephron. Histochemistry. 1988;89:437–442. doi: 10.1007/BF00492599. [DOI] [PubMed] [Google Scholar]
- 83.Freeman EJ, Chisolm GM, Ferrario CM, Tallant EA. Angiotensin-(1-7) inhibits vascular smooth muscle cell growth. Hypertension. 1996;28:104–108. doi: 10.1161/01.hyp.28.1.104. [DOI] [PubMed] [Google Scholar]
- 84.Frey EK, Kraut H, Werle E. In: Kallikrein Padutin [English transl. (1977) Vogel R, editor. Stuttgart: Ferdinand Enke Verlag; 1950. [Google Scholar]
- 85.Fumichi K, Gao JL, Horuk R, Wada T, Kaneko S, Murphy PM. Chemokine receptor CCR1 regulates inflammatory cell infiltration after renal ischemia-reperfusion injury. J Immunol. 2008;181:8670–8676. doi: 10.4049/jimmunol.181.12.8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Furuto-Kato S, Matsumoto A, Kitamura N, Nakanishi S. Primary structures of the mRNAs encoding the rat precursors for bradykinin and T-kinin. Structural relationship of kininogens with major acute phase protein and αl-cysteine proteinase inhibitor. J Biol Chem. 1985;260:12054–12059. [PubMed] [Google Scholar]
- 87.Gainer JV, Morrow JD, Loveland A, King DJ, Brown NJ. Effect of bradykinin-receptor blockade on the response to angiotensin-converting enzyme inhibitor in normotensive and hypertensive subjects. N Engl J Med. 1998;339:1285–1292. doi: 10.1056/NEJM199810293391804. [DOI] [PubMed] [Google Scholar]
- 88.Geiger R, Clausnitzer B, Fink E, Fritz H. Isolation of an enzymatically active glandular kallikrein from human plasma by immunoaffinity chromatography. Hoppe Seylers Z Physiol Chem. 1980;361:1795–1803. doi: 10.1515/bchm2.1980.361.2.1795. [DOI] [PubMed] [Google Scholar]
- 89.Gertz SD, Kurgan A. Tissue plasminogen activator and selective coronary vasodilation [letter] Am J Cardiol. 1988;62:173. doi: 10.1016/0002-9149(88)91404-x. [DOI] [PubMed] [Google Scholar]
- 90.Gohlke P, Kuwer I, Schnell A, Amann K, Mall G, Unger T. Blockade of bradykinin B2 receptors prevents the increase in capillary density induced by chronic angiotensin-converting enzyme inhibitor treatment in stroke-prone spontaneously hypertensive rats. Hypertension. 1997;29:478–482. doi: 10.1161/01.hyp.29.1.478. [DOI] [PubMed] [Google Scholar]
- 91.Gohlke P, Linz W, Schölkens BA, Kuwer I, Bartenbach S, Schnell A, Unger T. Angiotensin-converting enzyme inhibition improves cardiac function. Role of bradykinin. Hypertension. 1994;23:411–418. doi: 10.1161/01.hyp.23.4.411. [DOI] [PubMed] [Google Scholar]
- 92.Gohlke P, Pees C, Unger T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension. 1998;31:349–355. doi: 10.1161/01.hyp.31.1.349. [DOI] [PubMed] [Google Scholar]
- 93.Gorelik G, Carbini LA, Scicli AG. Angiotensin 1-7 induces bradykinin-mediated relaxation in porcine coronary artery. J Pharmacol Exp Ther. 1998;286:403–410. [PubMed] [Google Scholar]
- 94.Goto M, Liu Y, Yang X-M, Ardell JL, Cohen MV, Downey JM. Role of bradykinin in protection of ischemic preconditioning in rabbit hearts. Circ Res. 1995;11:611–621. doi: 10.1161/01.res.77.3.611. [DOI] [PubMed] [Google Scholar]
- 95.Griol-Charhbili V, Messadi-Laribi E, Bascands JL, Heudes D, Meneton P, Giudicelli JF, henc-Gelas F, Richer C. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. FASEB J. 2005;19:1172–1174. doi: 10.1096/fj.04-3508fje. [DOI] [PubMed] [Google Scholar]
- 96.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II-induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hall JM, Morton IKM. The pharmacology and immunopharmacology of kinin receptors. In: Farmer SG, editor. The Kinin System. San Diego, CA: Academic Press Inc; 1997. pp. 9–43. [Google Scholar]
- 98.Hansen JL, Hansen JT, Speerschneider T, Lyngso C, Erikstrup N, Burstein ES, Weiner DM, Walther T, Makita N, Iiri T, Merten N, Kostenis E, Sheikh SP. Lack of evidence for AT1 R/B2R heterodimerization in COS-7, HEK293, and NIH3T3 cells: How common is the AT1R/B2R heterodimer? J Biol Chem. 2009;284:1831–1839. doi: 10.1074/jbc.M804607200. [DOI] [PubMed] [Google Scholar]
- 99.Hara DB, Leite DF, Fernandes ES, Passos GF, Guimaraes AO, Pesquero JB, Campos MM, Calixto JB. The relevance of kinin B1 receptor upregulation in a mouse model of colitis. Br J Pharmacol. 2008;154:1276–1286. doi: 10.1038/bjp.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harrison DG, Guzik TJ, Goronzy J, Weyand C. Is hypertension an immunogic disease? Curr Cardiol Rep. 2009;10:464–469. doi: 10.1007/s11886-008-0073-6. [DOI] [PubMed] [Google Scholar]
- 101.Hartman JC, Wall TM, Hullinger TG, Shebuski RJ. Reduction of myocardial infarct size in rabbits by ramiprilat: reversal by the bradykinin antagonist HOE 140. J Cardiovasc Pharmacol. 1993;21:996–1003. doi: 10.1097/00005344-199306000-00022. [DOI] [PubMed] [Google Scholar]
- 102.Hashimoto K, Hamamoto H, Honda Y, Hirose M, Furukawa S, Kimura E. Changes in components of kinin system and hemodynamics in acute myocardial infarction. Am Heart J. 1978;95:619–626. doi: 10.1016/0002-8703(78)90304-6. [DOI] [PubMed] [Google Scholar]
- 103.Hecquet C, Tan F, Marcie BM, Erdös EG. Human bradykinin B2 receptor is activated by kallikrein and other serine proteases. Mol Pharmacol. 2000;58:828–836. doi: 10.1124/mol.58.4.828. [DOI] [PubMed] [Google Scholar]
- 104.Heitsch H. Bradykinin B2 receptor as a potential therapeutic target. Drug News Perspect. 2000;13:213–225. [PubMed] [Google Scholar]
- 105.Heitsch H. The therapeutic potential of bradykinin B2 receptor agonists in the treatment of cardiovascular disease. Expert Opin Investig Drugs. 2003;12:759–770. doi: 10.1517/13543784.12.5.759. [DOI] [PubMed] [Google Scholar]
- 106.Hilton SM. The physiological role of glandular kallikreins. In: Erdös EG, editor. Handbook of Experimental Pharmacology, Vol. 25: Bradykinin, Kallidin and Kallikrein. New York: Springer-Verlag; 1970. pp. 389–399. [Google Scholar]
- 107.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–R216. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Holland OB, Chud JM, Braunstein H. Urinary kallikrein excretion in essential and mineralocorticoid hypertension. J Clin Invest. 1980;65:347–356. doi: 10.1172/JCI109678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Holton FA, Holton P. The vasodilator activity of spinal roots. J Physiol. 1952;118:310–327. doi: 10.1113/jphysiol.1952.sp004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ignjatovic T, Stanisavljevic S, Brovkovych V, Skidgel RA, Erdös EG. Kinin B1 receptors stimulate nitric oxide production in endothelial cells: signaling pathways activated by angiotensin I-converting enzyme inhibitors and peptide ligands. Mol Pharmacol. 2004;66:1310–1316. doi: 10.1124/mol.104.001990. [DOI] [PubMed] [Google Scholar]
- 111.Ignjatovic T, Tan F, Brovkovych V, Skidgel RA, Erdös EG. Activation of bradykinin B1 receptor by ACE inhibitors. Int Immunopharmacol. 2002;2:1787–1793. doi: 10.1016/s1567-5769(02)00146-7. [DOI] [PubMed] [Google Scholar]
- 112.Ignjatovic T, Tan F, Brovkovych V, Skidgel RA, Erdös EG. Novel mode of action of angiotensin I converting enzyme inhibitors. Direct activation of bradykinin B1 receptor. J Biol Chem. 2002;277:16847–16852. doi: 10.1074/jbc.M200355200. [DOI] [PubMed] [Google Scholar]
- 113.Ishida H, Scicli AG, Carretero OA. Role of angiotensin converting enzyme and other peptidases in in vivo metabolism of kinins. Hypertension. 1989;14:322–327. doi: 10.1161/01.hyp.14.3.322. [DOI] [PubMed] [Google Scholar]
- 114.Jacobsen S. Separation of two different substrates for plasma kinin-forming enzymes. Nature. 1966;210:98–99. doi: 10.1038/210098a0. [DOI] [PubMed] [Google Scholar]
- 115.Jacobsen S. Substrates for plasma kmin-forming enzymes in human, dog and rabbit plasmas. Br J Pharmacol. 1966;26:403–411. doi: 10.1111/j.1476-5381.1966.tb01920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ju H, Venema VJ, Marrero MB, Venema RC. Inhibitory interactions of the bradykinin B2 receptor with endothelial nitric-oxide synthase. J Biol Chem. 1998;273:24025–24029. doi: 10.1074/jbc.273.37.24025. [DOI] [PubMed] [Google Scholar]
- 117.Kakoki M, Sullivan KA, Backus C, Hayes JM, Oh SS, Hua K, Gasim AM, Tomita H, Grant R, Nossov SB, Kim HS, Jennette JC, Feldman EL, Smithies O. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc Natl Acad Sci U S A. 2010;107:10190–10195. doi: 10.1073/pnas.1005144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kaman WE, Wolterink AF, Bader M, Boele LC, Van Der KD. The bradykinin B2 receptor in the early immune response against Listeria infection. Med Microbiol Immunol. 2009;198:39–46. doi: 10.1007/s00430-008-0103-4. [DOI] [PubMed] [Google Scholar]
- 119.Kaplan AP, Silverberg M. The coagulation-kinin pathway of human plasma. Blood. 1987;70:1–15. [PubMed] [Google Scholar]
- 120.Kaplan AP, Silverberg M, Ghebrehiwet B, Atkins P, Zweiman B. The kallikrein-kinin system in inflammation. Adv Exp Med Biol. 1989;247:125–136. doi: 10.1007/978-1-4615-9543-4_18. [DOI] [PubMed] [Google Scholar]
- 121.Kauker ML. Bradykinin action on the efflux of luminal 22Na in the rat nephron. J Pharmacol Exp Ther. 1980;214:119–123. [PubMed] [Google Scholar]
- 122.Kauker ML. Kallidin effect on renal tubular function in meclofenamateand vehicle-pretreated rats. Proc Soc Exp Biol Med. 1990;193:60–64. doi: 10.3181/00379727-193-42991. [DOI] [PubMed] [Google Scholar]
- 123.Kauker ML, Gisi PJ, Zawada ET. Renal kinins and sodium transport: Influence of a bradykinin receptor antagonist (BKRA) FASEB J. 1990;4:A990. [Google Scholar]
- 124.Keiser HR, Geller RG, Margolius HS, Pisano JJ. Urinary kallikrein in hypertensive animal models. Fed Proc. 1976;35:199–202. [PubMed] [Google Scholar]
- 125.Kellett DN. On the anti-inflammatory activity of protamine sulphate and of hexadimethrine bromide, inhibitors of plasma kinin formation. Br J Pharmacol Chemother. 1965;24:705–713. doi: 10.1111/j.1476-5381.1965.tb01626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kumamoto K, Stewart TA, Johnson AR, Erdos EG. Prolylcarboxypeptidase (angiotensinase C) in human lung and cultured cells. J Clin Invest. 1981;67:210–215. doi: 10.1172/JCI110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, Matsuura H, Chayama K, Teranishi Y, Iba O, Amano K, Matsubara H. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension. 2003;41:99–107. doi: 10.1161/01.hyp.0000050101.90932.14. [DOI] [PubMed] [Google Scholar]
- 128.Lahera V, Salom MG, Fiksen-Olsen MJ, Romero JC. Mediatory role of endothelium-derived nitric oxide in renal vasodilatory and excretory effects of bradykinin. Am J Hypertens. 1991;4:260–262. doi: 10.1093/ajh/4.3.260. [DOI] [PubMed] [Google Scholar]
- 129.Lako-Futo Z, Szokodi I, Sarman B, Foldes G, Tokola H, Ilves M, Leskinen H, Vuolteenaho O, Skoumal R, deChatel R, Ruskoaho H, Toth M. Evidence for a functional role of angiotensin II type 2 receptor in the cardiac hypertrophic process in vivo in the rat heart. Circulation. 2003;108:2414–2422. doi: 10.1161/01.CIR.0000093193.63314.D9. [DOI] [PubMed] [Google Scholar]
- 130.Langberg H, Bjørn C, Boushel R, Hellsten Y, Kjær M. Exercise-induced increase in interstitial bradykinin and adenosine concentrations in skeletal muscle and peritendinous tissue in humans. J Physiol. 2002;542:977–983. doi: 10.1113/jphysiol.2002.018077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lawton WJ, Proud D, Frech ME, Pierce JV, Keiser HR, Pisano JJ. Characterization and origin of immunoreactive glandular kallikrein in rat plasma. Biochem Pharmacol. 1981;30:1731–1737. doi: 10.1016/0006-2952(81)90002-2. [DOI] [PubMed] [Google Scholar]
- 132.Lewis GP. Plasma kinins and inflammation. Metabolism. 1964;13:1256–1263. doi: 10.1016/s0026-0495(64)80042-1. [DOI] [PubMed] [Google Scholar]
- 133.Liao T-D, Yang X-P, Liu YH, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, Carretero OA. Role of inflammation in the development of renal damage and dysfunction in angiotensin-II-induced hypertension. Hypertension. 2008;52:256–263. doi: 10.1161/HYPERTENSIONAHA.108.112706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lilja H. A kallikrein-like serine protease in prostatic fluid cleaves the predominant seminal vesicle protein. J Clin Invest. 1985;76:1899–1903. doi: 10.1172/JCI112185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Linz W, Martorana PA, Schölkens BA. Local inhibition of bradykinin degradation in ischemic hearts. J Cardiovasc Pharmacol. 1990;15(Suppl. 6):S99–S109. [PubMed] [Google Scholar]
- 136.Linz W, Schölkens BA. A specific B2-bradykinin receptor antagonist HOE 140 abolishes the antihypertrophic effect of ramipril. Br J Pharmacol. 1992;105:771–772. doi: 10.1111/j.1476-5381.1992.tb09054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Linz W, Wiemer G, Schölkens BA. ACE-inhibition induces NO-formation in cultured bovine endothelial cells and protects isolated ischemic rat hearts. J Mol Cell Cardiol. 1992;24:909–919. doi: 10.1016/0022-2828(92)91103-c. [DOI] [PubMed] [Google Scholar]
- 138.Linz W, Wiemer G, Schölkens BA. Role of kinins in the pathophysiology of myocardial ischemia. In vitro and in vivo studies. Diabetes. 1996;45(Suppl. 1):S51–S58. doi: 10.2337/diab.45.1.s51. [DOI] [PubMed] [Google Scholar]
- 139.Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, Gamper N. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl-channels. J Clin Invest. 2010;120:1240–1252. doi: 10.1172/JCI41084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liu X, Lukasova M, Zubakova R, Lewicka S, Hilgenfeldt U. Kallidinlike peptide mediates the cardioprotective effect of the ACE inhibitor Captopril against ischaemic reperfusion injury of rat heart. Br J Pharmacol. 2006;148:825–832. doi: 10.1038/sj.bjp.0706799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu YH, Xu J, Yang X-P, Yang F, Shesely EG, Carretero OA. Effect of ACE inhibitors and angiotensin II type 1 receptor antagonists on endothelial NO synthase knockout mice with heart failure. Hypertension. 2002;39:375–381. doi: 10.1161/hy02t2.102796. [DOI] [PubMed] [Google Scholar]
- 142.Liu YH, Yang X-P, Menta D, Bulagannawar M, Scicli GM, Carretero OA. Role of kinins in chronic heart failure and in the therapeutic effect of ACE inhibitors in kininogen-deficient rats. Am J Physiol Heart Circ Physiol. 2000;278:H507–H514. doi: 10.1152/ajpheart.2000.278.2.H507. [DOI] [PubMed] [Google Scholar]
- 143.Liu YH, Yang X-P, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99:1926–1935. doi: 10.1172/JCI119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu YH, Yang X-P, Sharov VG, Sigmon DH, Sabbah HN, Carretero OA. Paracrine systems in the cardioprotective effect of angiotensin-converting enzyme inhibitors on myocardial ischemia/reperfusion injury in rats. Hypertension. 1996;27:7–13. doi: 10.1161/01.hyp.27.1.7. [DOI] [PubMed] [Google Scholar]
- 145.Liu YH, Yang X-P, Shesely EG, Sankey SS, Carretero OA. Role of angiotensin II type 2 receptors and kinins in the cardioprotective effect of angiotensin II type 1 receptor antagonists in rats with heart failure. J Am Coll Cardiol. 2004;43:1473–1480. doi: 10.1016/j.jacc.2003.11.044. [DOI] [PubMed] [Google Scholar]
- 146.Loke KE, Curran CML, Messina EJ, Laycock SK, Shesely EG, Carretero OA, Hintze TH. Role of nitric oxide in the control of cardiac oxygen consumption in B2-kinin receptor knockout mice. Hypertension. 1999;34:563–567. doi: 10.1161/01.hyp.34.4.563. [DOI] [PubMed] [Google Scholar]
- 147.Madeddu P, Parpaglia PP, Demontis MP, Varani MV, Fattaccio MC, Glorioso N. Chronic inhibition of bradykinin B2-receptors enhances the slow vasopressor response to angiotensin II. Hypertension. 1994;23:646–652. doi: 10.1161/01.hyp.23.5.646. [DOI] [PubMed] [Google Scholar]
- 148.Madeddu P, Parpaglia PP, Demontis MP, Varani MV, Fattaccio MC, Tonolo G, Troffa C, Glorioso N. Bradykinin B2-receptor blockade facilitates deoxycorticosterone-salt hypertension. Hypertension. 1993;21:980–984. doi: 10.1161/01.hyp.21.6.980. [DOI] [PubMed] [Google Scholar]
- 149.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. lnterleukin 17 Promotes Angiotensin II-Induced Hypertension and Vascular Dysfunction. Hypertension. 2009;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Majima M, Katori M, Hanazuka M, Mizogami S, Nakano T, Nakao Y, Mikami R, Uryu H, Okamura R, Mohsin SSJ, Oh-Ishi S. Suppression of rat deoxycorticosterone-salt hypertension by kallikrein-kinin system. Hypertension. 1991;17:806–813. doi: 10.1161/01.hyp.17.6.806. [DOI] [PubMed] [Google Scholar]
- 151.Majima M, Mizogami S, Kuribayashi Y, Katori M, Oh-Ishi S. Hypertension induced by a nonpressor dose of angiotensin II in kininogen-deficient rats. Hypertension. 1994;24:111–119. doi: 10.1161/01.hyp.24.1.111. [DOI] [PubMed] [Google Scholar]
- 152.Majima M, Yoshida O, Mihara H, Muto T, Mizogami S, Kuribayashi Y, Katori M, Oh-Ishi S. High sensitivity to salt in kininogen-deficient Brown Norway Katholiek rats. Hypertension. 1993;22:705–714. doi: 10.1161/01.hyp.22.5.705. [DOI] [PubMed] [Google Scholar]
- 153.Manolis AJ, Marketou ME, Gavras I, Gavras H. Cardioprotective properties of bradykinin: role of the B(2) receptor. Hypertens Res. 2010;33:772–777. doi: 10.1038/hr.2010.82. [DOI] [PubMed] [Google Scholar]
- 154.Marceau F. Kinin B1 receptor induction and inflammation. In: Farmer SG, editor. The Kinin System. San Diego, CA: Academic Press Inc; 1997. pp. 143–156. [Google Scholar]
- 155.Marceau F, Hess JF, Bachvarov DR. The B1 receptors for kinins. Pharmacol Rev. 1998;50:357–386. [PubMed] [Google Scholar]
- 156.Marceau F, Regoli D. Therapeutic options in inflammatory bowel disease: Experimental evidence of a beneficial effect of kinin B1 receptor blockade. Br J Pharmacol. 2008;154:1163–1165. doi: 10.1038/bjp.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension. 2009;54:1384–1392. doi: 10.1161/HYPERTENSIONAHA.109.138305. [DOI] [PubMed] [Google Scholar]
- 158.Marcie BM, Erdös EG. Protein kinase C and phosphatase inhibitors block the ability of angiotensin I-converting enzyme inhibitors to resensitize the receptor to bradykinin without altering the primary effects of bradykinin. J Pharmacol Exp Ther. 2000;294:605–612. [PubMed] [Google Scholar]
- 159.Margolius HS, Horwitz D, Pisano JJ, Keiser HR. Urinary kallikrein excretion in hypertensive man. Relationships to sodium intake and sodium-retaining steroids. Circ Res. 1974;35:820–825. doi: 10.1161/01.res.35.6.820. [DOI] [PubMed] [Google Scholar]
- 160.Marin Grez M. The influence of antibodies against bradykinin on isotonic saline diuresis in the rat. Evidence for kinin involvement in renal function. Pflugers Arch. 1974;350:231–239. doi: 10.1007/BF00587802. [DOI] [PubMed] [Google Scholar]
- 161.Marin-Castaño ME, Schanstra JP, Neau E, Praddaude F, Pecher C, Ader J-L, Girolami J-P, Bascands J-L. Induction of functional bradykinin B1 -receptors in normotensive rats and mice under chronic angiotensin-converting enzyme inhibitor treatment. Circulation. 2002;105:627–632. doi: 10.1161/hc0502.102965. [DOI] [PubMed] [Google Scholar]
- 162.Marks ES, Bing RF, Thurston H, Swales JD. Vasodepressor property of the converting enzyme inhibitor Captopril (SQ 14 225): the role of factors other than renin-angiotensin blockade in the rat. Clin Sci. 1980;58:1–6. doi: 10.1042/cs0580001. [DOI] [PubMed] [Google Scholar]
- 163.Martorana PA, Kettenbach B, Breipohl G, Linz W, Schölkens BA. Reduction of infarct size by local angiotensin-converting enzyme inhibition is abolished by a bradykinin antagonist. Eur J Pharmacol. 1990;182:395–396. doi: 10.1016/0014-2999(90)90301-l. [DOI] [PubMed] [Google Scholar]
- 164.McCaa RE. Studies in vivo with angiotensin I converting enzyme (kininase II) inhibitors. Fed Proc. 1979;38:2783–2787. [PubMed] [Google Scholar]
- 165.McEachern AE, Shelton ER, Bhakta S, Obernolte R, Bach C, Zuppan P, Fujisaki J, Aldrich RW, Jarnagin K. Expression cloning of a rat B2 bradykinin receptor. Proc Natl Acad Sci USA. 1991;88:7724–7728. doi: 10.1073/pnas.88.17.7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Meini S, Maggi CA. Knee osteoarthritis: A role for bradykinin? Inflamm Res. 2008;57:351–361. doi: 10.1007/s00011-007-7204-1. [DOI] [PubMed] [Google Scholar]
- 167.Menasché P, Kevelaitis E, Mouas C, Grousset C, Piwnica A, Bloch G. Preconditioning with potassium channel openers. A new concept for enhancing cardioplegie protection? J Thorac Cardiovasc Surg. 1995;110:1606–1613. doi: 10.1016/S0022-5223(95)70020-X. [DOI] [PubMed] [Google Scholar]
- 168.Meneton P, Bloch-Faure M, Hagege AA, Ruetten H, Huang W, Bergaya S, Ceiler D, Gehring D, Martins I, Salmon G, Boulanger CM, Nussberger J, Crozatier B, Gasc J-M, Heudes D, Bruneval P, Doetschman T, Ménard J, Alhenc-Gelas F. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc Natl Acad Sci USA. 2001;98:2634–2639. doi: 10.1073/pnas.051619598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Messadi-Laribi E, Griol-Charhbili V, Gaies E, Vincent MP, Heudes D, Meneton P, henc-Gelas F, Richer C. Cardioprotection and kallikreinkinin system in acute myocardial ischaemia in mice. Clin Exp Pharmacol Physiol. 2008;35:489–493. doi: 10.1111/j.1440-1681.2008.04902.x. [DOI] [PubMed] [Google Scholar]
- 170.Milia AF, Gross V, Plehm R, De Silva JA, Jr, Bader M, Luft FC. Normal blood pressure and renal function in mice lacking the bradykinin B2 receptor. Hypertension. 2001;37:1473–1479. doi: 10.1161/01.hyp.37.6.1473. [DOI] [PubMed] [Google Scholar]
- 171.Milligan G, Kostenis E. Heterotrimeric G-proteins: a short history. Br J Pharmacol. 2006;147(Suppl 1):S46–S55. doi: 10.1038/sj.bjp.0706405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mindroiu T, Scicli G, Perini F, Carretero OA, Scicli AG. Identification of a new kinin in human urine. J Biol Chem. 1986;261:7407–7411. [PubMed] [Google Scholar]
- 173.Mombouli J-V, Illiano S, Nagao T, Scott-Burden T, Vanhoutte PM. Potentiation of endothelium-dependent relaxations to bradykinin by angiotensin I converting enzyme inhibitors in canine coronary artery involves both endothelium-derived relaxing and hyperpolarizing factors. Circ Res. 1992;71:137–144. doi: 10.1161/01.res.71.1.137. [DOI] [PubMed] [Google Scholar]
- 174.Monteiro AC, Schmitz V, Morrot A, de Arruda LB, Nagajyothi F, Granato A, Pesquero JB, Muller-Esterl W, Tanowitz HB, Scharfstein J. Bradykinin B2 Receptors of dendritic cells, acting as sensors of kinins proteolytically released by Trypanosoma cruzi, are critical for the development of protective type-I responses. PLoS Pathog. 2007;3:1730–1744. doi: 10.1371/journal.ppat.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Monteiro AC, Scovino A, Raposo S, Gaze VM, Cruz C, Svensjo E, Narciso MS, Colombo AP, Pesquero JB, Feres-Filho E, Nguyen KA, Sroka A, Potempa J, Scharfstein J. Kinin danger signals proteolytically released by gingipain induce Fimbriae-specific IFN-gamma- and IL-17-producing T cells in mice infected intramucosally with Porphyromonas gingivalis. J Immunol. 2009;183:3700–3711. doi: 10.4049/jimmunol.0900895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, Adam A. The kallikrein-kinin system: current and future pharmacological targets. J Pharmacol Sci. 2005;99:6–38. doi: 10.1254/jphs.srj05001x. [DOI] [PubMed] [Google Scholar]
- 177.Moreira CR, Schmaier AH, Mahdi F, da Motta G, Nader HB, Shariat-Madar Z. Identification of procarboxypeptidase as the cell matrix-associated prekallikrein activator. FEBS Lett. 2002;523:167–170. doi: 10.1016/s0014-5793(02)02980-0. [DOI] [PubMed] [Google Scholar]
- 178.Muller DN, Dechend R, Mervaala EMA, Park J-K, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC. NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension? 2000;35:193–201. doi: 10.1161/01.hyp.35.1.193. [DOI] [PubMed] [Google Scholar]
- 179.Murphey LJ, Gainer JV, Vaughan DE, Brown NJ. Angiotensinconverting enzyme insertion/deletion polymorphism modulates the human in vivo metabolism of bradykinin. Circulation. 2000;102:829–832. doi: 10.1161/01.cir.102.8.829. [DOI] [PubMed] [Google Scholar]
- 180.Nakagawa M, Nasjletti A. Plasma kinin concentration in deoxycorticosterone-salt hypertension. Hypertension. 1988;11:411–415. doi: 10.1161/01.hyp.11.5.411. [DOI] [PubMed] [Google Scholar]
- 181.Nakagawa M, Nasjletti A. Renal function as affected by inhibitors of kininase II and of neutral endopeptidase 24.11 in rats with and without desoxycorticosterone pretreatment. Adv Exp Med Biol. 1989;247:495–499. doi: 10.1007/978-1-4615-9546-5_82. [DOI] [PubMed] [Google Scholar]
- 182.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFβ -inducible antagonist of TGF-β signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 183.Nasjletti A, Colina-Chourio J, McGiff JC. Disappearance of bradykinin in the renal circulation of dogs. Effects of kininase inhibition. Circ Res. 1975;37:59–65. doi: 10.1161/01.res.37.1.59. [DOI] [PubMed] [Google Scholar]
- 184.Nasjletti A, McGiff JC, Colina-Chourio J. Interrelations of the renal kallikrein-kinin system and renal prostaglandins in the conscious rat. Influence of mineralocorticoids. Circ Res. 1978;43:799–807. doi: 10.1161/01.res.43.5.799. [DOI] [PubMed] [Google Scholar]
- 185.Ngo ML, Mahdi F, Kolte D, Shariat-Madar Z. Upregulation of prolylcarboxypeptidase (PRCP) in lipopolysaccharide (LPS) treated endothelium promotes inflammation. J Inflamm (Land) 2009;6:3. doi: 10.1186/1476-9255-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Noda K, Sasaguri M, Ideishi M, Ikeda M, Arakawa K. Role of locally formed angiotensin II and bradykinin in the reduction of myocardial infarct size in dogs. Cardiovasc Res. 1993;27:334–340. doi: 10.1093/cvr/27.2.334. [DOI] [PubMed] [Google Scholar]
- 187.Noda Y, Yamada K, Igic R, Erdös EG. Regulation of rat urinary and renal kallikrein and prekallikrein by corticosteroids. Proc Natl Acad Sci USA. 1983;80:3059–3063. doi: 10.1073/pnas.80.10.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nolly H, Carbini LA, Scicli G, Carretero OA, Scicli AG. A local kallikrein-kinin system is present in rat hearts. Hypertension. 1994;23:919–923. doi: 10.1161/01.hyp.23.6.919. [DOI] [PubMed] [Google Scholar]
- 189.Nolly H, Saed G, Carretero OA, Scicli G, Scicli AG. Adrenal kallikrein. Hypertension. 1993;21:911–915. doi: 10.1161/01.hyp.21.6.911. [DOI] [PubMed] [Google Scholar]
- 190.Nolly H, Scicli AG, Scicli G, Carretero OA. Characterization of a kininogenase from rat vascular tissue resembling tissue kallikrein. Circ Res. 1985;56:816–821. doi: 10.1161/01.res.56.6.816. [DOI] [PubMed] [Google Scholar]
- 191.Odya CE, Marinkovic DV, Hammon KJ, Stewart TA, Erdös EG. Purification and properties of prolylcarboxypeptidase (angiotensinase C) from human kidney. J Biol Chem. 1978;253:5927–5931. [PubMed] [Google Scholar]
- 192.Ohkubo I, Kurachi K, Takasawa T, Shiokawa H, Sasaki M. Isolation of a human cDNA for α2-thiol proteinase inhibitor and its identity with low molecular weight kininogen. Biochemistry. 1984;23:5691–5697. doi: 10.1021/bi00319a005. [DOI] [PubMed] [Google Scholar]
- 193.Okamoto H, Greenbaum LM. Kininogen substrates for trypsin and cathepsin D in human, rabbit and rat plasmas. Life Sci. 1983;32:2007–2013. doi: 10.1016/0024-3205(83)90052-8. [DOI] [PubMed] [Google Scholar]
- 194.Okamoto H, Greenbaum LM. Pharmacological properties of T-kinin (isoleucyl-seryl-bradykinin) from rat plasma. Biochem Pharmacol. 1983;32:2637–2638. doi: 10.1016/0006-2952(83)90039-4. [DOI] [PubMed] [Google Scholar]
- 195.Omata K, Carretero OA, Itoh S, Scicli AG. Active and inactive kallikrein in rabbit connecting tubules and urine during low and normal sodium intake. Kidney Int. 1983;24:714–718. doi: 10.1038/ki.1983.218. [DOI] [PubMed] [Google Scholar]
- 196.Ornata K, Carretero OA, Scicli AG, Jackson BA. Localization of active and inactive kallikrein (kininogenase activity) in the microdissected rabbit nephron. Kidney Int. 1982;22:602–607. doi: 10.1038/ki.1982.218. [DOI] [PubMed] [Google Scholar]
- 197.Omoro SA, Majid DSA, El-Dahr SS, Navar LG. Kinin influences on renal regional blood flow responses to angiotensin-converting enzyme inhibition in dogs. Am J Physiol. 1999;276:F271–F277. doi: 10.1152/ajprenal.1999.276.2.F271. [DOI] [PubMed] [Google Scholar]
- 198.Osterrieder W, Müller RKM, Powell JS, Clozel J-P, Hefti F, Baumgartner HR. Role of angiotensin II in injury-induced neointima formation in rats. Hypertension. 1991;18(Suppl II):13-60–11-64. doi: 10.1161/01.hyp.18.4_suppl.ii60. [DOI] [PubMed] [Google Scholar]
- 199.Overlack A, Stumpe KO, Heck I, Ressel C, Kühnert M, Krück F. Identification of angiotensin II- and kinin-dependent mechanisms in essential hypertension. In: Philipp T, Distler A, editors. Hypertension: Mechanisms and Management. Berlin: Springer-Verlag; 1980. pp. 183–191. [Google Scholar]
- 200.Oza NB, Schwartz JH, Goud HD, Levinsky NG. Rat aortic smooth muscle cells in culture express kallikrein, kininogen, and bradykininase activity. J Clin Invest. 1990;85:597–600. doi: 10.1172/JCI114479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009;31:5–22. doi: 10.1007/s00281-009-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pan H-L, Chen S-R, Scicli GM, Carretero OA. Cardiac interstitial bradykinin release during ischemia is enhanced by ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2000;279:H116–H121. doi: 10.1152/ajpheart.2000.279.1.H116. [DOI] [PubMed] [Google Scholar]
- 203.Parratt JR, Vegh A, Papp JG. Bradykinin as an endogenous myocardial protective substance with particular reference to ischemic preconditioning—a brief review of the evidence. Can J Physiol Pharmacol. 1995;73:837–842. doi: 10.1139/y95-114. [DOI] [PubMed] [Google Scholar]
- 204.Pasquié JL, Herizi A, Jover B, Mimran A. Chronic bradykinin infusion and receptor blockade in angiotensin II hypertension in rats. Hypertension. 1999;33:830–834. doi: 10.1161/01.hyp.33.3.830. [DOI] [PubMed] [Google Scholar]
- 205.Pees C, Unger T, Gohlke P. Effect of angiotensin AT2 receptor stimulation on vascular cyclic GMP production in normotensive Wistar Kyoto rats. Int J Biochem Cell Biol. 2003;35:963–972. doi: 10.1016/s1357-2725(02)00265-0. [DOI] [PubMed] [Google Scholar]
- 206.Pfeffer MA, Braunwald E, Moyé LA, Basta L, Brown EJ, Jr, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC, Klein M, Lamas GA, Packer M, Rouleau J, Rouleau JL, Rutherford J, Wertheimer JH, Hawkins CM, on behalf of the SAVE Investigators Effect of Captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. N Engl J Med. 1992;327:669–677. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- 207.Picard N, Eladari D, EI MS, Planes C, Bourgeois S, Houillier P, Wang Q, Burnier M, Deschenes G, Knepper MA, Meneton P, Chambrey R. Defective ENaC processing and function in tissue kallikrein-deficient mice. J Biol Chem. 2008;283:4602–4611. doi: 10.1074/jbc.M705664200. [DOI] [PubMed] [Google Scholar]
- 208.Picard N, Van AM, Campone C, Seiler M, Bloch-Faure M, Hoenderop JG, Loffing J, Meneton P, Bindeis RJ, Paillard M, henc-Gelas F, Houillier P. Tissue kallikrein-deficient mice display a defect in renal tubular calcium absorption. J Am Soc Nephrol. 2005;16:3602–3610. doi: 10.1681/ASN.2004110923. [DOI] [PubMed] [Google Scholar]
- 209.Pietrovski EF, Otuki MF, Regoli D, Bader M, Pesquero JB, Cabrini DA, Zampronio AR. The non-peptide kinin receptor antagonists FR 173657 and SSR 240612: Preclinical evidence for the treatment of skin inflammation. Regul Pept. 2009;152:67–72. doi: 10.1016/j.regpep.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 210.Pisano JJ, Corthorn J, Yates K, Pierce JV. The kallikrein-kinin system in the kidney. Contrib Nephrol. 1978;12:116–125. doi: 10.1159/000401659. [DOI] [PubMed] [Google Scholar]
- 211.Pixley RA, Colman RW. The kallikrein-kinin system in sepsis syndrome. In: Farmer SG, editor. The Kinin System. San Diego, CA: Academic Press Inc; 1997. pp. 173–186. [Google Scholar]
- 212.Pollock DM, Butterfield MI, Ader JL, Arendshorst WJ. Dissociation of urinary kallikrein activity and salt and water excretion in the rat. Am J Physiol. 1986;250:F1082–F1089. doi: 10.1152/ajprenal.1986.250.6.F1082. [DOI] [PubMed] [Google Scholar]
- 213.Pons S, Griol-Charhbili V, Heymes C, Fornes P, Heudes D, Hagege A, Loyer X, Meneton P, Giudicelli JF, Samuel JL, henc-Gelas F, Richer C. Tissue kallikrein deficiency aggravates cardiac remodelling and decreases survival after myocardial infarction in mice. Eur J Heart Fail. 2008;10:343–351. doi: 10.1016/j.ejheart.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 214.Pontieri V, Lopes OU, Ferreira SH. Hypotensive effect of Captopril. Role of bradykinin and prostaglandinlike substances. Hypertension. 1990;15(Suppl I):1-55–1-58. doi: 10.1161/01.hyp.15.2_suppl.i55. [DOI] [PubMed] [Google Scholar]
- 215.Powell JS, Müller RKM, Rouge M, Kuhn H, Hefti F, Baumgartner HR. The proliferative response to vascular injury is suppressed by angiotensin-converting enzyme inhibition. J Cardiovasc Pharmacol. 1990;16(Suppl 4):S42–S49. doi: 10.1097/00005344-199016004-00010. [DOI] [PubMed] [Google Scholar]
- 216.Powers CA, Nasjletti A. A major sex difference in kallikrein-like activity in the rat anterior pituitary. Endocrinology. 1984;114:1841–1844. doi: 10.1210/endo-114-5-1841. [DOI] [PubMed] [Google Scholar]
- 217.Prat A, Weinrib L, Becher B, Poirier J, Duquette P, Couture R, Antel JP. Bradykinin B1 receptor expression and function on T lymphocytes in active multiple sclerosis. Neurology. 1999;53:2087–2092. doi: 10.1212/wnl.53.9.2087. [DOI] [PubMed] [Google Scholar]
- 218.Pravenec M, Kren V, Kunes J, Scicli AG, Carretero OA, Simonet L, Kurtz TW. Cosegregation of blood pressure with a kallikrein gene family polymorphism. Hypertension. 1991;17:242–246. doi: 10.1161/01.hyp.17.2.242. [DOI] [PubMed] [Google Scholar]
- 219.Rabito SF, Scicli AG, Carretero OA. Immunoreactive glandular kallikrein in plasma. In: Gross F, Vogel G, editors. Enzymatic Release of Vasoactive Peptides. New York: Raven Press; 1980. pp. 247–256. [Google Scholar]
- 220.Rabito SF, Scicli AG, Kher V, Carretero OA. Immunoreactive glandular kallikrein in rat plasma: A radioimmunoassay for its determination. Am J Physiol. 1982;242:H602–H610. doi: 10.1152/ajpheart.1982.242.4.H602. [DOI] [PubMed] [Google Scholar]
- 221.Regoli D. Pharmacology of bradykinin and related kinins. Adv Exp Med Biol. 1983;156:569–584. [PubMed] [Google Scholar]
- 222.Regoli D, Barabe J. Pharmacology of bradykinin and related kinins. Pharmacol Rev. 1980;32:1–46. [PubMed] [Google Scholar]
- 223.Regoli D, Rhaleb N-E, Dion S, Drapeau G. New selective bradykinin receptor antagonists and bradykinin B2 receptor characterization. Trends Pharmacol Sci. 1990;11:156–161. doi: 10.1016/0165-6147(90)90067-I. [DOI] [PubMed] [Google Scholar]
- 224.Regoli D, Rhaleb N-E, Drapeau G, Dion S. Kinin receptor subtypes. J Cardiovasc Pharmacol. 1990;15(Suppl 6):S30–S38. [PubMed] [Google Scholar]
- 225.Regoli D, Rhaleb N-E, Drapeau G, Dion S, Tousignant C, D’Orl:eans-Juste P, Devillier P. Basic pharmacology of kinins: pharmacologic receptors and other mechanisms. Adv Exp Med Biol. 1989;247:399–407. doi: 10.1007/978-1-4615-9543-4_61. [DOI] [PubMed] [Google Scholar]
- 226.Ren Y, Garvin J, Carretero OA. Mechanism involved in bradykinin-induced efferent arteriole dilation. Kidney Int. 2002;62:544–549. doi: 10.1046/j.1523-1755.2002.00482.x. [DOI] [PubMed] [Google Scholar]
- 227.Rett K, Wicklmayr M, Dietze GJ, Häring HU. Insulin-induced glucose transporter (GLUT1 and GLUT4) translocation in cardiac muscle tissue is mimicked by bradykinin. Diabetes. 1996;45(Suppl. 1):S66–S69. doi: 10.2337/diab.45.1.s66. [DOI] [PubMed] [Google Scholar]
- 228.Rhaleb N-E, Peng H, Alfie M, Shesely EG, Carretero OA. Effect of ACE inhibitor on DOCA-salt- and aortic coarctation-induced hypertension in mice. Do kinin B2 receptors play a role? Hypertension. 1999;33:329–334. doi: 10.1161/01.hyp.33.1.329. [DOI] [PubMed] [Google Scholar]
- 229.Rhaleb N-E, Yang X-P, Nanba M, Shesely EG, Carretero OA. Effect of chronic blockade of the kallikrein-kinin system on the development of hvpertension in rats. Hypertension. 2001;37:121–128. doi: 10.1161/01.hyp.37.1.121. [DOI] [PubMed] [Google Scholar]
- 230.Rhaleb N-E, Yang X-P, Peng H, Cavasin MA, Liu YH, Yang F, Xu J, Carretero OA. Cardiovascular phenotype of male 129/SvEvTac, 129/SvJ and B2-KO mice [abstract] FASEB J. 2001;15:A101. [Google Scholar]
- 231.Rhaleb N-E, Yang X-P, Scicli AG, Carretero OA. Role of kinins and nitric oxide in the antihypertrophic effect of ramipril. Hypertension. 1994;23:865–868. doi: 10.1161/01.hyp.23.6.865. [DOI] [PubMed] [Google Scholar]
- 232.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, Macfadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Eng J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 233.Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, Johnson RJ, Pons HA. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol. 2002;282:F191–F201. doi: 10.1152/ajprenal.0197.2001. [DOI] [PubMed] [Google Scholar]
- 234.Roman RJ, Kaldunski ML, Scicli AG, Carretero OA. Influence of kinins and angiotensin II on the regulation of papillary blood flow. Am J Physiol. 1988;255:F690–F698. doi: 10.1152/ajprenal.1988.255.4.F690. [DOI] [PubMed] [Google Scholar]
- 235.Rubin LE, Levi R. Protective role of bradykinin in cardiac anaphylaxis. Coronary-vasodilating and antiarrhythmic activities mediated by autocrine/paracrine mechanisms. Circ Res. 1995;76:434–440. doi: 10.1161/01.res.76.3.434. [DOI] [PubMed] [Google Scholar]
- 236.Sabatini RA, Guimaraes PB, Fernandes L, Reis FC, Bersanetti PA, Mori MA, Navarro A, Hilzendeger AM, Santos EL, Andrade MC, Chagas JR, Pesquero JL, Casarini DE, Bader M, Carmona AK, Pesquero JB. ACE activity is modulated by kinin B2 receptor. Hypertension. 2008;51:689–695. doi: 10.1161/HYPERTENSIONAHA.107.091181. [DOI] [PubMed] [Google Scholar]
- 237.Saed GM, Carretero OA, MacDonald RJ, Scicli AG. Kallikrein messenger RNA in rat arteries and veins. Circ Res. 1990;67:510–516. doi: 10.1161/01.res.67.2.510. [DOI] [PubMed] [Google Scholar]
- 238.Saha JK, Sengupta JN, Goyal RK. Effect of bradykinin on opossum esophageal longitudinal smooth muscle: Evidence for novel bradykinin receptors. J Pharmacol Exp Ther. 1990;252:1012–1020. [PubMed] [Google Scholar]
- 239.Saitoh S, Scicli AG, Peterson E, Carretero OA. Effect of inhibiting renal kallikrein on prostaglandin E2, water, and sodium excretion. Hypertension. 1995;25:1008–1013. doi: 10.1161/01.hyp.25.5.1008. [DOI] [PubMed] [Google Scholar]
- 240.Salgado MCO, Rabito SF, Carretero OA. Blood kinin in one-kidney, one clip hypertensive rats. Hypertension. 1986;8(Suppl 1):I-110–I-113. [Google Scholar]
- 241.Sasaki N, Yamashita T, Takeda M, Shinohara M, Nakajima K, Tawa H, Usui T, Hirata K. Oral anti-CD3 antibody treatment induces regulatory T cells and inhibits the development of atherosclerosis in mice. Circulation. 2009;120:1996–2005. doi: 10.1161/CIRCULATIONAHA.109.863431. [DOI] [PubMed] [Google Scholar]
- 242.Schachter M, Longridge DJ, Wheeler GD, Mehta JG, Uchida Y. Immunocytochemical and enzyme histochemical localization of kallikrein-like enzymes in colon, intestine, and stomach of rat and cat. J Histochem Cytochem. 1986;34:927–934. doi: 10.1177/34.7.3519756. [DOI] [PubMed] [Google Scholar]
- 243.Scharfstein J, Monteiro AC, Schmitz V, Svensjo E. Angiotensinconverting enzyme limits inflammation elicited by Trypanosoma cruzi cysteine proteases: A peripheral mechanism regulating adaptive immunity via the innate kinin pathway. Biol Chem. 2008;389:1015–1024. doi: 10.1515/BC.2008.126. [DOI] [PubMed] [Google Scholar]
- 244.Scharfstein J, Schmitz V, Svensjo E, Granato A, Monteiro AC. Kininogens coordinate adaptive immunity through the proteolytic release of bradykinin, an endogenous danger signal driving dendritic cell maturation. Scand J Immunol. 2007;66:128–136. doi: 10.1111/j.1365-3083.2007.01983.x. [DOI] [PubMed] [Google Scholar]
- 245.Schmaier AH. Assembly, activation, and physiologic influence of the plasma kallikrein/kinin system. Int Immunopharmacot. 2008;8:161–165. doi: 10.1016/j.intimp.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Schmitz V, Svensjo E, Serra RR, Teixeira MM, Scharfstein J. Proteolytic generation of kinins in tissues infected by Trypanosoma cruzi depends on CXC chemokine secretion by macrophages activated via Toll-like 2 receptors. J Leukoc Biol. 2009;85:1005–1014. doi: 10.1189/jlb.1108693. [DOI] [PubMed] [Google Scholar]
- 247.Schoelkens BA, Linz W. Bradykinin-mediated metabolic effects in isolated perfused rat hearts. Agents Actions Suppl. 1992;38:36–42. [PubMed] [Google Scholar]
- 248.Schölkens BA, Linz W, Martorana PA. Experimental cardiovascular benefits of angiotensin-converting enzyme inhibitors: beyond blood pressure reduction. J Cardiovasc Pharmacol. 1991;18(Suppl. 2):S26–S30. [PubMed] [Google Scholar]
- 249.Schulze-Topphoff U, Prat A, Prozorovski T, Siffrin V, Paterka M, Herz J, Bendix I, Ifergan I, Schadock I, Mori MA, Van HJ, Schroter F, Smorodchenko A, Han MH, Bader M, Steinman L, Aktas O, Zipp F. Activation of kinin receptor B1 limits encephalitogenic T lymphocyte recruitment to the central nervous system. Nat Med. 2009;15:788–793. doi: 10.1038/nm.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Scicli AG, Carbini LA, Carretero OA. The molecular biology of the kallikrein-kinin system: II. The rat gene family. J Hypertens. 1993;11:775–780. doi: 10.1097/00004872-199308000-00002. [DOI] [PubMed] [Google Scholar]
- 251.Scicli AG, Carretero OA, Hampton A, Cortes P, Oza NB. Site of kininogenase secretion in the dog nephron. Am J Physiol. 1976;230:533–536. doi: 10.1152/ajplegacy.1976.230.2.533. [DOI] [PubMed] [Google Scholar]
- 252.Scicli AG, Gandolfi R, Carretero OA. Site of formation of kinins in the dog nephron. Am J Physiol. 1978;234:F36–F40. doi: 10.1152/ajprenal.1978.234.1.F36. [DOI] [PubMed] [Google Scholar]
- 253.Scicli AG, Mindroiu T, Scicli G, Carretero OA. Blood kinins, their concentration in normal subjects and in patients with congenital deficiency in plasma prekallikrein and kininogen. J Lab Clin Med. 1982;100:81–93. [PubMed] [Google Scholar]
- 254.Scicli AG, Ørstavik TB, Rabito SF, Murray RD, Carretero OA. Blood kinins after sympathetic nerve stimulation of the rat submandibular gland. Hypertension. 1983;5(Suppl I):I-101–I-106. doi: 10.1161/01.hyp.5.2_pt_2.i101. [DOI] [PubMed] [Google Scholar]
- 255.Scicli G, Nolly H, Carretero OA, Scicli AG. Glandular kallikrein-like enzyme in adrenal glands. Adv Exp Med Biol. 1989;247B:217–222. doi: 10.1007/978-1-4615-9546-5_36. [DOI] [PubMed] [Google Scholar]
- 256.Seino M, Abe K, Nushiro N, Omata K, Kasai Y, Yoshinaga K. Effects of a competitive antagonist of bradykinin on blood pressure and renal blood flow in anesthetized rats. J Hypertens. 1988;6:867–871. doi: 10.1097/00004872-198811000-00004. [DOI] [PubMed] [Google Scholar]
- 257.Seino M, Abe K, Otsuka Y, Saito T, Irokawa N, Yasujima M, Ciba S, Yoshinaga K. Urinary kallikrein excretion and sodium metabolism in hypertensive patients. Tohoku J Exp Med. 1975;116:359–367. doi: 10.1620/tjem.116.359. [DOI] [PubMed] [Google Scholar]
- 258.Seino M, Carretero OA, Albertini R, Scicli AG. Kinins in regulation of uteroplacental blood flow in the pregnant rabbit. Am J Physiol. 1982;242:H142–H147. doi: 10.1152/ajpheart.1982.242.2.H142. [DOI] [PubMed] [Google Scholar]
- 259.Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA. 2003;290:2945–2951. doi: 10.1001/jama.290.22.2945. [DOI] [PubMed] [Google Scholar]
- 260.Seyedi N, Xu X, Nasjletti A, Hintze TH. Coronary kinin generation mediates nitric oxide release after angiotensin receptor stimulation. Hypertension. 1995;26:164–170. doi: 10.1161/01.hyp.26.1.164. [DOI] [PubMed] [Google Scholar]
- 261.Shariat-Madar Z, Mahdi F, Schmaier AH. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem. 2002;277:17962–17969. doi: 10.1074/jbc.M106101200. [DOI] [PubMed] [Google Scholar]
- 262.Shariat-Madar Z, Mahdi F, Schmaier AH. Recombinant prolylcarboxypeptidase activates plasma prekallikrein. Blood. 2004;103:4554–4561. doi: 10.1182/blood-2003-07-2510. [DOI] [PubMed] [Google Scholar]
- 263.Sharma JN, Wirth KJ. Inhibition of rats adjuvant arthritis by a bradykinin antagonist Hoe 140 and its influence on kallikreins. Gen Pharmacol. 1996;27:133–136. doi: 10.1016/0306-3623(95)00078-x. [DOI] [PubMed] [Google Scholar]
- 264.Sinaiko AR, Glasser RJ, Gillum RF, Prineas RJ. Urinary kallikrein excretion in grade school children with high and low blood pressure. J Pediatr. 1982;100:938–940. doi: 10.1016/s0022-3476(82)80522-2. [DOI] [PubMed] [Google Scholar]
- 265.Siragy HM, Inagami T, Ichiki T, Carey RM. Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc Natl Acad Sci USA. 1999;96:6506–6510. doi: 10.1073/pnas.96.11.6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Siragy HM, Jaffa AA, Margolius HS. Stimulation of renal interstitial bradykinin by sodium depletion. Am J Hypertens. 1993;6:863–866. doi: 10.1093/ajh/6.10.863. [DOI] [PubMed] [Google Scholar]
- 267.Skidgel RA, Schulz WW, Tarn L-T, Erdös EG. Human renal angiotensin I converting enzyme and neutral endopeptidase. Kidney Int. 1987;31(Suppl 20):S-45–S-48. [PubMed] [Google Scholar]
- 268.Smith D, Gilbert M, Owen WG. Tissue plasminogen activator release in vivo in response to vasoactive agents. Blood. 1985;66:835–839. [PubMed] [Google Scholar]
- 269.Speechly-Dick ME, Mocanu MM, Yellon DM. Protein kinase C. Its role in ischemic preconditioning in the rat. Circ Res. 1994;75:586–590. doi: 10.1161/01.res.75.3.586. [DOI] [PubMed] [Google Scholar]
- 270.Stewart JM. Bradykinin antagonists as anti-cancer agents. Curr Pharm Des. 2003;9:2036–2042. doi: 10.2174/1381612033454171. [DOI] [PubMed] [Google Scholar]
- 271.Stewart JM, Gera L, York EJ, Chan DC, Whalley EJ, Bunn PA, Jr, Vavrek RJ. Metabolism-resistant bradykinin antagonists: development and applications. Biol Chem. 2001;382:37–41. doi: 10.1515/BC.2001.006. [DOI] [PubMed] [Google Scholar]
- 272.Stewart JM, Vavrek RJ. Bradykinin competitive antagonists for classical kinin systems. Adv Exp Med Biol. 1986;198:537–542. doi: 10.1007/978-1-4684-5143-6_71. [DOI] [PubMed] [Google Scholar]
- 273.Stoos BA, Carretero OA, Farhy RD, Scicli G, Garvin JL. Endofheliumderived relaxing factor inhibits transport and increases cGMP content in cultured mouse cortical collecting duct cells. J Clin Invest. 1992;89:761–765. doi: 10.1172/JCI115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Sueyoshi T, Enjyoji K, Shimada T, Kato H, Iwanaga S, Bando Y, Kominami E, Katunuma N. A new function of kininogens as thiolproteinase inhibitors: inhibition of papain and cathepsins B, H and L by bovine, rat and human plasma kininogens. FEBS Lett. 1985;182:193–195. doi: 10.1016/0014-5793(85)81182-0. [DOI] [PubMed] [Google Scholar]
- 275.Sugawara A, Kudo M, Saito A, Matsuda K, Uruno A, Ito S. Novel effects of beraprost sodium on vasculatures. Int Angiol. 2010;29:28–32. [PubMed] [Google Scholar]
- 276.Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosteroneinduced inflammation in the rat heart. Role of oxidative stress. Am J Pathol. 2002;161:1773–1781. doi: 10.1016/S0002-9440(10)64454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sundsmo JS, Fair DS. Relationships among the complement, kinin, coagulation and fibrinolytic systems in the inflammatory reaction. Clin Physiol Biochem. 1983;1:225–284. [PubMed] [Google Scholar]
- 278.Sustarsic DL, McPartland RP, Rapp JP, Schlager G, Tan SY. Urinary kallikrein and urinary prostaglandin E2 in genetically hypertensive mice. Proc Soc Exp Biol Med. 1980;163:193–199. doi: 10.3181/00379727-163-40746. [DOI] [PubMed] [Google Scholar]
- 279.Sybertz EJ, Chiu PJS, Vemulapalli S, Watkins R, Haslanger MF. Atrial natriuretic factor-potentiating and antihypertensive activity of SCH 34826. An orally active neutral metalloendopeptidase inhibitor. Hypertension. 1990;15:152–161. doi: 10.1161/01.hyp.15.2.152. [DOI] [PubMed] [Google Scholar]
- 280.Tiffany CW, Burch RM. Bradykinin stimulates tumor necrosis factor and interleukin-l release from macrophages. FEBS Lett. 1989;247:189–192. doi: 10.1016/0014-5793(89)81331-6. [DOI] [PubMed] [Google Scholar]
- 281.Tornita K, Pisano JJ. Binding of [3H]bradykinin in isolated nephron segments of the rabbit. Am J Physiol. 1984;246:F732–F737. doi: 10.1152/ajprenal.1984.246.5.F732. [DOI] [PubMed] [Google Scholar]
- 282.Tomiyama H, Scicli AG, Scicli GM, Carretero OA. Renal effects of Fab fragments of kinin antibodies on deoxycorticosterone acetate-salt-treated rats. Hypertension. 1990;15:761–766. doi: 10.1161/01.hyp.15.6.761. [DOI] [PubMed] [Google Scholar]
- 283.Tornei J, Madrid MI, García-Salom M, Wirth KJ, Fenoy FJ. Role of kinins in the control of renal papillary blood flow, pressure natriuresis, and arterial pressure. Circ Res. 2000;86:589–595. doi: 10.1161/01.res.86.5.589. [DOI] [PubMed] [Google Scholar]
- 284.Touyz RM. Molecular and cellular mechanisms in vascular injury in hypertension: role of angiotensin II. Curr Opin Nephrol Hyperlens. 2005;14:125–131. doi: 10.1097/00041552-200503000-00007. [DOI] [PubMed] [Google Scholar]
- 285.Trabold F, Pons S, Hagege AA, Bloch-Faure M, Alhenc-Gelas F, Giudicelli J-F, Richer-Giudicelli C, Meneton P. Cardiovascular phenotypes of kinin B2 receptor- and tissue kallikrein-deficient mice. Hypertension. 2002;40:90–95. doi: 10.1161/01.hyp.0000021747.43346.95. [DOI] [PubMed] [Google Scholar]
- 286.Trabold R, Eros C, Zweckberger K, Relton J, Beck H, Nussberger J, Muller-Esterl W, Bader M, Whalley E, Plesnila N. The role of bradykinin B(l) and B(2) receptors for secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2010;30:130–139. doi: 10.1038/jcbfm.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Tschöpe C, Gohlke P, Zhu Y-Z, Linz W, Schölkens B, Unger T. Antihypertensive and cardioprotective effects after angiotensin-converting enzyme inhibition: Role of kinins. J Card Fail. 1997;3:133–148. doi: 10.1016/s1071-9164(97)90047-6. [DOI] [PubMed] [Google Scholar]
- 288.Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mon Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest. 1999;104:925–935. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Uchiyama M, Otsuka T, Sakai K. Urinary kallikrein excretion in children of parents with essential hypertension. Arch Dis Child. 1985;60:974–975. doi: 10.1136/adc.60.10.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Ura N, Carretero OA, Erdös EG. Role of renal endopeptidase 24.11 in kinin metabolism in vitro and in vivo. Kidney Int. 1987;32:507–513. doi: 10.1038/ki.1987.239. [DOI] [PubMed] [Google Scholar]
- 291.Vane JR, Änggård EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med. 1990;323:27–36. doi: 10.1056/NEJM199007053230106. [DOI] [PubMed] [Google Scholar]
- 292.Vanhoutte PM. Endothelium and control of vascular function. State of the art lecture. Hypertension. 1989;13:658–667. doi: 10.1161/01.hyp.13.6.658. [DOI] [PubMed] [Google Scholar]
- 293.Vegh A, Szekeres L, Parratt JR. Protective effects of preconditioning of the ischaemic myocardium involve cyclo-oxygenase products. Cardiovasc Res. 1990;24:1020–1023. doi: 10.1093/cvr/24.12.1020. [DOI] [PubMed] [Google Scholar]
- 294.Vijayaraghavan J, Scicli AG, Carretero OA, Slaughter C, Moomaw C, Hersh LB. The hydrolysis of endothelins by neutral endopeptidase 24.11 (enkephalinase) J Biol Chem. 1990;265:14150–14155. [PubMed] [Google Scholar]
- 295.Vinci JM, Horwitz D, Zusman RM, Pisano JJ, Catt KJ, Keiser HR. The effect of converting enzyme inhibition with SQ20, 881 on plasma and urinary kinins, prostaglandin E and angiotensin II in hypertensive man. Hypertension. 1979;1:416–426. doi: 10.1161/01.hyp.1.4.416. [DOI] [PubMed] [Google Scholar]
- 296.Vio CP, Churchill L, Rabito SF, Terragno A, Carretero OA, Terragno NA. Renal kallikrein in venous effluent of filtering and non-filtering isolated kidneys. Adv Exp Med Biol. 1983;156B:897–905. [PubMed] [Google Scholar]
- 297.Virdis A, Schiffrin EL. Vascular inflammation: A role in vascular disease in hypertension? Curr Opin Nephrol Hyperlens. 2003;12:181–187. doi: 10.1097/00041552-200303000-00009. [DOI] [PubMed] [Google Scholar]
- 298.Wang H, Carretero OA, Garvin JL. Inhibition of apical Na+/H+ exchangers on the macula densa cells augments tubuloglomerular feedback. Hypertension. 2003;41:688–691. doi: 10.1161/01.HYP.0000048863.75711.B2. [DOI] [PubMed] [Google Scholar]
- 299.Wei M, Wang X, Kuukasjarvi P, Laurikka J, Rinne T, Honkonen EL, Tarkka M. Bradykinin preconditioning in coronary artery bypass grafting. Ann Thorac Surg. 2004;78:492–497. doi: 10.1016/j.athoracsur.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 300.Whalley ET, Hanson WL, Stewart JM, Gera L. Oral activity of peptide bradykinin antagonists following intragastric administration in the rat. Can J Physiol Pharmacol. 1997;75:629–632. [PubMed] [Google Scholar]
- 301.Wirth K, Hock FJ, Albus U, Linz W, Alpermann HG, Anagnostopoulos H, Henke S, Breipohl G, König W, Knolle J, Schölkens BA. Hoe 140 a new potent and long acting bradykinin-antagonist: In vivo studies. Br J Pharmacol. 1991;102:774–777. doi: 10.1111/j.1476-5381.1991.tb12249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Witherow FN, Helmy A, Webb DJ, Fox KAA, Newby DE. Bradykinin contributes to the vasodilator effects of chronic angiotensin-converting enzyme inhibition in patients with heart failure. Circulation. 2001;104:2177–2181. doi: 10.1161/hc4301.098252. [DOI] [PubMed] [Google Scholar]
- 303.Wolfrum S, Schneider K, Heidbreder M, Nienstedt J, Dominiak P, Dendorfer A. Remote preconditioning protects the heart by activating myocardial PKCɛ-isoform. Cardiovasc Res. 2002;55:583–589. doi: 10.1016/s0008-6363(02)00408-x. [DOI] [PubMed] [Google Scholar]
- 304.Wollheim E, Peterknecht S, Dees C, Wiener A, Wollheim CB. Defect in the excretion of a vasoactive polypeptide fraction A possible genetic marker of primary hypertension. Hypertension. 1981;3:574–579. doi: 10.1161/01.hyp.3.5.574. [DOI] [PubMed] [Google Scholar]
- 305.Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL, Sweeney JF, Peterson LE, Chan L, Smith CW, Ballantyne CM. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation. 2007;115:1029–1038. doi: 10.1161/CIRCULATIONAHA.106.638379. [DOI] [PubMed] [Google Scholar]
- 306.Xu J, Carretero OA, Liu YH, Shesely EG, Yang F, Kapke A, Yang X-P. Role of AT2 receptors in the cardioprotective effect of AT1 antagonists in mice. Hypertension. 2002;40:244–250. doi: 10.1161/01.hyp.0000029095.23198.ad. [DOI] [PubMed] [Google Scholar]
- 307.Xu J, Carretero OA, Liu YH, Yang F, Shesely EG, Oja-Tebbe N, Yang X-P. Dual inhibition of ACE and NEP provides greater cardioprotection in mice with heart failure. J Card Fail. 2004;10:83–89. [PubMed] [Google Scholar]
- 308.Xu J, Carretero OA, Shesely EG, Rhaleb N-E, Yang JJ, Bader M, Yang X-P. The kinin B1 receptor contributes to the cardioprotective effect of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in mice. Exp Physiol. 2009;94:322–329. doi: 10.1113/expphysiol.2008.045583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Yamaguchi T, Carretero OA, Scicli AG. A novel serine protease with vasoconstrictor activity coded by the kallikrein gene S3. J Biol Chem. 1991;266:5011–5017. [PubMed] [Google Scholar]
- 310.Yamaguchi T, Carretero OA, Scicli AG. A potent vasoconstrictor in the rat submandibular gland. Hypertension. 1991;17:101–106. doi: 10.1161/01.hyp.17.1.101. [DOI] [PubMed] [Google Scholar]
- 311.Yang X-P, Liu YH, Mehta D, Cavasin MA, Shesely EG, Xu J, Liu F, Carretero OA. Diminished cardioprotective response to inhibition of angiotensin-converting enzyme and angiotensin II type I receptor in B2 kinin receptor gene knockout mice. Circ Res. 2001;88:1072–1079. doi: 10.1161/hh1001.090759. [DOI] [PubMed] [Google Scholar]
- 312.Yang X-P, Liu YH, Scicli GM, Webb CR, Carretero OA. Role of kinins in the cardioprotective effect of preconditioning. Study of myocardial ischemia/reperfusion injury in B2 kinin receptor knockout mice and kininogen-deficient rats. Hypertension. 1997;30:735–740. doi: 10.1161/01.hyp.30.3.735. [DOI] [PubMed] [Google Scholar]
- 313.Yang X-P, Liu YH, Shesely EG, Bulagannawar M, Liu F, Carretero OA. Endothelial nitric oxide gene knockout mice. Cardiac phenotypes and the effect of angiotensin-converting enzyme inhibitor on myocardial ischemia/reperfusion injury. Hypertension. 1999;34:24–30. doi: 10.1161/01.hyp.34.1.24. [DOI] [PubMed] [Google Scholar]
- 314.Yin H, Chao L, Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad. 14-3-3 signaling pathways. J Biol Chem. 2005;280:8022–8030. doi: 10.1074/jbc.M407179200. [DOI] [PubMed] [Google Scholar]
- 315.Ytrehus K, Liu Y, Downey JM. Preconditioning protects ischemic rabbit heart by protein kinase C activation. Am J Physiol. 1994;266:H1145–H1152. doi: 10.1152/ajpheart.1994.266.3.H1145. [DOI] [PubMed] [Google Scholar]
- 316.Zhang H, Wang H, Bassel-Duby R, Maass DL, Johnston WE, Horton JW, Tao W. Role of interleukin-6 in cardiac inflammation and dysfunction after burn complicated by sepsis. Am J Physiol Heart Circ Physiol. 2007;292:H2408–H2416. doi: 10.1152/ajpheart.01150.2006. [DOI] [PubMed] [Google Scholar]
- 317.Zhou X, Ferraris JD, Cai Q, Agarwal A, Burg MB. Increased reactive oxygen species contribute to high NaCl-induced activation of the osmoregulatory transcription factor TonEBP/OREBP. Am J Physiol Renal Physiol. 2005;289:F377–F385. doi: 10.1152/ajprenal.00463.2004. [DOI] [PubMed] [Google Scholar]
- 318.Zhu L, Carretero OA, Liao T, Harding P, Li H, Sumners C, Yang X-P. Role of prolylcarboxypeptidase in angiotensin II type 2 receptor-mediated bradykinin release in mouse coronary artery endothelial cells. Hypertension. 2010;56:384–390. doi: 10.1161/HYPERTENSIONAHA.110.155051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Zimmerman BG, Raich PC, Vavrek RJ, Stewart JM. Bradykinin contribution to renal blood flow effect of angiotensin converting enzyme inhibitor in the conscious sodium-restricted dog. Circ Res. 1990;66:234–240. doi: 10.1161/01.res.66.1.234. [DOI] [PubMed] [Google Scholar]
- 320.Zimmermann A, Geiger R, Kortmann H. Similarity between a kininogenase (kallikrein) from human large intestine and human urinary kallikrein. Hoppe Seylers Z Physiol Chem. 1979;360:1767–1773. doi: 10.1515/bchm2.1979.360.2.1767. [DOI] [PubMed] [Google Scholar]
- 321.Zinner SH, Margolius HS, Rosner B, Keiser HR, Kass EH. Familial aggregation of urinary kallikrein concentration in childhood: relation to blood pressure, race and urinary electrolytes. Am J Epidemiol. 1976;104:124–132. doi: 10.1093/oxfordjournals.aje.a112282. [DOI] [PubMed] [Google Scholar]
- 322.Zuccollo A, Navarro M, Frontera M, Cueva F, Carattino M, Catanzaro OL. The involvement of kallikrein-kinin system in diabetes type 1 (insulitis) Immunopharmacology. 1999;45:69–74. doi: 10.1016/s0162-3109(99)00149-6. [DOI] [PubMed] [Google Scholar]
- 323.Zweckberger K, Plesnila N. Anatibant, a selective non-peptide bradykinin B2 receptor antagonist, reduces intracranial hypertension and histopathological damage after experimental traumatic brain injury. Neurosci Lett. 2009;454:115–117. doi: 10.1016/j.neulet.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 324.Ørstavik TB, Carretero OA, Johansen L, Scicli AG. Role of kallikrein in the hypotensive effect of Captopril after sympathetic stimulation of the rat submandibular gland. Circ Res. 1982;51:385–390. doi: 10.1161/01.res.51.3.385. [DOI] [PubMed] [Google Scholar]