

Abstract
Hemorrhagic shock resulting from blood loss directs the majority of the blood to the vital organs, dramatically reducing blood flow to the intestines and resulting in damage and inflammation. The excessive intestinal inflammatory response includes pro-inflammatory cytokines and complement activation, although the mechanism is not clear. Toll-like receptors play a vital role in the innate immune response and toll-like receptor 2 (TLR2) is required for intestinal ischemia/reperfusion-induced injury. We hypothesized that TLR2 plays an integral role in the intestinal inflammatory response after hemorrhage and subjected C57Bl/6 wild type and Tlr2-/- mice to atraumatic loss of ∼30% total blood volume. Two hours after blood removal, the intestinal injury and inflammation were assessed. We demonstrate that compared to wild type control mice, Tlr2-/- mice sustain less intestinal damage and inflammation. Importantly, TLR2 regulated eicosanoid and complement activation andIL-12 and TNFα secretions, indicating interactions between TLR2 and complement in response to significant blood loss.
Keywords: Cytokines, Complement, Intestine, tissue injury, immunohistochemistry
Introduction
Hemorrhage and the accompanying hemorrhagic shock result in damage and inflammation in bodily tissues that can lead to serious and immediate health complications. Most animals go into shock if 25-30% of their blood volume is rapidly removed; over 50% die if 30-40% is removed; and nearly all die if more than 40% is removed (1, 2). Hemorrhage results in decreased systemic perfusion, leading to a lack of blood in the intestines (3, 4). Splanchnic circulation accounts for 25-30% of total blood volume in the internal organs and decreased blood flow due to hemorrhage results in intestinal ischemia which may lead to sepsis (5). Many recent studies have suggested that both the inflammatory infiltrate and complement activation are involved in the intestinal damage in response to hemorrhage (6-9).
The inflammatory infiltrate is critical to hemorrhage induced tissue damage in multiple organs (6, 8, 10-12). Multiple aspects of the inflammatory response have been implicated in the tissue damage from neutrophils to toll-like receptors (TLRs) to complement activation. The neutrophilic infiltrate of the liver and lung produces an oxidative burst that is critical to the innate immune system's response to pathogen associated molecular patterns (PAMPs). Hemorrhage induces multiple pro-inflammatory cytokines, including TNFα, IL-6 and IL-12 in a TLR-dependent manner (8, 13). Multiple TLRs are up-regulated in response to hemorrhage (11-14) and TLR4 and TLR9 appear to have unique roles in hemorrhage (10, 12, 13, 15, 16). As a membrane protein molecule, TLR2 recognizes gram positive bacteria as well as damage-associated molecular pattern molecules (DAMPs). Importantly, TLR2 has unique inflammatory responses from other TLRs including increasing TNFα production (17) but requiring additional signals (MyD88 independent) for IL-6 production (18). In addition, activation of TLR2 leads to amplification of the complement cascade (19). Thus, it is likely that TLR2 may mediate the complement-induced intestinal tissue during hemorrhage.
The complement system consists of more than 30 proteins in the blood which remove pathogens and damaged tissue from the body. However, excessive complement activation mediates unnecessary damage in several animal models (reviewed in (20)) and specifically in hemorrhage (3, 7, 21). For example, in rat models, complement C3 depletion prevents clinical signs of hemorrhagic shock and complement activation is critical for tissue damage to occur as a result of hemorrhage (22). Importantly, complement inhibitors including C1 inhibitor (9), C5a receptor antagonist, (21), soluble complement receptor 1 (CR1) (23) and complement receptor 2-factor H (CR2-factor H) (8) attenuate hemorrhage-induced intestinal damage in wild type animals. Recent studies demonstrated that during hemorrhage complement induces IL-12p70 production and TLR2 is required for the complement mediated intestinal damage in response to ischemia/reperfusion injury. (24). However, it is unclear how these inflammatory components interact in response to hemorrhage.
Multiple complex animal models of hemorrhage or hemorrhagic shock exist, each mimicking various clinical conditions. However, it is difficult to determine which inflammatory responses are due to each of the components. To simplify the system and examine the inflammatory response to blood loss in the absence of traumatic tissue damage, we subjected wild type (C57BL/6) and Tlr2-/- mice to volume-controlled hemorrhage and evaluated the inflammatory responses including eicosanoids for recruitment of the inflammatory infiltrate as well as complement components. This allowed us to test the hypothesis that atraumatic blood loss alone activates TLR2 and subsequently complement cascade as measured by C3 and production of local complement inhibitors which leads to intestinal damage.
Materials and Methods
Mice
Male Tlr2-/- (B6.129-Tlr2tm1Kir/J) on the C57Bl/6 background and C57Bl/6J (wild type) mice (8-14 weeks old) were obtained from Jackson Laboratories and maintained in the Division of Biology at Kansas State University. All mice were kept in a 12 hour, light to dark, temperature controlled room and allowed water and food ad libitum. All mice were housed in a specific pathogen free facility (Helicobacter sp., mouse hepatitis virus, minute virus of mice, mouse parvovirus, Sendai virus, murine norovirus, Mycoplasma pulmonis, Theiler's murine encephalomyelitis virus, and endo- and ecto- parasites). All research was approved by the Institutional Animal Care and Use Committee (IACUC) and conducted in cooperation with the Animal Welfare Act and other federal statutes and policies that concern the animals.
Hemorrhage Protocol
Prior to bleeding, the mice were anesthetized with ketamine (16 mg/kg) and xylazine (80 mg/kg), and a drop of proparicaine hydrochloride ophthalmic solution was applied to the eye. The anesthetized hemorrhage treated mice were subjected to removal of 30% calculated total blood volume (body weight (g) × 0.03 (ml/g)) via the retro-orbital sinus. All bleeding was completed within 5-10 minutes. Mortality rate at 2h was less than 6% and animals that did not survive 2 h post bleeding were not included in the study. Sham mice were randomly assigned and subjected to similar anesthesia, but with no blood removal. All procedures were performed with the animals breathing spontaneously and body temperature maintained at 37°C using a water-circulating heating pad. After recovery for 2 hours post-bleeding, mice were euthanized via cervical dislocation and tissues and sera were collected for analysis. Midjejunal Intestinal tissues approximately 2 cm in length were formalin fixed for analysis of injury and additional sections were taken for immunohistochemistry and intestinal secretions.
Injury Score
Formalin fixed intestinal tissue samples were transversely sectioned and and stained with hematoxylin & eosin for scoring injury. The slides are scored using a 6-tiered scale to determine the degree of villus damage due to hemorrhage (25). Sections approximately 2 cm long yielded between 75-150 villi that were individually scored for injury by two blinded reviewers then averaged to determine a composite score for each mouse. On the scale, a score of zero indicates normal (undamaged) villi and damage severity increases with each number. Villi with distortion at the tips received a score of 1. Villi with Guggenheims space received a score of 2. Villi with visible disruptions in the epithelium of the villi received score of 3. Larger breaks in the epithelium with the lamina propria largely intact received a score of 4. Villi with lamina propria streaming out of the damaged villi received score of 5. Villi that were entirely displaying hemorrhage, or entirely denuded received score of 6.
Intestinal Secretions
Ex vivo intestinal supernatants were generated and used to determine the eicosanoid and total peroxidase production similar to previous studies (26). Mid-jejunal whole intestinal sections were minced, washed, and re-suspended in freshly oxygenated Tyrodes buffer. They were then incubated at 37° C for 20 min then stored at -80° C. The supernatants were analyzed for LTB4 and PGE2 by EIA (Cayman Chemical) and TNFα, IL-6, and IL-12p70 concentrations by Milliplex MAP immunoassay kit (Millipore) and read on a Milliplex Analyzer (Millipore). All cytokine and eicosanoid concentrations were normalized to intestinal protein concentration determined by bicinchoninic acid assay protein analysis. All secretions are reported as pg/mg intestinal tissue.
Real-Time PCR
Intestinal sections were homogenized in TRIzol (Invitrogen) and total RNA collected. RNA concentration was determined by Nanodrop and RNA integrity and DNA genomic contamination was assessed on a Bioanalyzer (Agilent). Only pure RNA samples with an RNA integrity number greater than 8.0 were used for further analysis. Total RNA was reverse transcribed into cDNA using random hexamers with Revert-aid MMuLV reverse transcriptase (Fermentas). Real-time PCR was performed in 25 ul volumes using a Mini-Opticon real time thermal cycler (Bio-Rad) and Perfecta SYBR Green Fastmix reagent (Quanta Biosciences) using the following protocol: 3m at 95°C; 50 cycles of 10s at 95°C, 20s at Tm (Table 1), 10s at 72°C; melt curve starting at 65°C, increasing 0.5°C every 5s up to 95°C. The target genes (CD55, Factor H, Factor B and C3, Table 1) Ct values were normalized to 18s, and the fold change compared to C57Bl/6 Sham was calculated as described previously (27). Melt-curve analysis of the PCR products ensured amplification of a single product.
Table 1. Real Time PCR Primer Sequences.
| Gene | Tm (°C)a | Sequence |
|---|---|---|
| C3 | 60 | FWD: CACCGCCAAGAATGCCTSC |
| REV: GATCAGGTGTTTCAGCCGC | ||
| CD55 | 58 | FWD: AATGTGGGGAGAGGGAAATC |
| REV: CTGTGGCGATTCTG TTACA | ||
| Cox-2 | 55 | FWD: ATTCAACACACTCTATCACTGGC |
| REV: TGGTCAAATCCTGTGCTCATACAT | ||
| Factor H | 55 | FWD: ACCACATGTGCCAAATGCTA |
| REV: TGTTGAGTCTCGGCACTTTG | ||
| Factor B | 58 | FWD: CCAGCATTTGGGTTTCAGTT |
| REV: CACACCTCCAGAGGAGAAGC | ||
| Ribosomal 18sb | 58 | FWD: CTGGTAATTCATCTCTCTGCTCTG |
| REV: GCGACCAAAGGAACCATAAC |
Annealing temperature
House-keeping gene to which genes of interest were normalized
Immunohistochemistry
Intestinal sections (8 μm) were cut, placed on slides, and blocked with 10% normal donkey serum (Jackson Immunoresearch) prior to staining with primary antibodies against target molecules. Anti-myeloperoxidase (Thermo-Fisher), anti-mouse F4/80 (BioLegend), and anti-mouse C3 (HyCult Biotechnologies) were used as primaries. After washing with PBS, the slides were incubated with the appropriate Texas-Red labeled secondary antibodies (Jackson ImmunoResearch). After additional washes, the slides were coverslipped and examined by fluorescent microscopy using a Nikon 80i microscope equipped with a CoolSnap CF camera (Photometrics) and MetaVue software (Molecular Devices).
Statistical Analysis
Data are presented as mean ± SEM and were compared by one-way analysis of variance with post hoc analysis using Newman-Kuels test (Graph Pad/Instat Software Inc. Philadelphia, PA). The difference between groups was considered significant when P < 0.05.
Results
Previous studies indicated that blood loss due to hemorrhage can induce intestinal damage and inflammation (8). To determine if TLR2 is required for this tissue damage, we removed 30% of the calculated blood volume from wild type (C57Bl/6) and Tlr2-/- mice and analyzed intestinal villus injury at 2h post-hemorrhage. Sham treatment resulted in minimal tissue damage in both strains of mice (Fig. 1). Similar to previous studies (8), wild type mice sustained significant intestinal damage at 2h post-hemorrhage (HEM). In contrast, Tlr2-/- mice received significantly less damage than wild type mice when subjected to hemorrhage. Representative H & E stained, intestinal villi from Sham and hemorrhage (HEM) treated mice are presented in Fig. 1B.
Figure 1. Tlr2-/- mice sustain significantly less intestinal damage than wild type C57Bl/6 mice when subjected to hemorrhage.
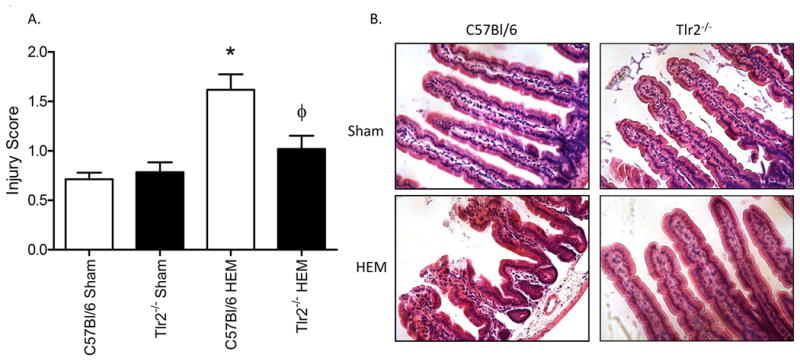
Mid-jejunal intestinal damage was determined 2 hours after bleeding by scoring 75-150 villi per intestinal section. Each bar (A) and photomicrograph (B) represents the mean ± SEM of 9-11 animals per treatment group. *Indicates significant difference (p<.05) between hemorrhage (HEM) and Sham treatments; Φ indicates p<0.05 difference between tissues from hemorrhage (HEM) treated C57Bl/6 and Tlr2-/- mice.
In intestinal ischemia/reperfusion, eicosanoids are critical for intestinal damage to occur, and they may be involved in hemorrhage-induced intestinal damage as well. Cyclooxygenase-2 (Cox-2) is an enzyme responsible for production of prostaglandins E2 (PGE2), which is an important mediator of the inflammatory response. We analyzed Cox-2 transcript levels to determine whether or not the inflammatory response is altered during hemorrhage. Compared to wild type mice, Cox 2 transcript is significantly lower in Tlr2-/- mice (Fig. 2A), suggesting that Cox-2-mediated PGE2 production may be mediated by TLR2 during hemorrhage. We also analyzed PGE2 levels in ex-vivo intestinal supernatants and found that Tlr2-/- mice produced significantly lower levels of PGE2 than wild type mice in response to hemorrhage (Fig.2B). Similarly, leukotriene B4 (LTB4), which is important for neutrophil recruitment during inflammation, was significantly decreased after hemorrhage in Tlr2-/- mice compared to wild type mice (Fig. 2C). Thus, TLR2 plays a critical role in eicosanoid production in response to hemorrhage.
Figure 2. TLR2 is required for hemorrhage-induced intestinal Cox-2, PGE2 and LTB4 production.
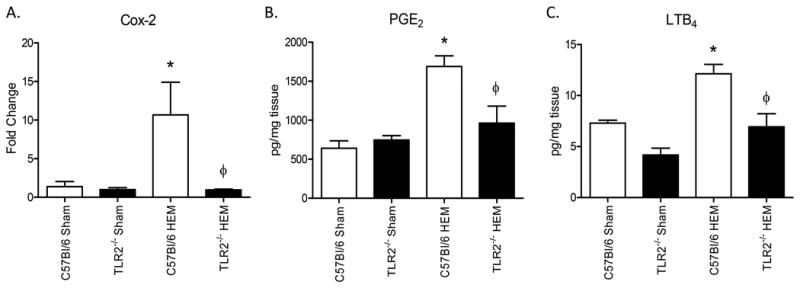
A) After isolating RNA, steady state Cox-2 transcripts were analyzed by Real-Time PCR and the Ct values normalized to 18s and fold change compared to tissues from C57Bl/6 Sham treated mice (N=5-6 animals/treatment group). B, C) Ex vivo intestinal PGE2 (B) and LTB4 (C) concentrations were determined 2 hours after bleeding (HEM) and normalized to mg protein. Each bar represents mean +/- SEM for 5-6 animals/treatment. *Indicates significant difference (p<.05) between hemorrhage (HEM) and Sham treatment; Φ indicates p<0.05 difference between tissues from hemorrhage-treated (HEM) C57Bl/6 and Tlr2-/- mice.
As LTB4 is a chemo-attractant for neutrophils, and previous studies demonstrated that hemorrhage induced macrophage infiltration (8), we examined intestines for the inflammatory infiltration. To examine neutrophil infiltration, we stained Sham and hemorrhage treated tissues from each strain of mice for myeloperoxidase, a neutrophil specific enzyme. Surprisingly, both wild type and TLr2-/- animals had a slight but not significant increase in myeloperoxidase stained cells suggesting a neutrophil infiltration of the villi in response to hemorrhage (Fig. 3A). In contrast, the macrophage infiltration as indicated by F4/80 staining was significantly elevated in response to hemorrhage in wild type mice but not Tlr2-/- mice (Fig. 3B). Importantly, tissues from wild type mice contained significantly more F4/80 positive cells than tissues from Tlr2-/- mice. Together these data suggest that TLR2 is critical for the macrophage inflammatory infiltrate into the intestine in response to hemorrhage.
Figure 3. Hemorrhage induced F4/80 expression but not MPO expression in a TLR2 dependent.
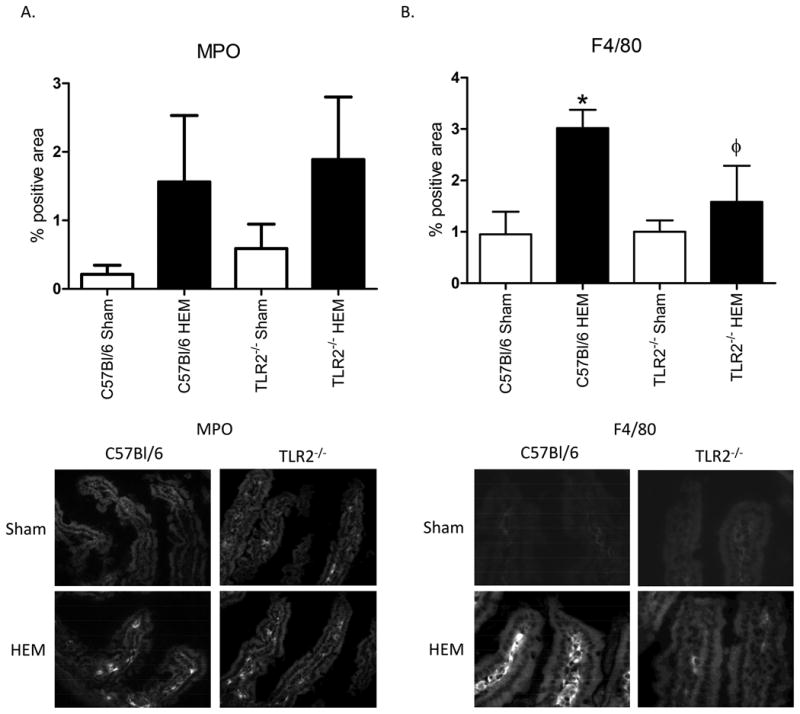
Intestinal tissues from Hemorrhage (HEM) or Sham treated C57Bl/6 or Tlr2-/- mice were stained for myeloperoxidase (MPO) (A) or F4/80 (B) by immunohistochemistry. Bar graphs represent photomicrographs analyzed with Image J. Photomicrographs (original magnification 200x) are representative of five photomicrographs of four to five animals stained in independent experiments. *Indicates significant difference (p<.05) between hemorrhage (HEM) and Sham treatment; Φ indicates p<0.05 difference between tissues from hemorrhage-treated (HEM) C57Bl/6 and Tlr2-/- mice.
Previous studies indicated that macrophage infiltration associates with hemorrhage-induced cytokines including TNFα, IL-6 and IL-12p70 (8, 12). As the macrophage infiltration was TLR2 dependent, we hypothesized that the pro-inflammatory cytokine profile would also be TLR2 dependent. As expected, compared to Sham treatment, all cytokines were elevated in the wild type tissues after hemorrhage (Fig. 4). In addition, TNFα and IL-12p70 were significantly reduced in the hemorrhage treated Tlr2-/- tissues compared to the wild type hemorrhaged tissues (Fig. 4A, B). However, secretion of IL-6 was elevated in hemorrhage-treated tissues from both strains of mice (Fig. 4C). Together, these data indicate that TNFα and IL-12p70 are TLR2 dependent, whereas IL-6 is TLR2 independent.
Figure 4. Hemorrhage-induced intestinal IL-12p70 and TNFα but not IL-6 are TLR2 dependent.
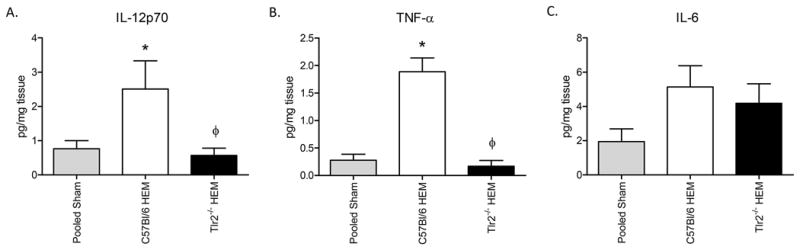
Ex vivo intestinal IL-12p70 (A), TNFα (B) and IL-6 (C) secretions were determined 2 hours after bleeding and normalized to mg protein. Each bar represents mean +/- SEM for 5-7 animals/treatment. *Indicates significant difference (p<.05) between hemorrhage (HEM) and Sham treatment; Φ indicates p<0.05 difference between tissues from hemorrhage-treated (HEM) C57Bl/6 and Tlr2-/- mice.
The cellular infiltrate is not the only form of inflammation in response to hemorrhage. Complement activation also leads to intestinal damage in mice subjected to hemorrhage (8). Although most complement components are produced by the liver (28), additional C3 and complement inhibitors can be produced in other tissues in response to stress (29). We initially examined the intestinal transcript of complement regulators CD55, Factor B and Factor H. As expected, hemorrhage increased intestinal steady state mRNA levels of all three complement regulators in wild type, C57Bl/6 mice (Fig. 5 and data not shown). After hemorrhage treatment, steady state levels of CD55 and Factor H transcript were significantly different from Sham treatment in Tlr2-/- mice (Fig. 5A-B). Despite the up-regulation of complement regulatory proteins, steady state C3 transcript was also elevated in wild type but not Tlr2-/- mice (Fig. 5C). Factor B transcript was increased in response to hemorrhage in both strains of mice (data not shown). To determine if TLR2 deficiency also regulated complement activation, tissues from hemorrhage and sham treated C57Bl/6 and Tlr2-/- mice were stained for C3 deposition. As indicated in Figure 6, hemorrhage induced significantly more C3 deposition within the intestine of wild type but not Tlr2-/- mice. These data suggest that TLR2 is required for complement activation and intestinal deposition as well as playing a role in extra-hepatic production of complement inhibitors.
Figure 5. Tlr2-/- mice produce less intestinal transcript of CD55, Factor H, and C3 than C57/Bl6 mice when subjected to hemorrhage.
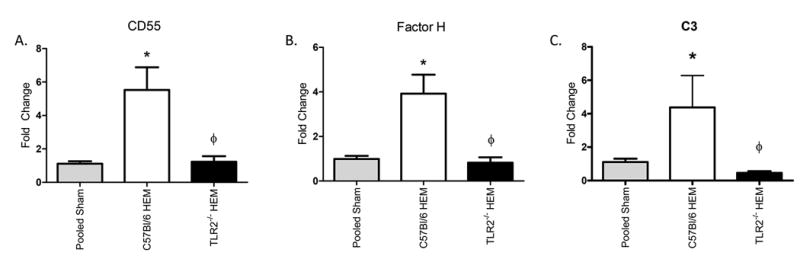
After isolating RNA, Real-Time PCR was used to analyze steady state CD55 (A), Factor H (B), and C3 (C) transcript expression. The target gene Ct values were normalized to ribosomal 18s RNA, and the dCt values of C57/Bl6 and Tlr2-/- hemorrhage (HEM) samples were normalized to C57/Bl6 Sham mice and the fold change calculated as ddCt. Each bar represents mean +/- SEM for 5-6 animals/treatment. * Indicates significant difference (p<0.05) from sham to hemorrhage. Φ indicates p<0.05 difference between tissues from hemorrhage-treated (HEM) C57Bl/6 and Tlr2-/- mice.
Figure 6. Hemorrhage induces intestinal C3 deposition in a TLR2 dependent manner.
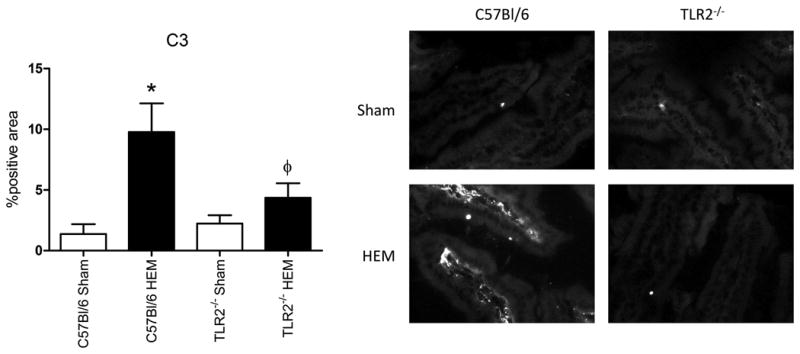
After hemorrhage (HEM) or Sham treatment, intestinal tissues from wild type or Tlr2-/- mice were stained for C3 deposition with immunohistochemistry. Bar graphs represent photomicrographs analyzed with Image J. Each photomicrograph (original magnification 200x) is representative of at least four animals (five-six photomicrographs from each) stained in at least four independent experiments. *Indicates significant difference (p<.05) between hemorrhage (HEM) and Sham treatment; Φ indicates p<0.05 difference between tissues from hemorrhage-treated (HEM) C57Bl/6 and Tlr2-/- mice.
Discussion
Multiple studies have examined the role of hemorrhage in conjunction with trauma and/or resuscitation with various solutions. Although these complex systems resemble the various clinical conditions, it is difficult to deduce which inflammatory responses are due to blood loss vs trauma or resuscitation. We utilized a volume controlled hemorrhage model to examine the inflammatory response to hemorrhage alone. Although this model may not induce severe shock and extreme serum inflammatory response (30), specific organs, such as the intestine, may be affected more than the systemic response. In addition, the non-invasive aspects of the model allows for investigation of the response to blood loss alone.
We demonstrate hemorrhage induces both a neutrophilic and macrophage inflammatory response as well as extrahepatic production of complement inhibitors. However, the production of complement inhibitors are not sufficient to prevent C3 deposition and intestinal damage. Although previous studies have suggested that the inflammatory response is largely TLR4 and/or TLR9 dependent, we demonstrate that the absence of TLR2 attenuates the macrophage infiltration, intestinal IL-12p70 and TNFα production, and complement deposition, suggesting that other TLRs are not sufficient for the entire inflammatory response. Additional limitations to the current study include the use of anesthesia prior to hemorrhage for humane reasons and the use of male mice only as female mice do not sustain similar intestinal epithelial damage (31). Together, we demonstrate that in hemorrhage, complement mediated intestinal damage is TLR2 dependent demonstrating the crosstalk between major components of the inflammatory pathways.
Pro-inflammatory cytokines such as IL-12p70, IL-6 and TNFα, are a key aspect of the inflammatory response. A previous study demonstrated that hemorrhage-induced intestinal inflammation is mediated by complement activation and macrophage production of IL-12p70 (8). The current study extended these results to demonstrate that the IL-12p70 and TNFα response but not IL-6 response is TLR2 dependent. A recent study demonstrated that TLR2-induced production of TNFα but not IL-6 requires activation of Bruton's tyrosine kinase (BTK) in human monocytes (17). As stimulation of macrophage TLR4 also releases all three cytokines, it is likely that IL-6 production is through TLR4 stimulation while TLR2 stimulation is BTK dependent.
During ischemia, the gut barrier becomes leaky and may allow the escape of bacterial products into the intestinal tissue or lymph. As hemorrhage is proposed to induce gut ischemia, it is possible that commenals or commensal products may activate TLR2 in response to hemorrhage. This hypothesis would correlate with the fact that antibiotic depletion of commensals decreased TLR2 expression within the intestine and decreased intestinal TNFα in response to ischemia/reperfusion (32). In addition, infection with heat-killed Brucella stimulates TLR2-mediated TNFα production and subsequently leads to TLR9-mediated increase in IL-12 (33). Another study indicated that hemorrhagic shock induced TLR2 expression on lung in a TLR4 dependent manner (16). Together, these data suggest that hemorrhage may induce commensal activation of TLR2 and other TLRs, resulting in cytokine production by the macrophages or endothelial cells.
As we examined the intestinal response to low blood flow, our data may be similar to that from studies on intestinal ischemia/reperfusion (IR). Our data indicated that TLR2 mediates hemorrhage-induced intestinal Cox-2, PGE2, TNFα and IL-12 but not IL-6 production. The role of TLR2 in mouse models of intestinal IR varies depending on the model. With a long ischemic time and 60 min reperfusion, TLR2 mediates TNFα and MPO but not Cox-2 or PGE2 (34). In contrast, 30 mins ischemia followed by 120 min reperfusion leads to a TLR2 dependent increase in Cox-2, PGE2, and TNFα (24). In addition, in a time course of trauma-hemorrhage, cytokines were not produced until 2 h post-injury (35). Together these data suggest that the current results may be due to the 120 min reperfusion time period. In other models of IR, the role of TLR2 in cytokine production appears to vary by organ type. For example, in kidney IR, TLR2 stimulation induces IL-6 and IL-12p40 and a TLR2 inhibitor OPN301 decreased the production of these cytokines (36). However, in myocardial IR, TLR2 is not required for IL-6 (37) and may or may not be required for TNFα production (37, 38). Finally, after intracerebral hemorrhage, HMGB-1 appears to stimulate TLR2 and TLR4 and provide a beneficial neurogenesis at later time points (39).
Multiple studies have demonstrated not only the importance of complement in hemorrhage (3, 7, 8, 21) but also immediate activation of complement as visualized within 3 min post injury in a porcine model of trauma-hemorrhage (35). Our previous studies indicated that CR2-fH inhibition of complement modulated hemorrhage-induced tissue damage and inflammatory cytokine and eicosanoid production. Similar to the previous study, the current study demonstrated that hemorrhage induced an increase in levels of complement C3 deposition while also increasing transcription of complement regulators CD55 and Factor H. In contrast, none of these complement components were elevated in the absence of TLR2 suggesting that TLR2 regulates complement activation and/or inhibition within the intestine. Although hemorrhage induced Factor B in the current study, the induction was not TLR2 dependent. Together, these data suggest that Factor H plays a role in hemorrhage in a TLR2-dependent manner.
Both the alternative and classical complement activation pathways have been implicated in related mouse models. The current study suggests that Factor B and the alternative pathway is elevated in a TLR2-independent manner. This differs from a mouse model of sepsis in which TLR2 signaling activated the alternative complement pathway via Factor B (40). TLR2 also regulates complement activation in the intestinal IR model by decreasing the classical pathway (24) and the TLR2 inhibitor, OPN301, inhibits C3 deposition in kidney IR (36). In vivo administration of zymosan, a TLR2 agonist, stimulated IL-6 and IL-12 production which was blocked by complement inhibition with cobra venom factor, demonstrating that complement also regulates TLR2 activation (41). Together these data indicate significant crosstalk between the innate immune components, TLR2 and complement in multiple clinical conditions, including blood loss.
In conclusion, we have demonstrated that hemorrhage induces both the cellular and humoral innate immune responses. The innate cellular response includes TLR2 dependent macrophage activation in addition to the traditional neutrophilic infiltration. Importantly, hemorrhage also induces a significant TLR2-dependent humoral innate response of complement activation despite the production of extra-hepatic of complement inhibitors. Together, these data demonstrate that during hemorrhage, TLR2 mediates crosstalk between the cellular and humoral innate immune response.
Acknowledgments
This work was supported by grants from National Institutes of Health (R21 AI107005 to S.D.F.) K-INBRE and Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103418, the Johnson Cancer Center, Confocal Microscopy Core Facility and Kansas State University.
Footnotes
There is no conflict of interest for any of the authors.
References
- 1.McGuill MW, Rowan AN. Biological Effects of Blood Loss Implications for Sampling Volumes and Techniques. ILAR Journal. 1989;31(4):5–20. [Google Scholar]
- 2.Kristensen AT, Feldman BF. General principles of small animal blood component administration. The Veterinary clinics of North America Small animal practice. 1995;25(6):1277–90. doi: 10.1016/s0195-5616(95)50154-8. [DOI] [PubMed] [Google Scholar]
- 3.Fruchterman TM, Spain DA, Wilson MA, Harris PD, Garrison RN. Complement inhbition prevents gut ischemia and endothelial cell dysfunction after hemorrhage/resuscitation. Surgery. 1998;124(4):782–792. doi: 10.1067/msy.1998.91489. [DOI] [PubMed] [Google Scholar]
- 4.Kvarstein G, Mirtheri P, Tonnessen TI. Detection of organ ischemia during hemorrhagic shock. Acta Anaesthesiologica Scandinavica. 2003:675–686. doi: 10.1034/j.1399-6576.2003.00134.x. [DOI] [PubMed] [Google Scholar]
- 5.Shea-Donohue T, Anderson J, Swiecki C. In: Ischemia/reperfusion injury. Tsokos GC, Atkins JL, editors. Totowa, New Jersey: Humana Press; 2003. pp. 219–248. [Google Scholar]
- 6.Hierholzer C, Kalff JC, Billiar TR, Bauer AJ, Tweardy DJ, Harbrecht BG. Induced nitric oxide promotes intestinal inflammation following hemorrhagic shock. Am J Physiol. 2004;286(2):G225–33. doi: 10.1152/ajpgi.00447.2002. [DOI] [PubMed] [Google Scholar]
- 7.Szebeni J, Baranyi L, Savay S, Gotze O, Alving CR, Bunger R, Mongan PD. Complement activation during hemorrhagic shock and resuscitation in swine. Shock. 2003;20(4):347–55. doi: 10.1097/01.shk.0000082444.66379.17. [DOI] [PubMed] [Google Scholar]
- 8.Hylton DJ, Hoffman SM, Rooijen NV, Tomlinson S, Fleming SD. Macrophage produced IL-12p70 mediates hemorrhage-induced damage in a complement dependent manner. Shock. 2011;35(2):134–140. doi: 10.1097/SHK.0b013e3181ed8ec9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalle Lucca JJ, Li Y, Simovic M, Pusateri AE, Falabella M, Dubick MA, Tsokos GC. Effects of C1 inhibitor on tissue damage in a porcine model of controlled hemorrhage. Shock. 2012;38(1):82–91. doi: 10.1097/SHK.0b013e31825a3522. [DOI] [PubMed] [Google Scholar]
- 10.Frink M, Hsieh YC, Thobe BM, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. TLR4 regulates Kupffer cell chemokine production, systemic inflammation and lung neutrophil infiltration following trauma-hemorrhage. Molecular Immunology. 2007;44(10):2625–30. doi: 10.1016/j.molimm.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Fan J. TLR Cross-Talk Mechanism of Hemorrhagic Shock-Primed Pulmonary Neutrophil Infiltration. Open Crit Care Med J. 2010;2:1–8. doi: 10.2174/1874828700902010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan J, Li Y, Vodovotz Y, Billiar TR, Wilson MA. Hemorrhagic shock-activated neutrophils augment TLR4 signaling-induced TLR2 upregulation in alveolar macrophages role in hemorrhage-primed lung inflammation. American Journal of Physiology. 2006;290(4):L738–L746. doi: 10.1152/ajplung.00280.2005. [DOI] [PubMed] [Google Scholar]
- 13.Thobe BM, Frink M, Hildebrand F, Schwacha MG, Hubbard WJ, Choudhry MA, Chaudry IH. The role of MAPK in Kupffer cell toll-like receptor (TLR) 2-, TLR4-, and TLR9-mediated signaling following trauma-hemorrhage. Journal of Cellular Physiology. 2007;210(3):667–75. doi: 10.1002/jcp.20860. [DOI] [PubMed] [Google Scholar]
- 14.Barsness KA, Arcaroli J, Harken AH, Abraham E, Banerjee A, Reznikov L, McIntyre RC. Hemorrhage-induced acute lung injury is TLR-4 dependent. American Journal of Physiology. 2004;287(3):R592–9. doi: 10.1152/ajpregu.00412.2003. [DOI] [PubMed] [Google Scholar]
- 15.Gill R, Ruan X, Menzel CL, Namkoong S, Loughran P, Hackam DJ, Billiar TR. Systemic inflammation and liver injury following hemorrhagic shock and peripheral tissue trauma involve functional TLR9 signaling on bone marrow-derived cells and parenchymal cells. Shock. 2011;35(2):164–70. doi: 10.1097/SHK.0b013e3181eddcab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Xiang M, Yuan Y, Xiao G, Zhang J, Jiang Y, Vodovotz Y, Billiar TR, Wilson MA, Fan J. Hemorrhagic shock augments lung endothelial cell activation role of temporal alterations of TLR4 and TLR2. Am J Physiol Regul Integr Comp Physiol. 2009;297(6):R1670–80. doi: 10.1152/ajpregu.00445.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horwood NJ, Page TH, McDaid JP, Palmer CD, Campbell J, Mahon T, Brennan FM, Webster D, Foxwell BM. Bruton's tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol. 2006;176(6):3635–41. doi: 10.4049/jimmunol.176.6.3635. [DOI] [PubMed] [Google Scholar]
- 18.Chang YL, Chen TH, Wu YH, Chen GA, Weng TH, Tseng PH, Hsieh SL, Fu SL, Lin CH, Chen CJ, Chu CL, Chio, Mak TW, Chen NJ. A novel TLR2-triggered signalling crosstalk synergistically intensifies TNF-mediated IL-6 induction. J Cell Mol Med. 2014;18(7):1344–57. doi: 10.1111/jcmm.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, Triantafilou M, Triantafilou K, Lambris JD, Hajishengallis G. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal. 2010;3(109):ra11. doi: 10.1126/scisignal.2000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fleming SD, Tsokos GC. In: Complement Inhibitors in Rheumatic Diseases. Tsokos GC, editor. Totowa, NJ: Humana Press; 2000. pp. 443–452. [Google Scholar]
- 21.Fleming SD, Phillips LM, Lambris JD, Tsokos GC. Complement component C5a mediates hemorrhage-induced intestinal damage. Journal of Surgical Research. 2008;150(2):196–203. doi: 10.1016/j.jss.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Younger JG, Sasaki N, Waite MD, Murray HN, Saleh EF, Ravage ZA, Hirschl RB, Ward PA, Till GO. Detrimental effects of complement activation in hemorrhagic shock. Journal of Applied Physiology. 2001;90:441–446. doi: 10.1152/jappl.2001.90.2.441. [DOI] [PubMed] [Google Scholar]
- 23.Spain DA, Fruchterman TM, Matheson PJ, Wilson MA, Martin AW, Garrison RN. Complement activation mediates intestinal injury after resuscitation from hemorrhagic shock. Journal of Trauma. 1999;46:224–233. doi: 10.1097/00005373-199902000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Pope MR, Fleming SD. TLR2 Modulates Antibodies Required for Intestinal Ischemia/Reperfusion-Induced Damage and Inflammation. J Immunol. 2015;194(3):1190–8. doi: 10.4049/jimmunol.1303124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu C-J, McArdle AH, Brown R, Scott HJ, Gurd FN. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Archives of Surgery. 1970;101(4):478–483. doi: 10.1001/archsurg.1970.01340280030009. [DOI] [PubMed] [Google Scholar]
- 26.Sjogren RW, Colleton C, Shea-Donohue TS. Intestinal myoelectric response in two different models of acute enteric inflammation. American Journal of Physiology. 1994;267:G329–G337. doi: 10.1152/ajpgi.1994.267.3.G329. [DOI] [PubMed] [Google Scholar]
- 27.Zhao A, Urban JF, Jr, Anthony RM, Sun R, Stiltz J, Rooijen NV, Wynn TA, Gause WC, Shea-Donohue T. Th2 cytokine-induced alterations in intestinal smooth muscle function depend on alternatively activated macrophages. Gastroenterology. 2008;135(1):217–225. doi: 10.1053/j.gastro.2008.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris KM, Goldberger G, Colten HR, Aden DP, Knowles BB. Biosynthesis and processing of a human precursor complement protein, pro-C3, in a hepatoma-derived cell line. Science. 1982;215(4531):399–400. doi: 10.1126/science.7199205. [DOI] [PubMed] [Google Scholar]
- 29.Pope MR, Hoffman SM, Tomlinson S, Fleming SD. Complement regulates TLR4-mediated inflammatory responses during intestinal ischemia reperfusion. Mol Immunol. 2010;48(1-3):356–364. doi: 10.1016/j.molimm.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeifer R, Lichte P, Schreiber H, Sellei RM, Dienstknecht T, Sadeghi C, Pape HC, Kobbe P. Models of hemorrhagic shock: differences in the physiological and inflammatory response. Cytokine. 2013;61(2):585–90. doi: 10.1016/j.cyto.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 31.Szabo A, Vollmar B, Boros M, Menger MD. Gender differences in ischemia-reperfusion-induced microcirculatory and epithelial dysfunctions in the small intestine. Life Sci. 2006;78(26):3058–65. doi: 10.1016/j.lfs.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Yoshiya K, Lapchak PH, Thai TH, Kanna L, Rani P, Dalle Lucca JJ, Tsokos GC. Depletion of gut commensal bacteria attenuates intestinal ischemia/reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2011;301(6):G1020–30. doi: 10.1152/ajpgi.00239.2011. [DOI] [PubMed] [Google Scholar]
- 33.Zhang CY, Bai N, Zhang ZH, Liang N, Dong L, Xiang R, Liu CH. TLR2 signaling subpathways regulate TLR9 signaling for the effective induction of IL-12 upon stimulation by heat-killed Brucella abortus. Cell Mol Immunol. 2012;9(4):324–33. doi: 10.1038/cmi.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe T, Tanigawa T, Kobata A, Takeda S, Nadatani Y, Otani K, Yamagami H, Shiba M, Tominaga K, Fujiwara Y, Arakawa T. Toll-like receptor 2 mediates ischemia-reperfusion injury of the small intestine in adult mice. PLoS One. 2014;9(10):e110441. doi: 10.1371/journal.pone.0110441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sillesen M, Rasmussen LS, Jin G, Jepsen CH, Imam A, Hwabejire JO, Halaweish I, DeMoya M, Velmahos G, Johansson PI, Alam HB. Assessment of coagulopathy, endothelial injury, and inflammation after traumatic brain injury and hemorrhage in a porcine model. J Trauma Acute Care Surg. 2014;76(1):12–9. doi: 10.1097/TA.0b013e3182aaa675. discussion 19-20. [DOI] [PubMed] [Google Scholar]
- 36.Farrar CA, Keogh B, McCormack W, O'Shaughnessy A, Parker A, Reilly M, Sacks SH. Inhibition of TLR2 promotes graft function in a murine model of renal transplant ischemia-reperfusion injury. Faseb J. 2012;26(2):799–807. doi: 10.1096/fj.11-195396. [DOI] [PubMed] [Google Scholar]
- 37.Favre J, Musette P, Douin-Echinard V, Laude K, Henry JP, Arnal JF, Thuillez C, Richard V. Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27(5):1064–71. doi: 10.1161/ATVBAHA.107.140723. [DOI] [PubMed] [Google Scholar]
- 38.Arslan F, Smeets MB, O'Neill LA, Keogh B, McGuirk P, Timmers L, Tersteeg C, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010;121(1):80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
- 39.Lei C, Wu B, Cao T, Zhang S, Liu M. Activation of the high-mobility group box 1 protein-receptor for advanced glycation end-products signaling pathway in rats during neurogenesis after intracerebral hemorrhage. Stroke. 2015;46(2):500–6. doi: 10.1161/STROKEAHA.114.006825. [DOI] [PubMed] [Google Scholar]
- 40.Zou L, Attuwaybi B, Kone BC. Effects of NF-kappa B inhibition on mesenteric ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2003;284(4):G713–21. doi: 10.1152/ajpgi.00431.2002. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, Wetsel RA, Miwa T, Song WC. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110(1):228–36. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]