

Abstract
Although a large part of the global domestic dog population is free-ranging and free-breeding, knowledge of genetic diversity in these free-breeding dogs (FBDs) and their ancestry relations to pure-breed dogs is limited, and the indigenous status of FBDs in Asia is still uncertain. We analyse genome-wide SNP variability of FBDs across Eurasia, and show that they display weak genetic structure and are genetically distinct from pure-breed dogs rather than constituting an admixture of breeds. Our results suggest that modern European breeds originated locally from European FBDs. East Asian and Arctic breeds show closest affinity to East Asian FBDs, and they both represent the earliest branching lineages in the phylogeny of extant Eurasian dogs. Our biogeographic reconstruction of ancestral distributions indicates a gradual westward expansion of East Asian indigenous dogs to the Middle East and Europe through Central and West Asia, providing evidence for a major expansion that shaped the patterns of genetic differentiation in modern dogs. This expansion was probably secondary and could have led to the replacement of earlier resident populations in Western Eurasia. This could explain why earlier studies based on modern DNA suggest East Asia as the region of dog origin, while ancient DNA and archaeological data point to Western Eurasia.
Keywords: Canis lupus familiaris, biogeographic reconstruction, dog origin, expansion wave, free-breeding dogs
1. Introduction
The global dog population has been estimated at 1 billion individuals [1], with about 75% of this population classified as free-ranging [2]. Free-ranging dogs may be owned but are not permanently restrained, semi-feral or feral [2]. Their common characteristic is that they are not artificially restricted in individual mate choice, i.e. are free-breeding (a term we use after [3]). While the population genetic structure of pure-breed dogs is defined by human breeding practices [4,5], the genetic structure of free-breeding dogs (FBDs) is expected to be largely defined by ecological and evolutionary processes (like dispersal patterns, mate choice and natural selection), while still being affected by certain human activities (e.g. translocations, introduction of non-native dog breeds). Unrestricted mate choice has thus major evolutionary implications.
Close breeding practices resulting in the development of modern dog breeds have only been introduced in the last few centuries [6], and the breed formation process was associated with severe bottlenecks and a large increase in linkage disequilibrium (LD) [7,8]. Therefore, FBDs that did not experience these breeding practices may be better suited to reconstruct events at earlier stages of dog history preceding the origin of modern breeds. However, this depends on whether they represent indigenous populations (i.e. deriving from ancestors native for a region they occupy) instead of being a recent admixture of modern breeds or originating from recent translocations.
The indigenous status of FBDs has been explicitly assessed in Africa [9,10], the Americas [10,11], and recently also in Oceania and southern parts of Europe and Asia [10]. African FBDs were shown to be a mosaic of indigenous dogs genetically distinct from non-African breed dogs, and non-native, mixed-breed individuals [9,10]. FBDs from South and North America (except for the Arctic regions) and from South Pacific mostly descend from European dogs, with indigenous American dogs contributing to only a small fraction of the modern gene pool [10,11]. In contrast, in FBDs from Central and South Asia, native ancestry components predominate [10].
Although Eurasia is a particularly important region in dog evolutionary history, being the continent where domestication took place [5,10,12–17], earlier studies focused mostly on FBD populations from southern parts of Asia [10,12,15–17], while little is known about FBDs from central and northern Eurasia. Recently, it has been shown that Arctic dog breeds trace a part of their ancestry to ancient Siberian wolves [18], implying that North Asia is an important region for the dog's evolutionary history. Therefore, for accurate reconstruction of this history, the analysis of genetic variability in populations from both southern and northern parts of Eurasia is required.
Understanding the ancestral status of Eurasian FBDs may also shed light on the origin of pure-breed dogs. Some breeds, mostly of non-European origin (electronic supplementary material, table S1), have been classified as ‘ancient’ based on their early branching in the phylogeny of pure-breed dogs [4,5], and it has been suggested that they ‘may be the best living representatives of the ancestral dog gene pool’ [4]. Alternatively, this branching pattern could simply reflect geographical isolation of these breeds and their consequent genetic differentiation from modern European breeds [6]. Reconstructing the phylogenetic relationships between these breeds and regional FBD populations may improve our understanding of relationships between different breeds and provide correct interpretation for the observed branching pattern.
In this study, we compared genome-wide SNP profiles of 200 FBDs from across Eurasia (figure 1) with 51 ‘ancient’ and modern breeds (combining newly generated and published datasets; [22]) in order to understand the relationships between these groups, assess the indigenous status of Eurasian FBDs, reconstruct their population genetic structure and infer past phylogeographic events in Eurasia.
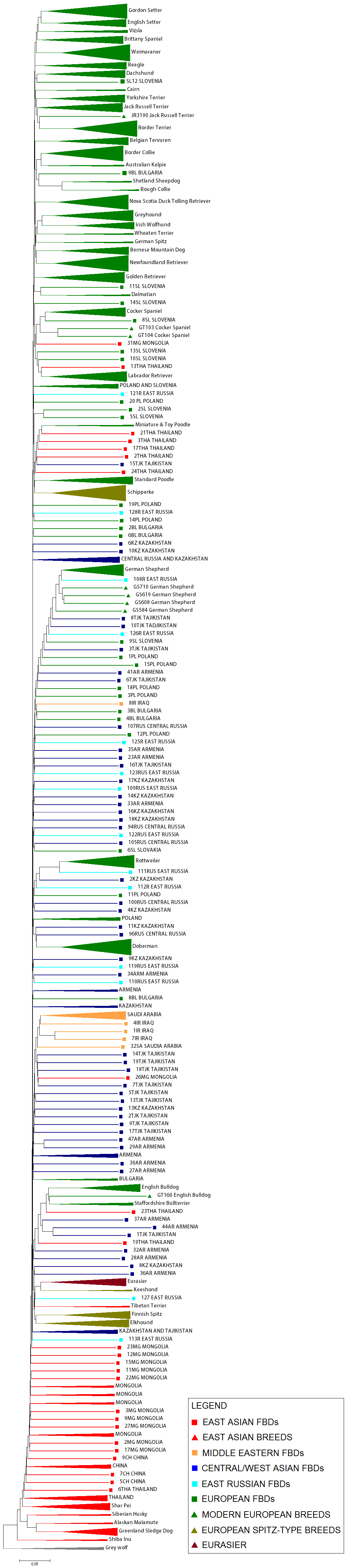
Figure 1.

Genetic differentiation in FBDs and dog expansion routes in Eurasia. (a) Distribution of sampling sites with their division into geographical regions, and dog expansion routes in Eurasia inferred using RASP. (b) Maximum-likelihood tree of genetic differentiation among FBDs and pure-breed dogs, constructed in TreeMix. Distribution of ancestral populations was inferred using RASP (with uncertainty assessed using BB MCMC) and is marked on nodes using colour codes. Black colour denotes undetermined distribution, and the colour codes are simplified compared with the original output (electronic supplementary material, figure S18a). In the RASP analysis, Arctic breeds were assigned to East Asia, according to their primary origin [19–21]. Bootstrap support (based on 1000 replicates) is marked with black stars if above 90% and with white stars if between 65 and 90%. (c) Population genetic structure in Eurasian FBDs, inferred using Admixture assuming two to four genetic clusters.
2. Material and methods
(a). Datasets
We collected blood samples from 234 free-breeding domestic dogs from 14 sites across Eurasia (figure 1a). Multiple samples were collected from each site (electronic supplementary material, table S2). All these samples were genotyped with CanineHD BeadChip (Illumina) at 167 989 autosomal SNP loci (referred to as 168 K) and 5660 X-chromosome SNP loci, together with four grey wolves from Armenia (the South Caucasus). We identified and removed closely related individuals from this dataset (see the electronic supplementary material), reducing it to 200 unrelated individuals. This dataset will be referred to as ‘FBD dataset’.
This dataset was complemented with two datasets of SNP genotypes of pure-breed dogs (electronic supplementary material, table S3). The first dataset consisted of 96 pure-breed or crossed-breed dogs collected from across the United Kingdom using Performagene saliva sample collection kits (DNA Genotek). These dogs represented 31 breeds (88 individuals, with one to nine individuals per breed; electronic supplementary material, table S3) and five types of crosses between two known breeds (eight individuals, with one to three individuals per cross type). This dataset will be referred to as ‘UK dataset’. The second dataset was a publicly available dataset from the LUPA project [22], which contained 446 pure-breed dogs representing 30 different breeds (with 10–26 individuals per breed). It will be referred to as the ‘LUPA dataset’.
These additional datasets were both generated using CanineHD BeadChip, the same as the FBD dataset, and therefore all three datasets could be merged without a reduction of the usable SNP set. Correct merging of the datasets was confirmed by the joint clustering of individuals representing the same breed, independent of whether they originated from the UK or LUPA datasets.
The initial set of 168 K autosomal loci was pruned using PLINK [23] from loci with minor allele frequency (MAF) below 0.01 and those with missing data for more than 10% of individuals. The X-chromosome loci were also removed from all datasets. This resulted in a set of 147 836 loci when the FBD dataset was analysed separately, and 147 485 loci when all the tree datasets were analysed together. For some analyses (highlighted throughout the text), a dataset pruned from loci in strong LD was required. It was obtained by further pruning the dataset from SNPs with an r2 < 0.5 within a 50 SNP sliding window, with a 10 SNPs step size (where r2 is a squared correlation in genotype frequencies between loci). The LD-pruning resulted in a set of 108 610 loci when the FBD dataset was analysed separately, and 104 769 loci when all three datasets were analysed together.
In LD analysis and principal components analysis (PCA), we also included a newly generated dataset of 79 grey wolves from different parts of Asia: the Caucasus (26 individuals), Mongolia (14 individuals), Saudi Arabia (two individuals) and Siberia (37 individuals). This dataset was also generated using CanineHD BeadChip, and therefore could be merged with the three dog datasets without reducing the usable SNP set. The combined dataset was pruned to remove loci with MAF < 0.01 and those with missing data for more than 10% of individuals as well as X-chromosome loci, which resulted in a set of 147 483 loci. The LD-pruning (r2 < 0.5) resulted in 110 112 loci.
(b). Analysis of genetic differentiation in Eurasian free-breeding dogs
We analysed population genetic structure using the LD-pruned FBD dataset. We used the Bayesian clustering methods with no prior population information as implemented in Admixture [24] and Structure [25]. In addition, we carried out a spatially explicit analysis of genetic structure using the software Geneland [26]. Because genetic clustering methods do not perform well in populations that exhibit an isolation-by-distance pattern of the spatial distribution of genetic diversity [27], we assessed whether such a pattern was present in the FBDs across Eurasia using a simple (univariate) Mantel test implemented in GenAlEx v. 6.5 [28]. We also used GenAlEx to carry out a spatial autocorrelation analysis based on pairwise FST values between 14 sampling sites. The details of all these analyses are described in the electronic supplementary material.
We calculated pairwise identity-by-state (IBS) distances between all individuals from the FBD dataset in PLINK and used a matrix of (1−IBS) values to construct a neighbour-joining tree representing genetic differentiation among individuals from different local populations, using the software MEGA6 [29]. To identify the dominant components of variability within the FBDs, we performed a PCA using the smartpca program from Eigensoft [30] package. Eigensoft was also used to estimate average divergence between and within 14 sampling sites, as well as pairwise FST between the sites.
(c). Heterozygosity, autozygosity and linkage disequilibrium analysis
Estimates of heterozygosity and LD are dependent on sample size. Therefore, we randomly selected nine unrelated individuals from all FBD populations that originally had larger numbers of samples. This way we obtained the equal sample size of nine individuals for all but two local populations (with sample sizes of four and five individuals).
Using Plink, we calculated observed and expected heterozygosity in each population. We also assessed autozygosity levels by identifying runs of homozygosity (ROHs), longer than 100 kb and spanning at least 25 SNPs, in individuals from each population. The LD-pruned dataset (with the threshold of r2 < 0.5) was used in this analysis to avoid identifying ROHs resulting from strong LD rather than from autozygosity.
For selected local populations representing each of the four main regions (East Asia, the Middle East, Central/West Asia and Europe), we calculated a genome-wide pairwise genotypic association coefficient r2 between all autosomal SNPs with MAF > 0.15, which provided us with an estimate of LD. In addition, we also analysed LD for grey wolf populations from Mongolia and Armenia, based on nine individuals each. We estimated effective population sizes (NE) based on the extent of LD, to compare the demographic trends between FBDs from different regions (see details in the electronic supplementary material).
(d). Analysis of genetic differentiation between free-breeding dogs and pure-breed dogs
For the combined SNP dataset consisting of FBDs and pure-breed dogs (LUPA and UK datasets), we constructed a PCA plot as well as neighbour-joining trees of both inter-population FST coefficients and inter-individual IBS distances, as described above. We also re-ran the PCA analysis with the addition of the grey wolf data.
Because uneven sample sizes of different populations may bias the PCA results [31], we also carried out the PCA after randomly removing individuals from some populations to obtain more even sample sizes: 93 FBDs (seven per sampling site except for two sites with lower sample number), 87 individuals representing European breeds (one or two individuals per breed), 31 individuals representing East Asian and Arctic breeds (all available), 12 Eurasiers (all available) and 83 grey wolves (all available).
(e). Analysis of admixture among dog breeds and free-breeding dogs
We analysed patterns of admixture among pure-breed and FBDs using the program TreeMix [32]. This analysis was carried out for the combined dataset consisting of free-breeding and pure-breed dogs (FBD, UK and LUPA datasets), with Caucasian grey wolves as an outgroup. The UK dataset included some known cross-breed individuals, which were removed from this analysis, because the presence of cross-breeds affected the tree topology by clustering parental breeds together (e.g. Labradors and other retrievers clustered with Poodles in the presence of Labradoodles; see electronic supplementary material, figure S10). We also removed dog breeds represented by one individual only, and a few pure-breed individuals that did not cluster with their alleged breed.
We constructed the maximum-likelihood trees containing both the pure-breeds and free-breeding populations assuming (i) no post-divergence gene flow among populations and (ii) 10 gene flow events. Although the LD-pruned dataset was used, to further account for LD we constructed the trees using blocks of 100 SNPs rather than individual SNPs. For the tree with no gene flow, we generated 1000 bootstrap replicates by re-sampling blocks of 100 SNPs. For the tree with 10 gene flow events, we generated only 100 replicates due to long computational times. For this tree, a jackknife analysis was used to assess whether the inclusion of each migration edge significantly improved the fit of this phylogenetic model to the data. We also constructed a tree assuming 15 gene flow events, to test whether the addition of more migration edges changes the tree topology.
(f). Reconstruction of distributions of ancestral dog populations
We used the software RASP [33], which estimates the occurrence of migration and vicariance events along a user-defined phylogenetic tree, to reconstruct the distribution of ancestral dog populations. For this purpose, we used the phylogenetic trees constructed in TreeMix (one assuming no post-divergence gene flow and the second assuming 10 gene flow events). The following distribution ranges were considered: Europe, Central/West Asia, Middle East, East Asia and East Russia. Arctic breeds were assigned to East Asia, according to their primary origin [19–21]. Eurasier was assigned to both Europe and East Asia. Grey wolf, used as an outgroup, occurs in all the distribution ranges considered, and therefore was uninformative in this analysis.
We used the parsimony-based statistical dispersal–vicariance analysis [34] and the Bayesian binary (BB) Markov chain Monte Carlo (MCMC) method [35] to estimate uncertainty in the reconstruction of ancestral distributions. A maximum of five geographical regions per node were considered, and an uninformative distribution was applied to the root. For the BB method, a total of 10 MCMC chains were run, nine of which were heated, with 10 000 000 iterations and 20% burn-in.
3. Results
(a). Genetic differentiation in Eurasian free-breeding dogs
Although genetic differentiation among Eurasian FBDs was relatively weak, the different population clustering methods we used supported a division into three large-scale regions: East Asia, the Middle East and Western Eurasia (figure 1). Western Eurasia was further sub-divided into Europe and Central/West Asia, based on both geographical proximity and genetic similarity of local populations (figure 1; electronic supplementary material, figures S1–S4). The population from East Russia, geographically belonging to East Asia, was genetically more similar to Central/West Asia. Therefore, it was considered as a separate region. We describe the results of population genetic and phylogenetic analyses that support this division in the electronic supplementary material.
Despite the weak differentiation, some meaningful patterns could be identified. Most East Asian dogs (from China, Thailand and Mongolia) branched from basal nodes of the IBS tree, even though they did not group into a single clade (electronic supplementary material, figure S3). Multiple individuals from Thailand and one from Mongolia grouped together with dogs from western Eurasia, which may reflect recent gene flow of ‘western’ dogs into East Asia. Individuals from Europe, West Asia and Central Asia did not group into clades consistent with geography, suggesting that they may constitute one genetic population (electronic supplementary material, figure S3). Dogs from East Russia were also part of this large admixed group in the IBS tree, and clustered with West Eurasia in all other analyses (electronic supplementary material, figures S1 and S2).
(b). Heterozygosity and linkage disequilibrium in Eurasian free-breeding dogs
The observed heterozygosity in the 14 sampling sites varied between 0.30 and 0.35, and no consistent differences in heterozygosity were found between the four main regions of Eurasia (electronic supplementary material, table S4). No consistent differences occurred between autozygosity levels, either (electronic supplementary material, figure S5 and table S5).
The Chinese FBD population had lower LD compared with other populations for all genetic distance classes (electronic supplementary material, figure S6a). FBD populations from Europe, Central/West Asia and the Middle East had similar LD levels for short distance classes (1.25–115 kb), supporting their common origin. The populations from Thailand and Mongolia had intermediate r2 values between the Chinese population and all other populations for distance classes between 1.25 and 60 kb, while their r2 values for larger distance classes were comparable with West Eurasian FBD populations (electronic supplementary material, figure S6b,c).
For short distance classes (1.25–40 kb), grey wolf populations had lower LD than FBDs (electronic supplementary material, figure S6b), as expected for an ancestral group. However, for long distance classes (275–1000 kb) LD was higher in wolves than in FBDs (electronic supplementary material, figure S6c), consistent with a long-term decline in wolf numbers in Eurasia [36,37]. LD decayed below r2 = 0.5 at 3.75 kb in Chinese FBDs, 5–7.5 kb in other FBD populations, and 2.5 kb in wolves. Chinese FBDs had higher NE estimates (inferred from LD) than any other FBD population throughout all the time periods assessed. The populations from Thailand and Mongolia had intermediate NE estimates between the Chinese population and all the remaining populations until about 2500 years ago (see electronic supplementary material, figure S7).
(c). Genetic differentiation between free-breeding dogs and pure-breed dogs
PCA placed FBDs in intermediate positions between groups of pure-breed dogs (electronic supplementary material, figure S8). The majority of modern European breeds were clustered together, and only a few free-breeding individuals grouped within this cluster, suggesting relatively low gene flow from pure-breed dogs into FBDs. FBDs from Slovenia and Poland were placed closer to the cluster of European breeds than any other FBD populations, which is consistent with a local origin of modern European breeds. German shepherd occupied an outlier position in the PCA (electronic supplementary material, figure S8a), which was unexpected, but consistent with other analyses in this study.
Breeds of East Asian (Shar Pei and Shiba Inu) and Arctic origin (Greenland Sledge Dog, Alaskan Malamute and Siberian Husky) were placed at the opposite end of PC1 relative to the European breeds cluster. The Arctic breeds originated in East Asia [19–21], so close clustering of these two groups of breeds reflects their common origin. FBDs were placed between these two extremes, with East Asian FBDs grouping closer to East Asian breeds, and European FBDs closer to European breeds (electronic supplementary material, figure S8c).
The inclusion of grey wolves into the PCA shows that dogs and wolves form clearly separated clusters, suggesting that gene flow between FBDs and wolves (revealed in our TreeMix analysis; see below) has not affected the genetic integrity of these populations (electronic supplementary material, figure S9a). At PC1, East Asian and Arctic breeds showed closest proximity to wolves of all the pure-breed dogs and FBDs. However, when this analysis was re-run with more balanced sample sizes for all groups (FBDs, breed dogs and wolves), East Asian and Middle Eastern FBDs showed a similar level of proximity to wolves at PC1 as East Asian and Arctic breeds (electronic supplementary material, figure S9b). Despite these differences, all the PCA plots were consistent in showing genetic distinctiveness of FBDs from breed dogs, and distinctiveness of East Asian and Arctic breeds from modern European breeds.
(d). Phylogenetic reconstruction of relationships between free-breeding dogs and pure-breed dogs
A maximum-likelihood tree of population divergence constructed in TreeMix and a neighbour-joining tree of FST-distances among FBDs and pure-breed dogs, consistently inferred the earliest divergence for East Asian and Arctic breeds, followed by East Asian FBDs (figure 1b; electronic supplementary material, figures S10 and S11). The early branching of East Asian breeds was inferred with 98–99% bootstrap support, and that of East Asian FBDs with 92–95% support (figure 1b; electronic supplementary material, figure S11). Modern European breeds were grouped in one clade, which also included Slovenian FBDs. All other FBDs were placed outside of this clade, with the Middle Eastern populations forming a distinct group from European and Central/West Asian populations.
Individual-based IBS distances produced a similar topology (electronic supplementary material, figure S12). Some individual FBDs clustered with particular dog breeds, suggesting their mixed-breed origin. However, most FBDs formed separate groups from breed dogs, indicating their genetic distinctiveness.
In an IBS tree including pure-breed dogs only, East Asian and Arctic breeds branched from basal nodes, showing the consistency in this branching pattern for phylogenies with and without FBDs. Spitz-type breeds of European origin (Keeshond, Elkhound, Finnish spitz, German spitz and Schipperke) or mixed European and East Asian ancestry (Eurasier) were placed outside of the modern European breed clade (electronic supplementary material, figure S13), suggesting their genetic distinctiveness.
(e). Admixture between free-breeding dogs, pure-breed dogs and grey wolves
The TreeMix analysis assuming 10 admixture events revealed post-divergence gene flow from grey wolves to Middle Eastern FBDs (electronic supplementary material, figure S14). We also identified gene flow from Keeshond to Eurasier, consistent with the origin of this last breed, which was developed by crossing Keeshond females with Chow Chow males [6]. Unexpectedly, we also detected gene flow from German shepherd to multiple FBD populations in Europe and Central/West Asia (electronic supplementary material, figure S14a), suggesting frequent mixing between this breed and FBDs.
Because the presence of the German shepherd prevented the detection of other admixture cases, we re-ran the analysis after removing this breed from the dataset. This analysis revealed additional gene flow events, including: (i) from modern breeds to FBDs in Thailand, East Russia and Europe, (ii) between Mongolian and Chinese FBDs, (iii) between Arctic and East Asian breeds, and (iv) between ancestral populations of modern European breeds (electronic supplementary material, figures S14b and S15). An analysis allowing 15 migration events instead of 10 further revealed gene flow from wolves to Greenland Sledge Dogs, and additional cases of gene flow between ancestral populations of modern European breeds (electronic supplementary material, figure S14c). The addition of migration edges significantly improved the fit of the phylogenetic model to the data (electronic supplementary material, figure S16).
Importantly, accounting for gene flow affected the topology of dog phylogeny. In TreeMix trees accounting for gene flow (assuming either 10 or 15 migration edges), Chinese and Thai FBDs formed the earliest branching clade together with East Asian breeds (electronic supplementary material, figures S14 and S15), suggesting that East Asian breeds and East Asian FBDs have a common origin. KimTree analysis (see the electronic supplementary material) provided higher support for this last topology than the topology where only pure-breeds branched from the most basal node (electronic supplementary material, figure S17).
(f). Reconstruction of the geographical distribution of ancestral dog populations
We used the software RASP [33] to reconstruct the geographical distribution of ancestral dog populations, based on the TreeMix trees both without and with gene flow. For both trees, this analysis indicated that the most recent common ancestor of extant dogs originated in East Asia (figure 1b; electronic supplementary material, figures S18 and S19). It also suggested a gradual westward expansion of dogs along two migration routes from East Asia (i) to the Middle East and (ii) to Europe through Central and West Asia (figure 1a,b). It is important to stress that this finding concerns the most recent common ancestor of extant dogs, rather than the most recent common ancestor all dogs shared with their ancestral grey wolf population.
4. Discussion
(a). Origin of free-breeding dogs in relation to pure-breed dogs
Our results show that Eurasian FBD populations are genetically distinct from pure-breed dogs. Although we found mixed-breed individuals among FBDs, they constituted a small fraction of the entire population. Another study has recently reached a similar conclusion for FBDs from South and Central Asia [10]. Taken together, these results suggest that most FBD populations in Asia represent lineages distinct from modern European breeds and probably native to their respective locations. Furthermore, we provided evidence for the long-term continuity of FBD lineages in East Asia by demonstrating their clustering with the dingo (electronic supplementary material, figure S20), which originated from East Asia and was isolated from other dog populations for at least 3500 years before the arrival of Europeans and their dogs to Australia [6,17,38]. This shows that East Asian FBD populations are indigenous; however, similar as FBDs in other parts of Asia, they include a small fraction of non-native mixed-breed individuals.
(b). Population structure and genetic diversity in Eurasian free-breeding dogs
Even though we sampled populations from discrete and distant locations (a sampling pattern that typically leads to overestimation of population structuring), we found no strong spatial genetic structure among Eurasian FBDs. Such a pattern may suggest a relatively recent common origin of all Eurasian FBD populations and/or intense admixture between regions. Declining spatial autocorrelation of genetic distances for geographical distance classes between 1000 and 4000 km (electronic supplementary material, figure S4b) shows the importance of geographical distance in shaping population differentiation of FBDs. However, higher genetic similarity of East Russian FBDs to dogs from Central Russia and other countries formerly belonging to USSR (Kazakhstan, Tajikistan and Armenia) than to geographically closer dogs from China suggests that genetic differentiation of FBDs is also shaped by cultural/political divisions in human populations.
The weak genetic differentiation among FBDs from different parts of Eurasia can explain the lack of consistent differences in autosomal genetic variability between the four main regions of Eurasia. This result is similar to that based on Y-chromosome data, which revealed comparably high diversity in Southwest Asia, Southeast Asia, Europe, Africa and Oceania [10,15]. However, mitochondrial DNA data showed instead the highest haplotype diversity in Southeast Asia [10,12,39]—a pattern which some studies interpreted as evidence for East Asian origin of the domestic dog [12,39].
Population genetic models of spatial expansion are typically based on a serial founder effect model, which assumes a continuous decline in diversity along a colonization route due to a series of bottlenecks, and no major phylogeographic changes after the initial colonization [40]. However, genetic clines may also result from alternative scenarios involving extensive post-colonization admixture [40]. Our TreeMix analysis revealed a number of admixture events among different FBD populations, which could have contributed to the contrasting diversity patterns between mtDNA and nuclear DNA, especially if admixture was sex-biased.
(c). Geographical patterns of free-breeding dogs expansion in Eurasia
We found that the Chinese FBD population had lower LD and higher NE estimates than other FBD populations, throughout all the time periods assessed. Such a pattern is expected from an ancestral population in comparison with derived populations, as illustrated by genetic studies on the origin of modern humans. An LD-based estimate of temporal NE changes in human populations showed a large reduction in NE in non-Africans compared to Africans lasting between 125 000 and 10 000 years ago, providing support for the ‘Out of Africa’ migration event [41]. Other East Asian FBD populations from Thailand and Mongolia had intermediate LD estimates between the Chinese populations and West Eurasian population for short distance classes. Increased LD and reduced NE in West Eurasian FBD populations as compared with East Asian populations are consistent with a migration event from East Asia westwards.
Identifying a precise geographical location of the source population for the inferred expansion would require denser sampling of Asian FBDs. Another study has recently found lowest LD at short inter-SNP distances in FBDs from Mongolia and Nepal [10], but did not include samples from China, and classified Mongolia as Central rather than East Asia.
In accordance with the inference from the LD pattern, we found that East Asian breeds and FBDs branch from basal nodes in the phylogeny of extant dogs (figure 1b; electronic supplementary material, figure S15). The biogeographic reconstruction of ancestral distributions using RASP showed a clear pattern of a gradual expansion of modern dogs from East Asia towards the Middle East and Europe, indicating that East Asia was a source population in a major migration event.
Patterns of Y-chromosome variability also suggest a large and rapid expansion of dogs from East Asia westwards [17]. This expansion was dated at between 4000 and 11 000 years ago (5800 years ago, s.e. 1750 or 8400 years ago, s.e. 2500, depending on the calibration; [17]), which is considerably later than current estimates of the time when the domestication process was initiated (approx. 19 000–40 000 years ago [13,18]). Such timing implies that this was a secondary rather than primary expansion wave, which could have led to the replacement of dog lineages that had earlier occupied western Eurasia [17], potentially diluting evidence for the primary expansion.
Although the dating of this expansion event is not precise, it could be linked to the neolithization process [17], and it could have occurred via trade and/or in association with spatial and demographic expansion of Neolithic humans [42,43]. The dogs from the new expansion wave could have admixed with earlier resident populations—in parallel with the admixture of expanding Neolithic humans with resident Mesolithic populations [42,43]. Alternatively, the new immigrants could have replaced earlier resident populations, similar to what was seen after the expansion of European dogs (and their human owners) in North America [11]. If Europe was the place of the primary dog origin [13], the replacement scenario is more likely, because in the case of admixture we should expect higher genetic diversity in Europe as compared with East Asia, as demonstrated in an example of the honeybee Apis mellifera, where diversity of admixed populations is higher compared with native populations [44].
(d). Integration with archaeological and ancient DNA data
The occurrence of the secondary expansion wave replacing earlier resident populations in western Eurasia can account for discrepancies between earlier studies based on modern DNA analysis, suggesting East or Central Asia as the region of dog origin [10,12,14], and evidence from archaeological and ancient DNA data, pointing instead to Europe or West Asia [6,13]. The occurrence of the secondary expansion event may also explain why ‘none of the ancient breeds derive from regions where the oldest archaeological remains have been found’ [6]. The early branching dog breeds from East Asia and the Arctic can be considered as ‘ancient’ in the sense that they likely represent lineages older than modern European breeds. However, this does not imply a direct line of descent from the first domesticated population, which may be extinct [18,36] or swamped by admixture.
We acknowledge that a major expansion from East Asia may not be the only scenario consistent with our data, however this conclusion is also supported by other independent datasets of modern Asian FBDs based on different types of genetic markers [10,12,15,17,39]. These earlier studies differ in the precise location of the source population (Southeast versus Central Asia) and in the interpretation of this expansion as a primary [10,12,15,39] or secondary [17] wave. In our opinion, this cannot be resolved without extensive analysis of archaeological dog samples from different parts of Asia.
(e). Admixture patterns
A recent study [18] provided evidence for introgression from a lineage of ancient Siberian wolves into Arctic and East Asian dog breeds (Siberian Husky, Greenland Sledge Dog and Shar-Pei). This past admixture with wolves could result in earlier branching of these breeds relative to East Asian FBDs in the phylogeny of Eurasian dogs. Therefore, we used the TreeMix approach to directly account for the post-divergence gene flow in the phylogenetic reconstruction. This resulted in a tree where Chinese and Thai FBDs formed the earliest branching clade together with East Asian breeds (electronic supplementary material, figure S15), implying that these two dog groups have a common origin, and lineages they represent are older than lineages of modern European breeds. Importantly, the TreeMix analysis revealed post-divergence gene flow from grey wolves to Greenland Sledge Dogs (electronic supplementary material, figure S14c), so the admixture event documented in [18] was accounted for.
The TreeMix analysis also revealed a number of other admixture events that may have an important effect on the inference of the dog evolutionary history. For example, it revealed post-divergence gene flow from grey wolves to Middle Eastern FBDs, consistent with the inference from whole-genome data [36]. Although gene flow in the opposite direction was also inferred from whole-genome data, it was less intense (6–9% versus 12–14%; [36]) and remained undetected here because of the limited number of migration events assumed (10 or 15).
Many gene flow events we detected were known from earlier studies or from breed histories, confirming that our results are accurate. For example, the cross-breed origin of Eurasier, resulting from an admixture between European and East Asian spitz-type dogs, was accurately inferred in our TreeMix analysis. Because the geographical origin of Eurasier was both in Europe and East Asia, this resulted in ambiguous inference of the geographical distribution for the common ancestor of Eurasier and East Asian dogs in the RASP analysis based on the TreeMix tree with 10 migration edges (see electronic supplementary material, figure S19a).
We also detected gene flow from modern breeds to FBDs in different parts of Eurasia. However, the tree of individual-based IBS distances showed that this is due to the presence of individual cross-breed dogs among FBDs. Most FBDs clustered separately from pure-breed dogs, further supporting our conclusion that FBDs are distinct genetic units rather than the result of ongoing admixture between breeds.
5. Conclusion
We presented here a large-scale assessment of genome-wide variability of Eurasian FBDs, showing that they are genetically distinct from pure-breed dogs, and their inclusion is necessary for a complete representation of genetic variability of extant dogs. We provided evidence that East Asian FBD populations are indigenous (although they include a fraction of mixed-breed individuals), while FBDs from West Asia and Europe derive from an ancient expansion of East Asian dogs. This expansion was probably secondary [17] and could have led to the replacement of earlier resident populations in Western Eurasia. The occurrence of such a secondary expansion wave can account for discrepancies between studies aimed at identifying the region of primary dog domestication based on modern DNA analysis and those based on archaeological and ancient DNA data. We also presented evidence for admixture between different FBD populations and for hybridization with wolves. The picture emerging from our results shows a very complex post-domestication history of the dog, which was as eventful as the history of humans.
Supplementary Material
Supplementary Material
{kind=link}
Acknowledgements
We thank colleagues who helped with collecting samples of free-breeding dogs: Malik H. Ali, Sergei V. Aramilev, Mika Bagramyan, Nikolay Baskakov, Sergey Belokobylskij, Mateusz Golan, Alexander V. Gromov, Maria Hołyńska, Thanapol Nongbua, Ksenija Praper, Akylbek Ryspaev, La-orsri Sanoamuang, Elena A. Sazhenova, Jacek Szwedo, Mikhail P. Tiunov, Elena Tsingarska and Odbayar Ts. We thank owners of pure-breed dogs who provided the samples used in this project. We also thank Eduard G. Yavruyan, Egor A. Nikolayev, Nikolay V. Mamayev and Odbayar Ts for samples of wolves. We thank Matthew Webster for his advice regarding the LUPA dataset. We are grateful to Robert D. Owen, Cassandra M. Miller-Butterworth, Oscar Gaggiotti and two anonymous referees for their helpful comments on the manuscript.
Ethics
Blood samples from free-ranging dogs were obtained by veterinarians or veterinary technicians. Samples from pure-breed dogs were obtained using saliva sample collection, with the owners’ consent. No animal was harmed for the purpose of this study. The study was approved by the National Science Centre in Poland and the Museum and Institute of Zoology, Polish Academy of Sciences.
Data accessibility
SNP genotypes generated in this study are available from Dryad: http://dx.doi.org/10.5061/dryad.078nc. Geographical locations of samples are provided in the electronic supplementary material.
Authors' contributions
M.P. participated in the design of the study, carried out the data analysis and wrote the manuscript; T.M. collected the samples and participated in the data analysis; A.E.M. carried out the data analysis and helped draft the manuscript; T.G. participated in the design of the study; K.O., A.R. and S.K. produced the SNP genotype data; F.R.F., A.N.A. and O.B.M. collected the samples and extracted DNA; D.S.M., G.K. and I.M.O. collected the samples; E.S. extracted DNA; W.B. participated in the design of the study, coordinated the project, collected the samples and helped draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Funding
This project was funded by the National Science Centre in Poland (grant no. 2011/01/B/NZ8/02978). Additional funding was provided by the University of Lincoln, UK (Returners Research Fund) and the Deanship of Scientific Research at the King Saud University, Saudi Arabia (project no. IRG-15-38).
References
- 1.Gompper ME. 2014. The dog–human–wildlife interface: assessing the scope of the problem. In Free-ranging dogs and wildlife conservation (ed. Gompper ME.), pp. 9–54. Oxford, UK: Oxford University Press. [Google Scholar]
- 2.Hughes J, Macdonald DW. 2013. A review of the interactions between free-roaming domestic dogs and wildlife. Biol. Conserv. 157, 341–351. ( 10.1016/j.biocon.2012.07.005) [DOI] [Google Scholar]
- 3.Boyko RH, Boyko AR. 2014. Dog conservation and the population genetic structure of dogs. In Free-ranging dogs and wildlife conservation (ed. Gompper ME.), pp. 185–210. Oxford, UK: Oxford University Press. [Google Scholar]
- 4.Parker HG, et al. 2004. Genetic structure of the purebred domestic dog. Science 304, 1160–1164. ( 10.1126/science.1097406) [DOI] [PubMed] [Google Scholar]
- 5.vonHoldt BM, et al. 2010. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464, 898–902. ( 10.1038/nature08837) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson G, et al. 2012. Rethinking dog domestication by integrating genetics, archeology, and biogeography. Proc. Natl Acad. Sci. USA 109, 8878–8883. ( 10.1073/pnas.1203005109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindblad-Toh K, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819. ( 10.1038/nature04338) [DOI] [PubMed] [Google Scholar]
- 8.Boyko AR, et al. 2010. A simple genetic architecture underlies quantitative traits in dogs. PLoS Biol. 8, e1000451 ( 10.1371/journal.pbio.1000451) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyko AR, et al. 2009. Complex population structure in African village dogs and its implications for inferring dog domestication history. Proc. Natl Acad. Sci. USA 106, 13 903–13 908. ( 10.1073/pnas.0902129106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shannon LM, et al. 2015. Genetic structure in village dogs reveals a Central Asian domestication origin. Proc. Natl Acad. Sci. USA 112, 13 639–13 644. ( 10.1073/pnas.1516215112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castroviejo-Fisher S, Skoglund P, Valadez R, Vila C, Leonard JA. 2011. Vanishing native American dog lineages. BMC Evol. Biol. 11, 73 ( 10.1186/1471-2148-11-73) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pang J-F, et al. 2009. mtDNA data indicate a single origin for dogs south of Yangtze river, less than 16,300 years ago, from numerous wolves. Mol. Biol. Evol. 26, 2849–2864. ( 10.1093/molbev/msp195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thalmann O, et al. 2013. Complete mitochondrial genomes of ancient canids suggest a European origin of domestic dogs. Science 342, 871–874. ( 10.1126/science.1243650) [DOI] [PubMed] [Google Scholar]
- 14.Wang G-D, et al. 2013. The genomics of selection in dogs and the parallel evolution between dogs and humans. Nat. Commun. 4, 1860 ( 10.1038/ncomms2814) [DOI] [PubMed] [Google Scholar]
- 15.Ding ZL, Oskarsson M, Ardalan A, Angleby H, Dahlgren L-G, Tepeli C, Kirkness E, Savolainen P, Zhang Y-P. 2012. Origins of domestic dog in southern East Asia is supported by analysis of Y-chromosome DNA. Heredity 108, 507–514. ( 10.1038/hdy.2011.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown SK, Pedersen NC, Jafarishorijeh S, Bannasch DL, Ahrens KD, Wu J-T, Okon W, Sacks BN. 2011. Phylogenetic distinctiveness of Middle Eastern and Southeast Asian village dog Y chromosomes illuminates dog origins. PLoS ONE 6, e28496 ( 10.1371/journal.pone.0028496) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sacks BN, Brown SK, Stephens D, Pedersen NC, Wu J-T, Berry O. 2013. Y chromosome analysis of dingoes and Southeast Asian village dogs suggests a Neolithic continental expansion from Southeast Asia followed by multiple Austronesian dispersals. Mol. Biol. Evol. 30, 1103–1118. ( 10.1093/molbev/mst027) [DOI] [PubMed] [Google Scholar]
- 18.Skoglund P, Ersmark E, Palkopoulou E, Dalén L. 2015. Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr. Biol. 25, 1–5. ( 10.1016/j.cub.2015.04.019) [DOI] [PubMed] [Google Scholar]
- 19.Brown SK, Darwent CM, Sacks BN. 2013. Ancient DNA evidence for genetic continuity in Arctic dogs. J. Archaeol. Sci. 40, 1279–1288. ( 10.1016/j.jas.2012.09.010) [DOI] [Google Scholar]
- 20.Brown SK, Darwent CM, Wictum EJ, Sacks BN. 2015. Using multiple markers to elucidate the ancient, historical and modern relationships among North American Arctic dog breeds. Heredity 115, 488–495. ( 10.1038/hdy.2015.49) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Asch B, Zhang A-B, Oskarsson MCR, Klütsch CFC, Amorim A, Savolainen P. 2013. Pre-Columbian origins of Native American dog breeds, with only limited replacement by European dogs, confirmed by mtDNA analysis. Proc. R. Soc. B 280, 20131142 ( 10.1098/rspb.2013.1142) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaysse A, et al. 2011. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 7, e1002316 ( 10.1371/journal.pgen.1002316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell S, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. ( 10.1086/519795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander DH, Novembre J, Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. ( 10.1101/gr.094052.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pritchard J, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guillot G, Santos F, Estoup A. 2008. Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 24, 1406–1407. ( 10.1093/bioinformatics/btn136) [DOI] [PubMed] [Google Scholar]
- 27.Meirmans PG. 2012. The trouble with isolation-by-distance. Mol. Ecol. 21, 2839–2846. ( 10.1111/j.1365-294X.2012.05578.x) [DOI] [PubMed] [Google Scholar]
- 28.Peakall R, Smouse PE. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539. ( 10.1093/bioinformatics/bts460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. ( 10.1093/molbev/mst197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patterson N, Price AL, Reich D. 2006. Population structure and eigenanalysis. PLoS Genet. 2, e190 ( 10.1371/journal.pgen.0020190) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McVean G. 2009. A genealogical interpretation of principal components analysis. PLoS Genet. 5, e1000686 ( 10.1371/journal.pgen.1000686) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickrell JK, Pritchard JK. 2012. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 ( 10.1371/journal.pgen.1002967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Harris AJ, Blair C, He X-J. 2015. RASP (reconstruct ancestral state in phylogenies): a tool for historical biogeography. Mol. Phylogenet. Evol. 87, 46–49. ( 10.1016/j.ympev.2015.03.008) [DOI] [PubMed] [Google Scholar]
- 34.Yu Y, Harris AJ, He X-J. 2010. S-DiVA (statistical dispersal–vicariance analysis): a tool for inferring biogeographic histories. Mol. Phylogenet. Evol. 56, 848–850. ( 10.1016/j.ympev.2010.04.011) [DOI] [PubMed] [Google Scholar]
- 35.Ronquist F. 2004. Bayesian inference of character evolution. Trends Ecol. Evol. 19, 475–481. ( 10.1016/j.tree.2004.07.002) [DOI] [PubMed] [Google Scholar]
- 36.Freedman AH, et al. 2014. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 10, e1004016 ( 10.1371/journal.pgen.1004016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilot M, Greco C, vonHoldt BM, Jędrzejewska B, Randi E, Jędrzejewski W, Sidorovich VE, Ostrander EA, Wayne RK. 2014. Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity 112, 428–442. ( 10.1038/hdy.2013.122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oskarsson MCR, Klütsch CFC, Boonyaprakob U, Wilton A, Tanabe Y, Savolainen P. 2012. Mitochondrial DNA data indicate an introduction through Mainland Southeast Asia for Australian dingoes and Polynesian domestic dogs. Proc. R. Soc. B 279, 967–974. ( 10.1098/rspb.2011.1395) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duleba K, Skonieczna K, Bogdanowicz W, Malyarchuk B, Grzybowski T. 2015. Complete mitochondrial genome database and standardized classification system for Canis lupus familiaris. Forensic Sci. Int. Genet. 19, 123–129. ( 10.1016/j.fsigen.2015.06.014) [DOI] [PubMed] [Google Scholar]
- 40.Pickrell JK, Reich D. 2014. Toward a new history and geography of human genes informed by ancient DNA. Trends Genet. 30, 377–389. ( 10.1016/j.tig.2014.07.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McEvoy BP, Powell JE, Goddard ME, Visscher PM. 2011. Human population dispersal ‘Out of Africa’ estimated from linkage disequilibrium and allele frequencies of SNPs. Genome Res. 21, 821–829. ( 10.1101/gr.119636.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazaridis I, et al. 2014. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 513, 409–413. ( 10.1038/nature13673) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haak W, et al. 2015. Migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211. ( 10.1038/nature14317) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harpur BA, Minaei S, Kent CF, Zayed A. 2012. Management increases genetic diversity of honey bees via admixture. Mol. Ecol. 21, 4414–4421. ( 10.1111/j.1365-294X.2012.05614.x) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SNP genotypes generated in this study are available from Dryad: http://dx.doi.org/10.5061/dryad.078nc. Geographical locations of samples are provided in the electronic supplementary material.