

Abstract

A series of red-shifted azobenzene amino acids were synthesized in moderate-to-excellent yields via a two-step procedure in which tyrosine derivatives were first oxidized to the corresponding quinonoidal spirolactones followed by ceric ammonium nitrate-catalyzed azo formation with the substituted phenylhydrazines. The resulting azobenzene–alanine derivatives exhibited efficient trans/cis photoswitching upon irradiation with a blue (448 nm) or green (530 nm) LED light. Moreover, nine superfolder green fluorescent protein (sfGFP) mutants carrying the azobenzene–alanine analogues were expressed in E. coli in good yields via amber codon suppression with an orthogonal tRNA/PylRS pair, and one of the mutants showed durable photoswitching with the LED light.
Until recently, the use of azobenzene photoswitches in biological systems typically required short-wavelength UV light in order to induce isomerization of the azo bond, which is undesirable because of the phototoxicity to cells. To minimize phototoxicity and reduce light scattering, recent efforts have focused on the design of highly substituted red-shifted azobenzenes.1 On the basis of an earlier observation2 that bulky substituents at the ortho positions of the azo bond caused a red shift in the n → π* band and a blue shift in the π → π* band, Woolley3 and Bléger4 designed tetra-o-methoxy- and tetra-o-fluoro-substituted, red-shifted azobenzenes, respectively, that showed robust photoswitching properties with the use of visible light. On the other hand, three strategies have been employed to introduce the azobenzenes into proteins site-selectively for the photoregulation of protein function in vivo. These include the design of the cysteine-reactive tethered ligands,5,6 the genetic encoding of the azobenzene amino acids in E. coli(7) and mammalian cells,8 and the use of bioorthogonal ligation with genetically encoded bioorthogonal reporters.9 Among them, the genetic encoding of azobenzene amino acids is particularly attractive as it offers the highest incorporation efficiency in vivo without interference from intracellular thiols as well as potential side reactions found in bioorthogonal reactions. In addition, a genetically encoded azo bridge for proteins was reported recently, showing potentially improved photo control of protein function in vivo.10
To expand genetically encodable azobenzene photoswitches for photochemical control of protein function in vivo, efficient synthetic methods need to be developed. Previously, the azobenzene amino acids were prepared by treating anilines with Oxone to generate the nitrosobenzene intermediates, which reacted with the p-aminophenylalanine to afford the protected azobenzene amino acids in moderate yields (Scheme 1a).7,8 Since the nitroso intermediates are unstable and the p-aminophenylalanine analogues are not widely available, alternative synthetic methods are highly desirable. Herein, we report the facile synthesis of the highly substituted azobenzene amino acids through the reaction of quinonoidal spirolactones, which can be prepared in one step from the tyrosines, and phenylhydrazines in the presence of ceric ammonium nitrate (CAN) catalyst (Scheme 1b). Furthermore, nine new azobenzene amino acids were incorporated into the superfolder green fluorescent protein (sfGFP) in E. coli using a reported orthogonal tRNA/PylRS pair. One of the resulting azo-sfGFP showed durable photoswitching with the LED light.
Scheme 1. Approaches for Synthesis of Azobenzene Amino Acids.

In search of efficient methods for the synthesis of azobenzene amino acids, we were intrigued by a report from Carreño and co-workers where they showed that the azobenzenes can be prepared in good-to-excellent yields from quinone bisacetals and arylhydrazines in the presence of a catalytic amount of CAN.11 To examine whether this method can be extended to the quinone analogue derived from tyrosine, we prepared the quinonoidal spirolactone 1a (structure in Table 1 scheme) in 37% yield by treating N-Boc-l-tyrosine with a slight excess amount of phenyliodonium diacetate (PIDA).12 Separately, the phenylhydrazine derivatives are either commercially available or can be readily prepared from appropriate anilines through the diazonium salt formation followed by reduction with SnCl2 in HCl.13
Table 1. Synthesis of N-Boc-Protected Azobenzene–Alanine Analogues.
| entry | X | R | product | yielda (%) |
|---|---|---|---|---|
| 1 | H | 4-Me (2a) | 3a | 73 |
| 2 | H | 2-OMe (2b) | 3b | 72 |
| 3 | H | 4-CN (2c) | 3c | 72 |
| 4 | H | 3-CN (2d) | 3d | 85 |
| 5 | H | 3-CH=CH2 (2e) | 3e | 68 |
| 6 | H | 3-C≡CH (2f) | 3f | 83b |
| 7 | H | 2,6-F2 (2g) | 3g | 95 |
| 8 | H | 2,4,6-F3(2h) | 3h | 91 |
| 9 | H | 2,6-F2-4-CONH2 (2i) | 3i | 94c |
| 10 | H | 2,6-F2-4-I (2j) | 3j | 82d,e |
| 11 | H | 2,3,4,5,6-F5 (2k) | 3k | 89 |
| 12 | F | 2,6-F2 (2g) | 3l | 72 |
| 13 | F | 2,4,6-F3 (2h) | 3m | 72 |
| 14 | F | 2,3,4,5,6-F5 (2k) | 3n | 60 |
| 15 | Cl | 2,6-F2 (2g) | 3o | 68e,f |
| 16 | Cl | 2,4,6-F3 (2h) | 3p | 67f,g |
| 17 | Cl | 2,3,4,5,6-F5 (2k) | 3q | 63 |
Isolated yields were reported. Unless noted otherwise, reactions were carried out by stirring a solution of spirolactone (1a–c) with 1.1 equiv of the substituted phenylhydrazine in MeCN at room temperature for 12–17 h.
24 h reaction time.
MeOH/MeCN (1:4) was used as solvent.
1.5 equiv of the substituted phenylhydrazine was used.
72 h reaction time.
2.5 equiv of the substituted phenylhydrazine was used.
48 h reaction time.
With the substrates in hand, we performed the azo formation reaction in acetonitrile in the presence of 3 mol % of CAN, and the results are summarized in Table 1. For monosubstituted phenylhydrazines 2a–f, the N-Boc-protected trans-dominant azobenzene–alanines were obtained in 68–85% yield (entries 1–6, Table 1) with no obvious electronic and steric effects. The vinyl group at the meta position in the azo product 3e might provide a potential cross-linking site for a proximal cysteine (e.g., at the i+7 position) through the thiol–ene click reaction as reported.14 Similarly, the acetylene moiety at the meta position of 3f offers a bioorthogonal handle for conjugation with an azide-containing ligand via either copper-catalyzed click chemistry15 or copper-catalyzed Glaser–Hays bioconjugation reaction.16 Since Bléger et al. showed that o-fluorine substitution leads to superior photoswitching properties,4 we investigated the reactivity of a series of fluorine-substituted phenylhydrazines 2g–k in the CAN-catalyzed azo formation reaction. To our satisfaction, the fluorinated azobenzene–alanine analogues were obtained in good to excellent yields (entries 7–11, Table 1), irrespective of the number of fluorines as well as additional substituents.
Since the ortho-substituted azobenzenes show red-shifted absorbance and improved photophysical properties compared to the azobenzene parent, we chose two commercially available ortho-halogenated l-tyrosine derivatives and prepared their corresponding quinonoidal spirolactones, 1b and 1c, using the PIDA method (structures are shown in the Table 1 scheme). To maximize potential improvement in photophysical properties, these two spirolactones were reacted with the ortho-difluorinated phenylhydrazines 2g, 2h, and 2k. All six halogenated azobenzene–alanine products 3l–q were obtained uneventfully in moderate yields (entries 12–17, Table 1), though longer incubation times were required for the two chlorinated azo products, presumably due to the bulky chlorine substituent that slows down the nucleophilic addition.
Because the ortho-fluorinated azobenzenes were reported to show good separation of trans/cis n → π* bands, and as a result achieve high cis and trans photostationary state (pss),4 we investigated photoswitching properties of nine fluorinated azobenzene–alanine analogues we synthesized. The simplest difluorinated azobenzene analogue, 3g, showed a wavelength separation of 8 nm and 66% cis and 62% trans pss after 30 min photoirradiation with green (530 nm) and blue (448 nm) LED light, respectively (Table 2). Adding additional fluorine on one side of the azo bond increased the wavelength separation but led to lower cis-pss (compare 3h and 3k to 3g in Table 2). When a fluorine was introduced in the ortho position of the internal benzene ring, all three tri-ortho-fluorinated azobenzene–alanine analogues (3l, 3m, and 3n) showed greater wavelength separation (12–16 nm) than 3g; most strikingly, 3m exhibited 76% cis-pss after irradiation with green light. However, when a chlorine was placed at the ortho position of the internal benzene ring, divergent behaviors were observed for the resulting three azobenzene–alanine analogs (3o, 3p, and 3q). For di- and trifluorinated analogues (3o and 3p), there was hardly any separation for the n → π* bands, but 3o showed the highest cis-pss (77%) detected in the series along with the second highest trans-pss (70%). For 3q, despite the largest bathochromic shift in the trans n → π* band (448 nm) and improved wavelength separation compared to 3g, the cis-pss remained unchanged while the trans-pss showed a modest improvement. It is noteworthy that the photostationary states were reached after only 5 min photoirradiation in a time–course study with 3h (Table S1, Supporting Information) and that the cis-pss of 3h did not show measurable reduction after 4 days of dark adaptation at room temperature (Figure S1, Supporting Information). Taken together, there was no apparent correlation between the wavelength separation and the pss, suggesting an alternative mechanism may be needed to account for this discrepancy. Unexpectedly, the o-chlorine-substituted di-o-fluoroazobenzene–alanine analogues 3o and 3p exhibited essentially no wavelength separation but highest cis- and trans-pss values, the parameters most relevant in photochemical control of protein function in biological system.
Table 2. Photophysical Properties of N-Boc-Protected Azobenzene–Alanine Analogues.
| compd | X | R | trans λmax n→π* (nm) | cis λmax n→π* (nm) | n→π* separationa (nm) | trans:cis 530 nm | trans:cis 448 nm |
|---|---|---|---|---|---|---|---|
| 3g | H | 2,6-F2 | 435 | 427 | 8 | 34:66b | 62:38b |
| 3h | H | 2,4,6-F3 | 439 | 429 | 10 | 38:62c | 61:39c |
| 3k | H | 2,3,4,5,6-F5 | 446 | 428 | 18 | 43:57c | 64:36c |
| 3l | F | 2,6-F2 | 442 | 426 | 16 | 36:64b | 60:40b |
| 3m | F | 2,4,6-F3 | 436 | 424 | 12 | 24:76b | 61:39b |
| 3n | F | 2,3,4,5,6-F5 | 442 | 429 | 13 | 35:65c | 62:38c |
| 3o | Cl | 2,6-F2 | 428 | 426 | 2 | 23:77b | 70:30b |
| 3p | Cl | 2,4,6-F3 | 425 | 425 | 0 | 28:72b | 71:29b |
| 3q | Cl | 2,3,4,5,6-F5 | 448 | 434 | 15 | 34:66b | 67:33b |
Wavelength separation was calculated using the following equation: Δ = λmax,trans – λmax,cis.
Trans/cis ratio was determined by 19F NMR.
Trans/cis ratio was determined by 1H NMR. Sample was dissolved in DMSO-d6 in an NMR tube to obtain a concentration of 15–22 mM. The photoirradiation was performed by placing the NMR tube on top of an LED light in an enclosed compartment for 30 min.
Since several substituted azobenzene–alanines have been site-specifically incorporated into proteins using amber codon suppression with the orthogonal MmPylRS/tRNACUA pairs,8 we surmised that our new azobenzene–alanine analogues could be similarly incorporated into proteins using the same system. Accordingly, pEvol-PylT-MmPylRS plasmid encoding a Pyl-tRNACUA and a PylRS variant carrying four mutations (A302T, L309S, N346V, and C348G) in its active site was prepared and then cotransformed into BL21(DE3) cells along with the pET-sfGFP-S2TAG reporter plasmid. The resulting transformants were grown in 10 mL LB medium supplemented with 1 mM azobenzene–alanine (Aba) analogue at 37 °C, and the Aba-encoded sfGFP proteins were isolated through Ni–NTA affinity chromatography. Among the azobenzene–alanine analogues tested, small substituents on the distal benzene ring such as cyano and fluorine are generally well accepted by the synthetase with expression yields reaching as high as 32.0 mg L–1 (Table 3), along with high fidelity evidenced by mass spectrometry (see the Supporting Information). One exception is pentafluoro analogue 4k, which produced a lower yield of 5.4 mg L–1 with 36% near-cognate Gln-suppression product,17 indicating the m-fluoro substituents are not well tolerated by the synthetase. When the o-fluorine was introduced into the internal benzene ring, the azobenzene–alanine derivatives (4l, 4m, and 4n) showed lower expression yields, along with the erosion of fidelity. A similar phenomenon was also observed for the ortho-substituted azobenzene–alanine analogues 4o and 4p, presumably due to the repulsion between the internal halogen and the distal azo-nitrogen, which twists the azo bond out of coplanarity with the internal benzene ring4 resulting in poorer substrate properties. With these observations, it appears that new PylRS variants are warranted in order to efficiently charge the twisted tri-o-halogenated azobenzene–alanines with high fidelity in E. coli.
Table 3. Genetic Incorporation of Azobenzene–Alanine Analogues into Superfolder GFP.
| compd | X | R | calcd mass, Da | obsd mass, Da | yield (mg/L) | % Gln incorpa |
|---|---|---|---|---|---|---|
| 4d | H | 3-CN | 27856.3 | 27857.6 ± 1.3 | 32.0 | 0 |
| 4g | H | 2,6-F2 | 27868.1 | 27867.6 ± 1.0 | 22.3 | 0 |
| 4h | H | 2,4,6-F3 | 27886.1 | 27885.2 ± 0.9 | 31.6 | 0 |
| 4k | H | 2,3,4,5,6-F5 | 27921.3 | 27920.3 ± 1.8 | 5.4 | 36 |
| 4l | F | 2,6-F2 | 27886.1 | 27886.0 ± 1.1 | 10.2 | 4 |
| 4m | F | 2,4,6-F3 | 27903.3 | 27901.9 ± 1.6 | 3.9 | 21 |
| 4n | F | 2,3,4,5,6-F5 | 27941.1 | 27940.3 ± 1.7 | 4.8 | 50 |
| 4o | Cl | 2,6-F2 | 27902.1 | 27904.6 ± 1.6 | 7.8 | 68 |
| 4p | Cl | 2,4,6-F3 | 27920.5 | 27920.3 ± 1.2 | 4.4 | 27 |
The Gln incorporation into sfGFP in the purified protein samples was calculated on the basis of ion counts using the following equation: Gln % = IsfGFP-S2Q/(IsfGFP-S2Q + IsfGFP-S2→Aba), where IsfGFP-S2Q and IsfGFP-S2→Aba are the ion counts of sfGFP-S2Q and sfGFP-S2 → Aba, respectively, in the deconvoluted mass spectra.
To examine whether the genetically encoded azobenzenes in proteins can respond to visible-light induced photoswitching, we subjected the highly expressed sfGFP mutant sfGFP–4h to alternating 5 min green-blue irradiation cycles and monitored the π → π* bands (Figure S2, Supporting Information) because of their relatively high intensity in the UV–vis spectra. Closer examination revealed rhythmic changes in absorbance at 340 nm over 10 cycles, consistent with reversible photoswitching between the trans (high absorbance at 340 nm) and cis (low absorbance at 340 nm) form of the azo bond with no sign of “fatigue”, the erosion of cis ratio during photoswitching cycles (Figure 1). The photoswitching durability of the trifluorinated azobenzene in sfGFP–4h also highlighted an exceptional photostability for the fluorinated azobenzene system.4
Figure 1.
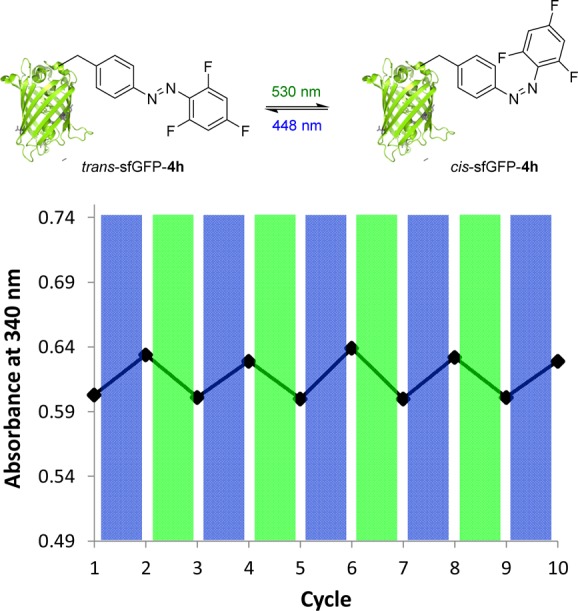
Reversible photoswitching of sfGFP–4h in PBS buffer with alternating 530/448 nm LED photoirradiation.
In summary, we have synthesized a series of red-shifted azobenzene–alanine analogues from the substituted tyrosine and phenylhydrazine derivatives via a two-step procedure. The route involves storable spirolactone intermediates and enables facile unsymmetrical azo formation with the phenylhydrazines in the presence of the ceric ammonium nitrate catalyst. The photophysical properties of the fluorinated azobenzene-alanine analogues were characterized, with the o-chlorodifluoroazobenzene 3o reaching the highest photostationary states (77% cis-pss after 530 nm photoirradiation and 70% trans-pss after 448 nm photoirradiation). In addition, nine azobenzene–alanine amino acids were genetically incorporated into proteins in E. coli with good-to-excellent yields and varying degrees of fidelity via amber codon suppression with an orthogonal tRNA/PylRS pair; eight of them are reported here for the first time. One azobenzene-containing protein, sfGFP–4h, showed durable photoswitching upon photoirradiation with alternating green-blue LED light. The successful development of the spirolactone approach to azobenzene amino acid synthesis, in conjunction with continuous evolution of additional azobenzene–alanine-specific tRNA/PylRS pairs, should greatly enhance our capability to harness the power of azobenzene photoswitches for the photochemical regulation of protein function in vivo with a high spatiotemporal precision.
Acknowledgments
We gratefully acknowledge the National Institutes of Health (GM 85092) for financial support. We thank Prof. Wenshe Liu at Texas A&M University for providing the pEvol-MmPylRS and pET-sfGFPS2TAG plasmids and Tracey Lewandowski in the Q.L. lab at SUNY Buffalo for assistance with cloning.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.5b03268.
Supplemental figures, experimental procedure, and characterization of all new compounds (PDF)
Author Present Address
† Zhejiang University, College of Pharmaceutical Sciences, China.
The authors declare no competing financial interest.
Supplementary Material
References
- Dong M.; Babalhavaeji A.; Samanta S.; Beharry A. A.; Woolley G. A. Acc. Chem. Res. 2015, 48, 2662–2670 10.1021/acs.accounts.5b00270. [DOI] [PubMed] [Google Scholar]
- Forber C. L.; Kelusky E. C.; Bunce N. J.; Zerner M. C. J. Am. Chem. Soc. 1985, 107, 5884–5890 10.1021/ja00307a009. [DOI] [Google Scholar]
- Beharry A. A.; Sadovski O.; Woolley G. A. J. Am. Chem. Soc. 2011, 133, 19684–19687 10.1021/ja209239m. [DOI] [PubMed] [Google Scholar]
- Bléger D.; Schwarz J.; Brouwer A. M.; Hecht S. J. Am. Chem. Soc. 2012, 134, 20597–20600 10.1021/ja310323y. [DOI] [PubMed] [Google Scholar]
- Bartels E.; Wassermann N. H.; Erlanger B. F. Proc. Natl. Acad. Sci. U. S. A. 1971, 68, 1820–1823 10.1073/pnas.68.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banghart M.; Borges K.; Isacoff E.; Trauner D.; Kramer R. H. Nat. Neurosci. 2004, 7, 1381–1386 10.1038/nn1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose M.; Groff D.; Xie J.; Brustad E.; Schultz P. G. J. Am. Chem. Soc. 2006, 128, 388–389 10.1021/ja055467u. [DOI] [PubMed] [Google Scholar]
- Hoppmann C.; Lacey V. K.; Louie G. V.; Wei J.; Noel J. P.; Wang L. Angew. Chem., Int. Ed. 2014, 53, 3932–3936 10.1002/anie.201400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y.-H.; Essig S.; James J. R.; Lang K.; Chin J. W. Nat. Chem. 2015, 7, 554–561 10.1038/nchem.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppmann C.; Maslennikov I.; Choe S.; Wang L. J. Am. Chem. Soc. 2015, 137, 11218–11221 10.1021/jacs.5b06234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreño M. C.; Mudarra G. F.; Merino E.; Ribagorda M. J. Org. Chem. 2004, 69, 3413–3416 10.1021/jo0498011. [DOI] [PubMed] [Google Scholar]
- Rama Rao A. V.; Gurjar M. K.; Sharma P. A. Tetrahedron Lett. 1991, 32, 6613–6616 10.1016/0040-4039(91)80236-Y. [DOI] [Google Scholar]
- Hunsberger I. M.; Shaw E. R.; Fugger J.; Ketcham R.; Lednicer D. J. Org. Chem. 1956, 21, 394–399 10.1021/jo01110a004. [DOI] [Google Scholar]
- Hoppmann C.; Kühne R.; Beyermann M. Beilstein J. Org. Chem. 2012, 8, 884–889 10.3762/bjoc.8.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran V.; Lindhorst T. K. Chem. Commun. 2012, 48, 7519–7521 10.1039/c2cc33542e. [DOI] [PubMed] [Google Scholar]
- Lampkowski J. S.; Villa J. K.; Young T. S.; Young D. D. Angew. Chem., Int. Ed. 2015, 54, 9343–9346 10.1002/anie.201502676. [DOI] [PubMed] [Google Scholar]
- Odoi K. A.; Huang Y.; Rezenom Y. H.; Liu W. R. PLoS One 2013, 8, e57035. 10.1371/journal.pone.0057035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.