

Abstract
Despite advances in precision medicine approaches over the past decade, the majority of nonsmall cell lung cancers (NSCLCs) are refractory to treatment with targeted small molecule inhibitors. Previous work has identified mutations in the Discoidin Domain Receptor 2 (DDR2) kinase as potential therapeutic targets in NSCLCs. While DDR2 is potently targeted by several multitargeted kinase inhibitors, most notably dasatinib, toxicity has limited the clinical application of anti-DDR2 therapy. Here, we have characterized compound 1 and other tool compounds demonstrating selectivity for DDR2 and show that while these compounds inhibit DDR2 in lung cancer model systems, they display limited antiproliferative activity in DDR2 mutated cell lines as compared to dual DDR2/SRC inhibitors. We show that DDR2 and SRC are binding partners, that SRC activity is tied to DDR2 activation, and that dual inhibition of both DDR2 and SRC leads to enhanced suppression of DDR2 mutated lung cancer cell lines. These results support the further evaluation of dual SRC/DDR2 targeting in NSCLC, and we report a tool compound, compound 5, which potently inhibits both SRC and DDR2 with a distinct selectivity profile as compared to dasatinib.
Lung cancer is the leading cause of cancer-related mortality in the United States with approximately 160 000 deaths per year.1 The most common type of lung cancer, nonsmall cell lung cancer (NSCLC), accounts for 85% of cases carrying a poor prognosis.2 The majority of patients present with locally advanced or metastatic disease and require treatment with systemic therapies.
For patients with lung adenocarcinoma, the most common subtype of NSCLC, the discovery of oncogenic drivers and effective targeted therapeutics have resulted in significant survival improvements in certain patient subsets, notably those carrying alterations in EGFR(3−5) and ALK.(6,7) Compared to standard treatment with platinum-based chemotherapy, these targeted therapies are both more efficacious and less toxic in selected populations and have led to a paradigm shift in the management of lung adenocarcinoma.2 In contrast, for patients with lung squamous cell carcinoma (lung SqCC), the second most common subtype of NSCLC, targeted therapies developed for lung adenocarcinoma are not very effective, and standard chemotherapy remains the standard of care given the lack of overlapping targetable genomic alterations with lung adenocarcinoma.8,9
Recently, genomic profiling studies of SqCC have been completed and have identified a number of new potential therapeutic targets for this disease. Notably, lung SqCCs do not harbor at appreciable frequencies the genomic alterations associated with response to targeted therapies in patients with lung adenocarcinoma.10 In lung SqCC, preclinical data have described amplification, mutations, and translocations of Fibroblast Growth Factor Receptors (FGFRs),11−15 copy number increases and/or mutations in PIK3CA,16 Discoidin Domain Receptor Tyrosine kinase 2 (DDR2) mutations,10,17 and kinase-inactivating BRAF mutations18,19 as potential therapeutic targets. FGFR alterations and DDR2 mutation have been associated with the response to targeted agents in both preclinical models and in early phase clinical trials, and several selective inhibitors of FGFR kinases are moving forward clinically.20,21
DDR2 is a receptor tyrosine kinase which was found to be mutated in approximately 4% of patients with lung SqCC in studies utilizing both Sanger sequencing and next-generation sequencing approaches.10,17DDR2 mutations have also been reported in lung adenocarcinoma, gastric cancer, breast cancer, and brain cancers.22−24 DDR2 is a receptor for extracellular collagens, and previous work has shown that DDR2, following collagen binding, activates a complex signaling network involving SHP-2 as well as SRC and MAP kinases.25−27 DDR2 regulates epithelial-mesenchymal transitions (EMT), and a subset of mutations in DDR2 are oncogenic in cellular model systems.17,26,28,29
DDR2 is potently targeted by FDA-approved multitargeted kinase inhibitors including dasatinib, imatinib, nilotinib, and ponatinib, and these agents suppress the proliferation of DDR2 mutated cancer cell lines.30−32 Dasatinib, the most potent of these inhibitors, has been studied in multiple lung cancer clinical trials, including studies focused on subjects with DDR2 mutations.33,34 While two responses to dasatinib have been reported in patients with the DDR2 S768R mutation, the highly multitargeted nature of dasatinib and its associated toxicity have limited its clinical development in lung cancer.17,33
Given the paucity of effective targeted therapeutics for patients with lung SqCC with DDR2 mutations,22 we sought to develop potent and selective inhibitors of DDR2 that could be used to pharmacologically address the impact of inhibiting the kinase activity of DDR2. We previously generated and characterized selective DDR1 inhibitors; however, these compounds did not display appreciable activity against DDR2.31 Novel potent DDR2 inhibitors have been reported by others,32 but these compounds have not been explored in cellular models, nor do they exhibit the same degree of selectivity for DDR2 as compared to selective DDR1 inhibitors.
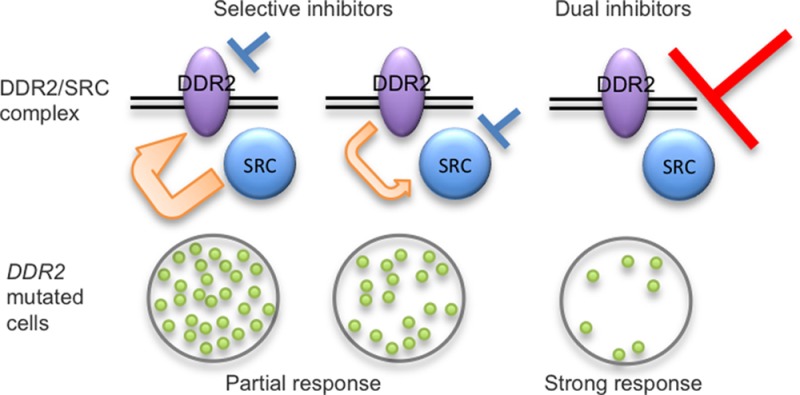
We report here the characterization of compound 1, a molecule previously characterized for its ability to inhibit Ephrin-family kinases,35 as a potent inhibitor of DDR2. In addition, we also characterize additional potent DDR2 inhibitors 2,363, and 4. We show that these DDR2 inhibitors decrease DDR2 kinase activity in vitro and in cellular systems with comparable potency and with a greater degree of specificity as compared to previously characterized DDR2 inhibitors. Using these compounds, we show that DDR2 activation is intimately linked to SRC function, that SRC phosphorylates DDR2 in a complex, and that SRC activity is dominant to DDR2 in maintaining the survival of DDR2 mutated cancer cell lines. Further, we show that either selective SRC or DDR2 inhibition is potentiated by inhibition of the other kinase, suggesting a coordinated role of SRC and DDR2 in mediating the survival of cells with DDR2 mutations. Additionally, we present a dual SRC/DDR2 inhibitor, compound 5, which suppresses DDR2 mutated lung cancer models. Our results indicate that selective inhibition of DDR2 will likely not be a successful sole therapeutic strategy to target tumors with DDR2 mutations in contrast to dual SRC/DDR2 inhibition.
Results and Discussion
Development of a Selective Inhibitor of the Discoidin Domain Receptor 2 Kinase
To identify novel and potent DDR2 inhibitors, we screened a previously generated “type-II” kinase inhibitor library that was designed to conform to a type II inhibitor pharmacophore model.37,38 We constructed a library of inhibitors based on the well-established pharmacophore of type II kinase inhibitors and performed kinome-wide selectivity profiling in an effort to identify new inhibitors and the kinases that might be susceptible to inhibition by type II inhibitors. A library of approximately 100 potential type II inhibitors was screened against a panel of over 350 kinases using the KinomeScanTM approach. The structure of 1 presents a typical pharmacophore for type-II kinase inhibitors: 5-substituted nicotinamide as a “head” motif which interacts with the hinge region with hydrogen bonds, a 1,3,5-substituted phenyl ring as a “linker” motif which is often vertical to the “head” and traverses the area proximal to the gatekeeper residue of kinases, and 3-substituted (trifluoromethyl)benzene as the “tail” motif that occupies the hydrophobic pocket around the “DFG” motif of kinases and prevents the “DFG-in” conformation which is favorable for activated kinases (Figure 1A). As one of our type-II inhibitors, compound 1 was screened against a panel of over 350 kinases (KinomeScan, DiscoverX);39,40 the results suggested a small number of kinases were potential hits, and DDR2 was the top hit among them (Figure 1C). Compound 1 exhibits a S(1) score of 0.02, which compares favorably to other selective inhibitors.41 As the KinomeScan assays measure binding, we also performed enzymatic assays for potential targets (SelectScreen, Life Technologies),42 which demonstrated that compound 1 exhibited potent inhibition of DDR1/2 with two-digit nanomolar IC50s, whereas it showed micromolar IC50’s against most other kinases (Figure 1). With its excellent overall kinase selectivity and apparent potency for DDR2, we further characterized the inhibition of DDR2 signaling and related proliferation by compound 1 in a cellular context.
Figure 1.

Characterization of compound 1, a selective DDR2 inhibitor. (A) Structure of the DDR2 inhibitor compound 1. (B) KinomeScan kinase selectivity profiles for 1. Compound 1 was profiled at a concentration of 1 μM against a diverse panel of 353 kinases by DiscoverX. (C) Top hits of compound 1 against 353 kinases. Shown here are the subset of kinases that exhibited a score of 10 or below (score is percent relative to DMSO control, smaller numbers indicate stronger binding). Biochemical kinase IC50’s (performed at ATP concentrations equal to the apparent Km) were determined for some kinase targets using enzymatic assays and are reported in nanomolar concentrations.
Compound 1 Decreases DDR2 Phosphorylation but Has Little Effect on the Proliferation of DDR2 Mutated Cancer Cell Lines
To investigate the biochemical and cellular potency of compound 1, we first utilized HEK293T (293T) cells transfected with wild type DDR2. This cell type does not display detectable endogenous DDR2 protein levels by immunoblot. After overnight treatment with dasatinib or DMSO, immunoprecipitation of cell lysates was performed using a polyclonal anti-DDR2 antibody, followed by Western blotting with anti-DDR2 or antiphosphotyrosine (4G10) to measure DDR2 phosphorylation status. We observed that compound 1 decreased p-DDR2 in a dose-dependent fashion similar to dasatinib but with less potency (Figure 2A). Given that we had previously observed an antiproliferative effect of dasatinib in two lung cancer cell lines with DDR2 mutations, NCI-H2286 and HCC-366, we treated these cell lines with compound 1 to assess whether this compound might display a similar phenotype. Unexpectedly, compound 1 displayed very modest potency as compared to dasatinib in these cell lines in cell proliferation assays (Figure 2B).
Figure 2.

Comparative analysis of compound 1 and dasatinib. (A) DDR2 was transiently expressed in 293T cells by the pCMV6 expression vector. Cells were treated with depicted concentrations of compound 1 or dasatinib. Cell lysates were immunoprecipitated with anti-DDR2, followed by Western blotting with anti-DDR2 or antiphosphotyrosine. (B) Proliferation of NCI-H2286 and HCC-366 grown for 5 days in the presence of compound 1 or dasatinib. Graph shows mean ± SD from a single experiment representative of three independent experiments with three replicates per treatment per experiment. (C) Effects of dasatinib and compound 1 treatment on p-SFK levels in NCI-H2286 and HCC-366. Cells were treated for 3 h with 0.5 μM of each drug.
Prior work has shown that SRC phosphorylates DDR2, which, in turn, drives DDR2 autophosphorylation, and that overexpression of SRC increases DDR2 kinase activity.43 SRC family kinases (SFKs) are potently inhibited by dasatinib, and we reasoned that dasatinib might result in more complete inhibition of DDR2-dependent signaling as compared to compound 1 given that it targets both DDR2 and SRC family kinases. To confirm that dasatinib but not compound 1 could inhibit SFK phosphorylation, we treated NCI-H2286 and HCC-366 with either drug and observed a robust decrease in p-SFK with dasatinib but not compound 1 (Figure 2C), consistent with the in vitro kinase profiles of these drugs.
Characterization of Additional Selective DDR2 Inhibitors
Given the unexpected result that compound 1 led to a decrease in p-DDR2 with very little associated antiproliferative activity in DDR2 mutated lung cancer cells, we characterized three additional selective DDR2 inhibitors, compounds 2,363, and 4, to determine whether the phenotypes observed with compound 1 would be observed with other selective DDR2 inhibitors (Figure 3A, Supporting Information Figure 1 and Table 1). Compounds 3 and 4 were developed in an effort to identify an analog with improved kinome selectivity and mouse microsomal stability to enable eventual use in vivo. Compounds 3 and 4 were also designed to display orthogonal off targets as compared to compound 1. These inhibitors with distinctive hinge binders or DFG-pocket-occupying tails displayed potency against DDR2, whereas they were weak or inactive against SRC in biochemical assays (Figure 3B).44,45 Similar to compound 1, compounds 2, 3, and 4 displayed very modest potency against DDR2 mutated cell lines (Figure 3B). In contrast to this, two dual DDR2/SRC inhibitors, compounds 5 and 6,35 were more potent in suppressing DDR2 mutated cell lines (Figure 3B). Compound 5 was generated in an attempt to improve the selectivity profile of 6 while retaining potent DDR2 and SRC inhibitory activity (Supporting Information Figure 1 and Table 1).
Figure 3.

Development and characterization of additional DDR2 inhibitors. (A) Structures of DDR2 inhibitors 2, 3, 4, 5, and 6. (B) Enzymatic activities against DDR2 and SRC kinases and antiproliferative activity of DDR2 inhibitors against DDR2-mutated lung cancer cell lines.
To assess whether these inhibitors might display differential activity against WT versus mutated DDR2, we generated 293T cells expressing DDR2 L239R or DDR2 I638F, the DDR2 mutations found in HCC-366 and NCI-H2286, respectively. We observed that compounds 1 and 5 decreased p-DDR2 with similar potency with mutated DDR2 as compared to wild-type DDR2 (Supporting Information Figure 2).
Combination Therapy with Selective DDR2 Inhibitors and Saracatinib Displays Additive Effects on DDR2 Kinase Activity and Cell Viability of DDR2 Mutated Cells
From these results, we hypothesized that SFK activity and DDR2 activity might coordinately drive proliferation of DDR2 mutated cancer cell lines. To investigate the relationship between SFKs and DDR2 in more detail, we first tested a selective SFK inhibitor, saracatinib, in 293T cells expressing wild-type DDR2. Saracatinib inhibits SRC and other SFKs with low nanomolar potency and displays an IC50 for DDR2 of 291 nM.17 Immunoprecipitation/Western blotting showed that compound 1 decreased p-DDR2, and this was further decreased upon the addition of saracatinib (Figure 4A), consistent with prior work placing SRC upstream of DDR2 and catalyzing DDR2 autophosphorylation.43 The observed incomplete inhibition of phosphorylation also suggests that DDR2 is phosphorylated by other upstream kinases. We next treated DDR2 mutated cancer cell lines with saracatinib and observed a greater inhibition of cell proliferation as compared to compound 1 in viability assays (Figure 4B). This effect was dominant to inhibition of DDR2, as the addition of compound 1 to saracatinib did not display a synergistic effect, but rather an additive effect, on cell viability.
Figure 4.

SFK inhibition decreasing DDR2 phosphorylation and suppressing DDR2 mutated cancer cell lines. (A) DDR2 was ectopically expressed in 293T cells, and phosphorylation was measured by Western blotting with antiphosphotyrosine (4G10) after immunoprecipitation with an anti-DDR2 antibody. Cells were treated with the indicated concentration of 1 in combination with or without 1 μM of saracatinib. (B) Proliferation of NCI-H2286 and HCC-366 cells grown for 5 days in the presence of compound 1 in combination with DMSO or 1 μM of saracatinib or in the presence of different concentrations of saracatinib with DMSO. Graph shows mean ± SD from a single experiment representative of three independent experiments with three replicates per treatment per experiment. (C) DDR2 WT or SRC WT was ectopically expressed in 293T cells. Cells were treated with saracatinib. Receptor phosphorylation was measured by Western blotting with antiphosphotyrosine after immunoprecipitation with an anti-DDR2 antibody. Western blotting for anti-DDR2, anti-t-SRC, anti-p-SFK, and antiactin was performed with the same lysate. (D) DDR2 WT, DDR2 GK, SRC WT, or SRC GK was ectopically expressed in 293T cells. Cells were treated with a vehicle or dasatinib. Western blotting with antiphosphotyrosine, anti-DDR2, or total SRC was performed after immunoprecipitation with an anti-DDR2 antibody.
To confirm these results with chemically distinct DDR2 inhibitors, we also probed compounds 2, 3, and 4, agents which similarly displayed very modest antiproliferative potency as single agents against DDR2 mutated cell lines but did lead to a comparable decrease of p-DDR2 in treated cells (Supporting Information Figures 3A and 3B). Similar to compound 1, compounds 2, 3, and 4 did not inhibit SFK activity (Supporting Information Figure 4) but did inhibit p-DDR2 (Supporting Information Figure 3B). Inhibition of p-DDR2 was increased by cotreatment with saracatinib (Supporting Information Figure 3B). This contrasted with dasatinib, and compounds 5 and 6, all of which displayed potent inhibition of both SFK (Supporting Information Figure 4) and DDR2 phosphorylation (Supporting Information Figure 5B) as well as suppression of the DDR2 mutated cell lines NCI-H2286 and HCC-366 (Supporting Information Figure 5A).
To further examine the role of SRC in DDR2 activity, we used 293T cells cotransfected with SRC and DDR2. We observed that SRC and DDR2 form a complex that can be immunoprecipitated (Figure 4C and D) and that saracatinib decreases both p-SRC and p-DDR2 in this complex in a dose-dependent fashion (Figure 4C). In this experiment, immunobloting for SRC after immunoprecipitation with a DDR2 antibody indicated that SRC and DDR2 could form a complex and that the SFK activity in this complex was likely SRC itself. Interestingly, we observed an increase in total SRC with saracatinib treatment, perhaps representing a feedback mechanism in response to the antiproliferative effects of saracatinib.
To examine whether SRC activity directly contributes to dasatinib-sensitive DDR2 phosphorylation, we used 293T cells transfected with a SRC or DDR2 transgene with a mutation in the gatekeeper (GK) residue (T341 M or T654I), respectively. Immunoprecipitation followed by immunoblotting showed that DDR2 or SRC GK mutants blocked the effects of dasatinib on p-DDR2 to a substantial degree (Figure 4D). In this experiment, we also observed an increase of total SRC in immunoblotting after immunoprecipitation with dasatinib treatment similar to what was observed with saracatinib in Figure 4C.
DDR2 Mutant Cell Lines Are Rescued from Dasatinib Suppression by SRC Gatekeeper Mutation
To confirm the effect of SRC on DDR2 phosphorylation, we performed Western blotting for phosphotyrosine after immunoprecipitation with an anti-DDR2 antibody in 293T cells treated with dasatinib. In this experiment, 293T cells were transfected with SRC WT or SRC GK. We observed that the phosphorylation of SRC bound to DDR2 was maintained after dasatinib treatment in 293T cells with expression of the SRC GK mutation (Figure 5A). We also observed that SRC GK could partially rescue p-DDR2, consistent with the results in Figure 3 (Figure 5A). To elucidate whether SRC activity was a relevant target of dasatinib in DDR2 mutated cancer cell lines, we introduced the SRC transgene with gatekeeper mutations into NCI-H2286, HCC-366, and A549 cells. Cell proliferation assays showed that DDR2 mutated cell lines (NCI-H2286 and HCC-366) were rescued from dasatinib induced cell death by SRC GK mutation to a degree comparable to what we previously observed with DDR2 GK mutation.17 We also observed the expected lack of activity of saracatinib in NCI-H2286 and HCC-366 with expression of the SRC GK mutation (Supporting Information Figure 6). We observed no effect on viability in the non-DDR2 mutated A549 cells expressing the SRC GK mutation when treated with dasatinib, though the intrinsic sensitivity to dasatinib in this line was modest (Figure 5B).
Figure 5.

DDR2 mutated cell lines rescued from dasatinib by SRC gatekeeper mutation. (A) Western blotting for phosphotyrosine was performed after immunoprecipitation with a DDR2 antibody. 293T cells were transfected with DDR2 and/or SRC with GK mutations and treated with dasatinib. (B) Cell proliferation assay performed with cells expressing ectopic SRC GK mutation as compared to parental cell lines. Graph shows mean ± SD from a single experiment representative of three independent experiments with three replicates per treatment per experiment.
Inhibition of SRC or DDR2 Increases the Sensitivity of DDR2 Mutated Cell Lines to Selective DDR2 Inhibition or Selective SRC Inhibition
To determine if decreased expression of SRC or DDR2 themselves, but not simply decreased kinase activity, might affect the sensitivity of DDR2 mutated cell lines to a selective DDR2 inhibitor or selective SFK inhibitor respectively, we performed RNAi knock down experiments. At first, we used a previously described shRNA for DDR2 and confirmed that this shRNA potently knocked down DDR2 protein levels and strongly inhibited cell viability in NCI-H2286 and HCC-366 (Supporting Information Figure 7A–C). Western blotting also showed that expression of this shRNA was associated with p-SRC depletion, suggesting a scaffold effect of DDR2 on SRC. The potency of this shRNA was so great that experiments assessing the additional benefit of selective SFK inhibition were challenging to interpret.
To overcome this limitation, we used siRNAs targeting DDR2. We identified two independent siRNAs which decreased both DDR2 mRNA and protein, though to a degree less than what we observed with the shRNA used above (Supporting Information Figure 8A,B). siRNA depletion of DDR2 itself had little effect on cell growth, similar to what was observed with compound 1 and treatment with other selective DDR2 inhibitors. However, it increased the potency of selective SFK inhibition on DDR2 mutated cell lines (Supporting Information Figure 8C). Similarly, two independent siRNAs strongly depleted SRC mRNA and protein and decreased proliferation of NCI-H2286 and HCC-366 cells to a degree comparable to saracatinib treatment. The addition of compound 1 to SRC siRNA also led to more killing (Supporting Information Figure 8D). These results suggested that the kinase activity of SRC was dominant as compared to the kinase activity of DDR2 and also suggested a role for DDR2 itself in regulating SRC activity. Consistent with this, we observed that overexpression of DDR2, WT, or kinase dead (KD) in DDR2 mutated cells increased p-SRC levels (Supporting Information Figure 9).
Discussion
Here, we have presented biochemical and cellular characterization of DDR2 inhibitors (compounds 1–4) with improved potency and selectivity profiles as compared to currently available DDR2 inhibitors. These compounds inhibit p-DDR2 in a dose-dependent manner but have very modest potency as a single agent in DDR2 mutated lung cancer cells. Among them, compounds 1 and 3 are most potent and selective against DDR2 (Figure 1, 3), and they have the potential to serve as tools to interrogate the biological functions of DDR2 kinase activity in cellular assays. Compound 3 additionally displayed murine microsomal stability with a T1/2 of 21 min, whereas compound 1 was less stable with a T1/2 of 3.3 min, suggesting that it may be the most promising of these tool compounds for in vivo studies if its pharmacokinetic properties are acceptable.
We have observed that combination therapy with selective DDR2 inhibitors and the selective SFK inhibitor saracatinib demonstrates additive effects on DDR2 kinase activity and cell viability of DDR2 mutated cancer cells with the effects on SRC dominant to those on DDR2. Further, we have shown that inhibition of SRC or DDR2 increases the sensitivity of DDR2 mutated cell lines to selective DDR2 inhibition or selective SRC inhibition, respectively, suggesting a coordinated role of SRC and DDR2 in maintaining proliferation of DDR2 mutated lung cancer cell lines and present a dual SRC/DDR2 inhibitor, compound 5.
Small molecules targeting tumor specific genomic alterations in cancer can be not only more effective but also less toxic than cytotoxic chemotherapy. However, many targeted agents also display unique toxicities which limit their clinical application despite their efficacy.46 While some of the toxicities associated with targeted agents are due to inhibition of their intended target, such as rash and diarrhea with EGFR inhibitors, some toxicities can be limited by increasing the selectivity of targeted agents to avoid off-target toxicities.
Given that the kinase inhibitors reported to potently target DDR2, such as dasatinib, are known to inhibit a number of kinases with low nanomolar potency, we reasoned that a selective DDR2 inhibitor might retain the antitumor efficacy of dasatinib and reduce the toxicity associated with this agent, especially if the toxicities associated with dasatinib therapy were due to inhibition of targets other than DDR2. However, the data we report here do not suggest that selective DDR2 inhibition will be a fruitful therapeutic strategy and instead indicate that dual SRC/DDR2 inhibition is superior to inhibiting either kinase alone in DDR2 mutated lung cancer cell lines. In this way, these studies are similar to several recently reported cancer models in which combinations of SRC inhibitors and other selective kinase inhibitors appear superior to the use of the kinase inhibitor alone. Combined treatment with RAF inhibitors and SRC inhibitors has been shown to be superior to the use of selective RAF inhibitors in BRAFV600E positive thyroid cancer cells.47 Combinations of saracatinib and the MEK inhibitor selumetinib have also been reported to more effectively suppress the growth and invasion of melanoma cells as compared to either agent alone.48 Further, off-target effects of BTK inhibitors on SRC family kinases also appear to be important for the efficacy of this class of drugs.49,50 We feel that these studies, in combination with our data, suggest that there are likely to be many cancer contexts in which SRC family kinase inhibitors can potentiate the effects of selective kinase inhibitors or in which the use of less selective kinase inhibitors may be advantageous either for initial efficacy or to suppress adaptive resistance mechanisms.
Our data suggest that dual targeting of SRC and DDR2 may be more effective than selective inhibition of either kinase alone in DDR2 mutated lung cancer models and indicates that a combination of these two activities may be sufficient to target DDR2 mutated tumors. Such an approach could be better tolerated than the use of dasatinib and warrants further investigation with the compounds described here (e.g., compound 5) or with other selective DDR2 and SRC inhibitors. Given that some lung cancer patients with DDR2 mutations appear to benefit from dasatinib therapy, identifying treatment regimens which maintain efficacy but spare as much toxicity as possible will be important to develop and explore in appropriate models moving forward.
Methods
Enzymatic Assays
The enzymatic activities DDR2 were tested in LanthaScreen binding assays, the activities against SRC were tested in Z′-Lyte assays with ATP concentrations of Km (50 μM). All the protocols are available from Life Technologies. Z′-Lyte assays: http://www.lifetechnologies.com/content/dam/LifeTech/migration/files/drug-discovery/pdfs.par.60256.file.dat/20130430%20ssbk%20customer%20protocol%20and%20assay%20conditions.pdf. TTK assay: http://tools.lifetechnologies.com/content/sfs/manuals/TTK_LanthaScreen_Binding.pdf
Cell Culture and Proliferation Assays
Lung cancer cell lines (NCI-H2286, HCC-366, and A549) were obtained from the ATCC and maintained in RPMI 1640 (Life Technologies) plus 10% fetal calf serum (Gibco). Cell proliferation was measured with the Cell-Titer-Glo reagent (Promega) per the manufacturer’s instructions. Cells were plated in clear-bottomed 96-well plates at a density of 1500 cells per well. The drug was added the following day, and cell proliferation was measured 5 days later using a standard 96-well plate luminometer. Relative proliferation at a given drug concentration was determined by comparing the luminescence at the concentration to that of DMSO treated cells of the same cell type. The IC50 was defined as the drug concentration that induced 50% cell viability in comparison with DMSO control, which was calculated by nonlinear regression analysis (Prism, GraphPad 6.0). Eight different doses of drug were added for calculating IC50’s. All experiments were performed in triplicate independent assays.
Vectors
DDR2 retroviral expression vectors (p-Wzl-Blast-DDR2 wild-type (WT), p-Wzl-Blast-DDR2 T654I (gatekeeper mutant), p-Wzl-Blast-DDR2 K608E (kinase dead mutant), p-Wzl-Blast-DDR2 L239R, and p-Wzl-Blast-DDR2 I638F) were described previously.17 pCMV6-DDR2 was obtained from Origene. For SRC lentiviral expression vectors, wild-type pDONR223-SRC was obtained from Addgene. pDONR223-SRC T341M (gatekeeper mutant) was made by site-directed mutagenesis using a QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). pLX301-SRC WT and PLX301-SRC T341M lentiviral vectors were made from pDONR223-SRC WT and pDONR223–SRC T341M, respectively, by performing LR clonase reactions with the pLX301 destination vector (Life Technologies).
Reagents
Compounds 1, 2, and 6 were synthesized as previously described,35,36 and the structures and synthetic procedures of compounds 3, 4, and 5 are available in the Supporting Information. Dasatinib was obtained from LC laboratories. Saracatinib was purchased from Selleckchem.com.
Immunoprecipitation and Immunoblotting
Cellular lysates were prepared using RIPA buffer supplemented with protease and phosphatase inhibitors. The concentration of lysates was determined by Bradford assay (Bio-Rad). Immunoblotting was performed using the Nupage system (Life Technologies) with 100 μg of lysate. The primary antibodies used were DDR2 (Bethyl Laboratories), p-TYR clone 4G10 (Millipore), t-Src (Cell Signaling Technologies), p-Src family kinase (Tyr416; Cell Signaling Technologies), β-actin (Cell Signaling Technologies), and vinculin (Sigma). Immunoprecipitation was performed by incubating 1 mg of lysate with 5 μL of antibody for 1 h at 4 °C with shaking. Fifty microliters of protein A magnetic beads (Milipore) was then added, followed by a 2 h incubation at 4 °C with shaking. The beads were washed three times with RIPA buffer and resuspended in SDS sample buffer (Boston BioProducts). Immunoblotting was then performed using the Nupage system (Life Technologies).
Quantitative RT-PCR
Total cellular RNA was prepared from the cells by using an RNeasy Mini Kit (Qiagen), and 1.0 μg of the RNA was then reverse transcribed to cDNA using the High Capacity RNA to c-DNA kit (Life Technologies). For quantitative RT-PCR analysis, we used an ABI Prism 7300 Sequence Detection System (Life Technologies). Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization of input cDNA.
siRNA for DDR2 and SRC
NCI-H2286 and HCC-366 cell lines were transfected with DDR2 siRNA, SRC siRNA, or negative control siRNA (DDR2; #s9761 for DDR2 siRNA #1 and #s9762 for DDR2 siRNA #2, SRC; #s13413 for SRC siRNA #1 and #s13414 for SRC siRNA #2, control; silencer select negative control #2 for control siRNA). For Western-blotting, NCI-H2286 was transfected with a final concentration of 20 nM DDR2 siRNA or control siRNA; for the other experiments, cells were transfected with a final concentration of 5 nM DDR2 siRNA, SRC siRNA, or control siRNA. These concentrations were determined based on titration experiments to identify the lowest siRNA concentrations providing measurable target knock-down. Silencer select and negative control silencer select (Ambion) were used according to the manufacturer’s instructions. For transfection, Lipofectamine 2000 (Life Technologies) was used according to the manufacturer’s protocol. Knockdown of DDR2 and SRC expression was confirmed using quantitative reverse transcription-PCR and Western-blotting. For the viability assays, cells were seeded in 96-well plates at 1500 cells per well; the following day, cells were transfected with DDR2 siRNA #1–2, SRC siRNA #1–2, or control siRNA.
After 24 h of incubation, cells were treated with five different doses of compound 1 or saracatinib for an additional 96 h. Cell proliferation was measured using a standard 96-well plate luminometer. Relative proliferation at a given drug concentration was determined by comparing the luminescence of DMSO treated to siRNA transfected cells. All experiments were performed as independent triplicates.
shRNA for DDR2
shRNA vectors targeting DDR2 were originally obtained from the RNAi Consortium (TRCN0000121117) at the Broad Institute. The use of this vector was described previously.17 Briefly, lentiviral infections were performed with 293T cells transfected with the combination of 1 μg of pLKO plasmid, 100 ng of VSVG, and 900 ng of delta 8.9. Virus was collected and used to infect the lung cancer cell lines in the presence of Polybrene. Stable cell lines were generated using puromycin selection at a concentration of 2 μg/mL for NCI-H2286 and 4 μg/mL for HCC-366.
Acknowledgments
This work was supported in part by NCI P01 CA154303 (M.M. and N.S.G.) and K08 CA163677 (P.S.H.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00655.
Synthetic procedures of compounds 3, 4, and 5; supplementary figures; and kinase selectivity profiles of all compounds (PDF)
Author Present Address
∇ (Q.L.) High Magnetic Field Laboratory Chinese Academy of Sciences Hefei, Anhui, P.R. China
Author Contributions
# (H.T., L.T.) These authors contributed equally to this work.
The authors declare the following competing financial interest(s): M.M. and P.S.H. are patent holders on methods for treating patients with squamous cell lung cancers with DDR2 mutations (US 20130261017), and P.S.H. reports consulting fees from Molecular MD and support for clinical trials from Bristol-Myers Squibb. M.M. is a founder of and equity holder in Foundation Medicine.
Supplementary Material
References
- Lennes I. T.; Lynch T. J. (2009) Quality indicators in cancer care: development and implementation for improved health outcomes in non-small-cell lung cancer. Clin. Lung Cancer 10, 341–346 10.3816/CLC.2009.n.046. [DOI] [PubMed] [Google Scholar]
- West H.; Harpole D.; Travis W. (2009) Histologic considerations for individualized systemic therapy approaches for the management of non-small cell lung cancer. Chest 136, 1112–1118 10.1378/chest.08-2484. [DOI] [PubMed] [Google Scholar]
- Pao W.; Miller V.; Zakowski M.; Doherty J.; Politi K.; Sarkaria I.; Singh B.; Heelan R.; Rusch V.; Fulton L.; Mardis E.; Kupfer D.; Wilson R.; Kris M.; Varmus H. (2004) EGF receptor gene mutations are common in lung cancers from ″never smokers″ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. U. S. A. 101, 13306–13311 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez J. G.; Janne P. A.; Lee J. C.; Tracy S.; Greulich H.; Gabriel S.; Herman P.; Kaye F. J.; Lindeman N.; Boggon T. J.; Naoki K.; Sasaki H.; Fujii Y.; Eck M. J.; Sellers W. R.; Johnson B. E.; Meyerson M. (2004) EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 304, 1497–1500 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Lynch T. J.; Bell D. W.; Sordella R.; Gurubhagavatula S.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Haserlat S. M.; Supko J. G.; Haluska F. G.; Louis D. N.; Christiani D. C.; Settleman J.; Haber D. A. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Soda M.; Choi Y. L.; Enomoto M.; Takada S.; Yamashita Y.; Ishikawa S.; Fujiwara S.; Watanabe H.; Kurashina K.; Hatanaka H.; Bando M.; Ohno S.; Ishikawa Y.; Aburatani H.; Niki T.; Sohara Y.; Sugiyama Y.; Mano H. (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Shaw A. T.; Yeap B. Y.; Solomon B. J.; Riely G. J.; Gainor J.; Engelman J. A.; Shapiro G. I.; Costa D. B.; Ou S. H.; Butaney M.; Salgia R.; Maki R. G.; Varella-Garcia M.; Doebele R. C.; Bang Y. J.; Kulig K.; Selaru P.; Tang Y.; Wilner K. D.; Kwak E. L.; Clark J. W.; Iafrate A. J.; Camidge D. R. (2011) Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 12, 1004–1012 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R. G.; Watanabe H.; Meyerson M.; Hammerman P. S. (2012) Targeted therapy for squamous cell lung cancer. Lung Cancer Manage. 1, 293–300 10.2217/lmt.12.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A.; Rekhtman N.; Ladanyi M.; Paik P. (2012) Squamous-cell carcinomas of the lung: emerging biology, controversies, and the promise of targeted therapy. Lancet Oncol. 13, e418–e426 10.1016/S1470-2045(12)70291-7. [DOI] [PubMed] [Google Scholar]
- (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J.; Sos M. L.; Seidel D.; Peifer M.; Zander T.; Heuckmann J. M.; Ullrich R. T.; Menon R.; Maier S.; Soltermann A.; Moch H.; Wagener P.; Fischer F.; Heynck S.; Koker M.; Schottle J.; Leenders F.; Gabler F.; Dabow I.; Querings S.; Heukamp L. C.; Balke-Want H.; Ansen S.; Rauh D.; Baessmann I.; Altmuller J.; Wainer Z.; Conron M.; Wright G.; Russell P.; Solomon B.; Brambilla E.; Brambilla C.; Lorimier P.; Sollberg S.; Brustugun O. T.; Engel-Riedel W.; Ludwig C.; Petersen I.; Sanger J.; Clement J.; Groen H.; Timens W.; Sietsma H.; Thunnissen E.; Smit E.; Heideman D.; Cappuzzo F.; Ligorio C.; Damiani S.; Hallek M.; Beroukhim R.; Pao W.; Klebl B.; Baumann M.; Buettner R.; Ernestus K.; Stoelben E.; Wolf J.; Nurnberg P.; Perner S.; Thomas R. K. (2010) Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2, 62ra93. 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutt A.; Ramos A. H.; Hammerman P. S.; Mermel C.; Cho J.; Sharifnia T.; Chande A.; Tanaka K. E.; Stransky N.; Greulich H.; Gray N. S.; Meyerson M. (2011) Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One 6, e20351. 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R. G.; Jung J.; Tchaicha J.; Wilkerson M. D.; Sivachenko A.; Beauchamp E. M.; Liu Q.; Pugh T. J.; Pedamallu C. S.; Hayes D. N.; Gray N. S.; Getz G.; Wong K. K.; Haddad R. I.; Meyerson M.; Hammerman P. S. (2013) Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 73, 5195–5205 10.1158/0008-5472.CAN-12-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski I. J.; Mittempergher L.; Davidson N. M.; Bosma A.; Willems S. M.; Horlings H. M.; de Rink I.; Greger L.; Hooijer G. K.; Peters D.; Nederlof P. M.; Hofland I.; de Jong J.; Wesseling J.; Kluin R. J.; Brugman W.; Kerkhoven R.; Nieboer F.; Roepman P.; Broeks A.; Muley T. R.; Jassem J.; Niklinski J.; van Zandwijk N.; Brazma A.; Oshlack A.; van den Heuvel M.; Bernards R. (2013) Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred RNA sequencing. Journal of pathology 230, 270–276 10.1002/path.4209. [DOI] [PubMed] [Google Scholar]
- Wu Y. M.; Su F.; Kalyana-Sundaram S.; Khazanov N.; Ateeq B.; Cao X.; Lonigro R. J.; Vats P.; Wang R.; Lin S. F.; Cheng A. J.; Kunju L. P.; Siddiqui J.; Tomlins S. A.; Wyngaard P.; Sadis S.; Roychowdhury S.; Hussain M. H.; Feng F. Y.; Zalupski M. M.; Talpaz M.; Pienta K. J.; Rhodes D. R.; Robinson D. R.; Chinnaiyan A. M. (2013) Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discovery 3, 636–647 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H.; Shigematsu H.; Nomura M.; Lockwood W. W.; Sato M.; Okumura N.; Soh J.; Suzuki M.; Wistuba II; Fong K. M.; Lee H.; Toyooka S.; Date H.; Lam W. L.; Minna J. D.; Gazdar A. F. (2008) PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 68, 6913–6921 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman P. S.; Sos M. L.; Ramos A. H.; Xu C.; Dutt A.; Zhou W.; Brace L. E.; Woods B. A.; Lin W.; Zhang J.; Deng X.; Lim S. M.; Heynck S.; Peifer M.; Simard J. R.; Lawrence M. S.; Onofrio R. C.; Salvesen H. B.; Seidel D.; Zander T.; Heuckmann J. M.; Soltermann A.; Moch H.; Koker M.; Leenders F.; Gabler F.; Querings S.; Ansen S.; Brambilla E.; Brambilla C.; Lorimier P.; Brustugun O. T.; Helland A.; Petersen I.; Clement J. H.; Groen H.; Timens W.; Sietsma H.; Stoelben E.; Wolf J.; Beer D. G.; Tsao M. S.; Hanna M.; Hatton C.; Eck M. J.; Janne P. A.; Johnson B. E.; Winckler W.; Greulich H.; Bass A. J.; Cho J.; Rauh D.; Gray N. S.; Wong K. K.; Haura E. B.; Thomas R. K.; Meyerson M. (2011) Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discovery 1, 78–89 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haura E. B.; Tanvetyanon T.; Chiappori A.; Williams C.; Simon G.; Antonia S.; Gray J.; Litschauer S.; Tetteh L.; Neuger A.; Song L.; Rawal B.; Schell M. J.; Bepler G. (2010) Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J. Clin. Oncol. 28, 1387–1394 10.1200/JCO.2009.25.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen B.; Peng S.; Tang X.; Erickson H. S.; Galindo H.; Mazumdar T.; Stewart D. J.; Wistuba I.; Johnson F. M. (2012) Kinase-impaired BRAF mutations in lung cancer confer sensitivity to dasatinib. Sci. Transl. Med. 4, 136ra70. 10.1126/scitranslmed.3003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R.; Rodon J.; Prat A.; Perez-Garcia J.; Adamo B.; Felip E.; Cortes J.; Iafrate A. J.; Nuciforo P.; Tabernero J. (2014) Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO 25, 552–563 10.1093/annonc/mdt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J.; LoRusso P. M.; Camidge R. D.; Perez J. M.; Tabernero J.; Hidalgo M.; Schuler M.; Tian G. G.; Soria J. C.; Delord J. P.; Campone M.; Bachelot T.; van der Noll R.; Ringeisen F. P.; Nogova L.; Sequist L. V.; Schellens J. H. M. (2012) Abstract LB-122: A phase I dose escalation study of NVP-BGJ398, a selective pan FGFR inhibitor in genetically preselected advanced solid tumors. Cancer Res. 72, LB-122–LB-122 10.1158/1538-7445.AM2012-LB-122. [DOI] [Google Scholar]
- Valiathan R. R.; Marco M.; Leitinger B.; Kleer C. G.; Fridman R. (2012) Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 31, 295–321 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E.; Gao J.; Dogrusoz U.; Gross B. E.; Sumer S. O.; Aksoy B. A.; Jacobsen A.; Byrne C. J.; Heuer M. L.; Larsson E.; Antipin Y.; Reva B.; Goldberg A. P.; Sander C.; Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery 2, 401–404 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Aksoy B. A.; Dogrusoz U.; Dresdner G.; Gross B.; Sumer S. O.; Sun Y.; Jacobsen A.; Sinha R.; Larsson E.; Cerami E.; Sander C.; Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signaling 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitinger B. (2003) Molecular analysis of collagen binding by the human discoidin domain receptors, DDR1 and DDR2. Identification of collagen binding sites in DDR2. J. Biol. Chem. 278, 16761–16769 10.1074/jbc.M301370200. [DOI] [PubMed] [Google Scholar]
- Leitinger B. (2014) Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 310, 39–87 10.1016/B978-0-12-800180-6.00002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrador J. P.; Azcoitia V.; Tuckermann J.; Lin C.; Olaso E.; Manes S.; Bruckner K.; Goergen J. L.; Lemke G.; Yancopoulos G.; Angel P.; Martinez C.; Klein R. (2001) The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. 2, 446–452 10.1093/embo-reports/kve094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaso E.; Labrador J. P.; Wang L.; Ikeda K.; Eng F. J.; Klein R.; Lovett D. H.; Lin H. C.; Friedman S. L. (2002) Discoidin domain receptor 2 regulates fibroblast proliferation and migration through the extracellular matrix in association with transcriptional activation of matrix metalloproteinase-2. J. Biol. Chem. 277, 3606–3613 10.1074/jbc.M107571200. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Corsa C. A.; Ponik S. M.; Prior J. L.; Piwnica-Worms D.; Eliceiri K. W.; Keely P. J.; Longmore G. D. (2013) The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 15, 677–687 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day E.; Waters B.; Spiegel K.; Alnadaf T.; Manley P. W.; Buchdunger E.; Walker C.; Jarai G. (2008) Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 599, 44–53 10.1016/j.ejphar.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lu X.; Ren X.; Ding K. (2015) Small Molecule Discoidin Domain Receptor Kinase Inhibitors and Potential Medical Applications. J. Med. Chem. 58, 3287. 10.1021/jm5012319. [DOI] [PubMed] [Google Scholar]
- Richters A.; Nguyen H. D.; Phan T.; Simard J. R.; Grutter C.; Engel J.; Rauh D. (2014) Identification of type II and III DDR2 inhibitors. J. Med. Chem. 57, 4252–4262 10.1021/jm500167q. [DOI] [PubMed] [Google Scholar]
- Brunner A. M.; Costa D. B.; Heist R. S.; Garcia E.; Lindeman N. I.; Sholl L. M.; Oxnard G. R.; Johnson B. E.; Hammerman P. S. (2013) Treatment-related toxicities in a phase II trial of dasatinib in patients with squamous cell carcinoma of the lung. J. Thorac. Oncol. 8, 1434–1437 10.1097/JTO.0b013e3182a47162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitini V.; Arrigo C.; Di Mirto C.; Mondello P.; Altavilla G. (2013) Response to dasatinib in a patient with SQCC of the lung harboring a discoid-receptor-2 and synchronous chronic myelogenous leukemia. Lung Cancer 82, 171–172 10.1016/j.lungcan.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Choi Y.; Syeda F.; Walker J. R.; Finerty P. J. Jr.; Cuerrier D.; Wojciechowski A.; Liu Q.; Dhe-Paganon S.; Gray N. S. (2009) Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 19, 4467–4470 10.1016/j.bmcl.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning P.; Tan L.; Chu K.; Lee S. W.; Gray N. S.; Bullock A. N. (2014) Structural mechanisms determining inhibition of the collagen receptor DDR1 by selective and multi-targeted type II kinase inhibitors. J. Mol. Biol. 426, 2457–2470 10.1016/j.jmb.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Gray N. S. (2006) Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358–364 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Wu H.; Wang L.; Liu Y.; Knapp S.; Liu Q.; Gray N. S. (2014) Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery?. ACS Chem. Biol. 9, 1230–1241 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Lebakken C. S.; Riddle S. M.; Singh U.; Frazee W. J.; Eliason H. C.; Gao Y.; Reichling L. J.; Marks B. D.; Vogel K. W. (2009) Development and applications of a broad-coverage, TR-FRET-based kinase binding assay platform. J. Biomol. Screening 14, 924–935 10.1177/1087057109339207. [DOI] [PubMed] [Google Scholar]
- Ikeda K.; Wang L. H.; Torres R.; Zhao H.; Olaso E.; Eng F. J.; Labrador P.; Klein R.; Lovett D.; Yancopoulos G. D.; Friedman S. L.; Lin H. C. (2002) Discoidin domain receptor 2 interacts with Src and Shc following its activation by type I collagen. J. Biol. Chem. 277, 19206–19212 10.1074/jbc.M201078200. [DOI] [PubMed] [Google Scholar]
- Green T. P.; Fennell M.; Whittaker R.; Curwen J.; Jacobs V.; Allen J.; Logie A.; Hargreaves J.; Hickinson D. M.; Wilkinson R. W.; Elvin P.; Boyer B.; Carragher N.; Ple P. A.; Bermingham A.; Holdgate G. A.; Ward W. H.; Hennequin L. F.; Davies B. R.; Costello G. F. (2009) Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol. Oncol. 3, 248–261 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare T.; Walters D. K.; Stoffregen E. P.; Jia T.; Manley P. W.; Mestan J.; Cowan-Jacob S. W.; Lee F. Y.; Heinrich M. C.; Deininger M. W.; Druker B. J. (2005) In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 65, 4500–4505 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- Liu S.; Kurzrock R. (2014) Toxicity of targeted therapy: Implications for response and impact of genetic polymorphisms. Cancer Treat. Rev. 40, 883–891 10.1016/j.ctrv.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Vanden Borre P.; Gunda V.; McFadden D. G.; Sadow P. M.; Varmeh S.; Bernasconi M.; Parangi S. (2014) Combined BRAF(V600E)- and SRC-inhibition induces apoptosis, evokes an immune response and reduces tumor growth in an immunocompetent orthotopic mouse model of anaplastic thyroid cancer. Oncotarget 5, 3996–4010 10.18632/oncotarget.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson J.; Arozarena I.; Ehrhardt M.; Wellbrock C. (2013) Combination of MEK and SRC inhibition suppresses melanoma cell growth and invasion. Oncogene 32, 86–96 10.1038/onc.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. (2010) The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U. S. A. 107, 13075–13080 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Wang W.; Liu F.; Weisberg E. L.; Tian B.; Chen Y.; Li B.; Wang A.; Wang B.; Zhao Z.; McMillin D. W.; Hu C.; Li H.; Wang J.; Liang Y.; Buhrlage S. J.; Liang J.; Liu J.; Yang G.; Brown J. R.; Treon S. P.; Mitsiades C. S.; Griffin J. D.; Liu Q.; Gray N. S. (2014) Discovery of a potent, covalent BTK inhibitor for B-cell lymphoma. ACS Chem. Biol. 9, 1086–1091 10.1021/cb4008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.