

Biological processes are exquisitely well controlled on a spatial and temporal scale, and this has driven interest in drug delivery devices that can be altered on-demand to adapt the release profile in real time. However, systems designed to release payload in response to extracorporeal or environmental cues typically exhibit considerable leakiness in drug release. We hypothesized that a more complete On/Off switch could be achieved with physical entrapment of nanoparticles within hydrogels, exploiting steric hindrance to reduce baseline release, and that the microarchitecture of the system could be reversibly adapted using ultrasound to enable switchable release. To test this, the release of PEGylated gold-nanoparticles from ionically crosslinked alginate hydrogels was first examined and demonstrated a dramatic increase in release rate in response to ultrasound. Bone morphogenetic protein-2 (BMP-2) conjugated gold nanoparticles could also be released from hydrogels with ultrasound, and maintained bioactivity following alginate encapsulation and ultrasound release. This approach to increasing control over local bioagent delivery should afford researchers and clinicians the ability to mimic and drive natural temporal responses.
Natural biological processes (e.g., embryological development, bone generation and angiogenesis) are intricately controlled in the temporal and spatial domain, and systems that enable this type of signaling control could provide powerful research and clinical tools. One successful strategy to obtain spatial control is polymer-based drug delivery, as these allow local delivery at a specific anatomic site. These delivery systems are engineered to exhibit temporal control by sustaining release of bioagents over a defined period.[1] Despite the success and clinical translation of some of these strategies, the advantages of more precise release initiation or intermittent release profiles is becoming clear both in pathologic[2, 3] and tissue engineering applications.[4] In addition, the majority of monolithic polymeric systems exhibit an initial burst release.[5] A high initial drug concentration may be undesirable, and may also be wasteful as this coincides with the timing of the initial inflammatory response – a potentially harsh environment. Systems that can be instructed to deliver their payload on-demand are favorable in many situations. For example, delayed delivery of BMP-2 can enhance fracture healing, when compared with immediate delivery.[6] Furthermore, increased control of a delivery system may allow a reduction in the bioagent payload, which could improve safety while reducing cost.
Drug delivery devices can alter the drug release rate by taking information from their environment (e.g., temperature, pH)[7] or from non-invasive, externally modulated energy sources such as heat[8], magnetic[9], electrical[10], light[11] or by wirelessly communicating with implanted microchips.[12] Ultrasound, which is commonly employed in the clinic for diagnostic and therapeutic purposes, has previously been demonstrated to accelerate release of bioactive agents from biomaterials.[13, 14] These systems typically alter their structure permanently (i.e., ultrasound destruction of the material), which results in a more permanent increase in release rate. However, inspired by sonophoresis[15], self-healing ionically crosslinked alginate hydrogels that return to a baseline release rate following the removal of the ultrasound stimulus were recently demonstrated.[2] A common limitation of all these systems is that, similar to most polymeric controlled drug delivery strategies, there can be relatively high baseline release rate from the material. There are many reports of responsive nanoparticles[16] that can respond to stimuli such as those listed above or that are embedded within matrices to effect a change on the matrix, which in turn releases a drug payload; however, we are unaware of reports that specifically deliver bioactive nanoparticles in response to a stimulus.
This project was based on the hypothesis that incorporation of nanoparticles into an ultrasound responsive hydrogel would largely eliminate baseline release due to steric hindrance, and that release of the nanoparticles could be triggered in response to ultrasound. The pore size of alginate hydrogels is typically in the range of several nm[17], which was expected to lead to physical entrapment of nanoparticles larger than 10 nm. This system can additionally exploit the favorable physicochemical properties of nanoparticles, including their ability to co-deliver agents and their ability to enhance bioactivity.[18, 19] This approach could also overcome the challenge of localizing nanoparticles at defect sites, as the hydrogel depot can be physically placed in the desired anatomic location. The first aim of the study was to explore, using a model nanoparticle, the release rate of gold nanoparticles (AuNPs) in response to ultrasound. Next, BMP-2 was selected as a model therapeutic due to its clinical use and prior demonstrations of its enhanced efficacy when delivered in a delayed manner in a femoral fracture critical sized defect model in rats.[6] BMP-2 was conjugated to the gold nanoparticles and the ability of these particles to be released from the hydrogels in response to ultrasound, in a bioactive form, was analyzed in vitro.
Release of PEG-AuNPs from hydrogels
Gold nanoparticles were prepared at four different sizes as previously described [20] and demonstrated a tight monophasic size distribution, with hydrodynamic diameters of: seed = 19 nm; ‘30 nm’ = 28 nm; ‘60 nm’ = 68 nm; and ‘100 nm’ = 99 nm (polydispersity index <0.1; Figure 1). As these particles are not stable in ionic media[21], they were initially coupled with 5 kDa poly(ethylene glycol) via thiol end groups on the PEG; this increased the hydrodynamic radius of particles by ~20 nm. The absorption spectra were recorded for each particle as a function of concentration and time to confirm the stability of the resultant particles and that there was a linear relationship between concentration and absorbance at 518 nm within the ranges used in the study (see methods in Supporting Information and Figure S1 for more information). Following mixing with 2% w v−1 alginate, gels were ionically crosslinked with calcium ions. The resultant gels were homogenously red in color suggesting an even distribution of nanoparticles throughout the gels (Figure 1b). Overnight, these gels released <1% of their nanoparticle payload (Figure S2). As the zeta potential of gold nanoparticles are negative-to-neutral with PEG [22], no interaction between the nanoparticles and negatively charged alginate was anticipated; the very low baseline release was attributed to steric hindrance alone. Next, these alginate discs were stimulated with ultrasound (2.5 min every hr, for 5 hr, at 9.6mW cm−2) and demonstrated a 6-fold increase in cumulative release over the 5 hrs when compared with diffusion only controls. The release rate during the 2.5 min ultrasound period was accelerated 110-fold. The gels showed only a moderate, and non-significant effect of nanoparticle size on release rate (Figure 1C) and remained macroscopically intact.
Figure 1.

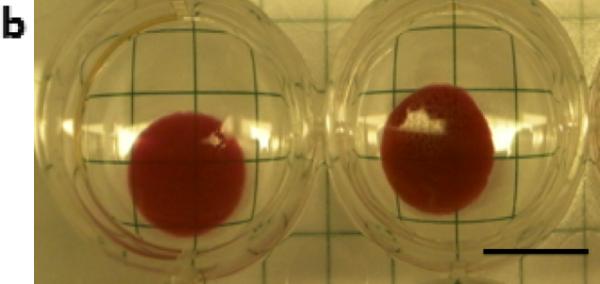

Ultrasound releases pegylated gold nanoparticles on-demand. (a) Dynamic light scattering measurements of hydrodynamic diameters of gold nanoparticles (AuNPs) following fabrication. (b) Alginate hydrogel discs showed a characteristic red color when loaded with pegylated AuNPs (PEG-AuNPs; scalebar = 1cm). (c) Absorbance spectroscopy was used to evaluate the concentration of PEG-AuNPs in the media following ultrasound stimulated release (control = 0 mW cm−2 ; US = 9.6 mW cm−2 ; 2.5 mins per hr; 5 hrs total) from ionically crosslinked alginate hydrogels. *** = p<0.001.
We next hypothesized that increasing the surface-to-volume ratio of the hydrogels would increase the release rate with ultrasound, as individual nanoparticles would have a shorter distance to navigate prior to being freed from the gel. Calcium crosslinked alginate microbeads (d ~ 250 μm) containing PEG-AuNP were prepared using a nebulizer[23], and following overnight diffusion (<1% release), microbeads were treated with ultrasound. These gels demonstrated macroscopically visible release, with the microbeads remaining intact following ultrasound stimulation (Figure 2). The beads released ~10-fold greater amount of PEG-AuNPs following ultrasound when compared with diffusion over the duration of the study, and the release rate during US stimulation increased ~200-fold over the diffusion-only rate (Figure 2c). TEM was used to image the release medium to determine whether individual AuNPs were being released or whether the gels were fracturing. Released nanoparticles were generally in isolation (Figure 2b) with no indication of gel fragments in the medium, suggesting that the nanoparticles were being ‘freed’ from the alginate gel in response to ultrasound.
Figure 2.
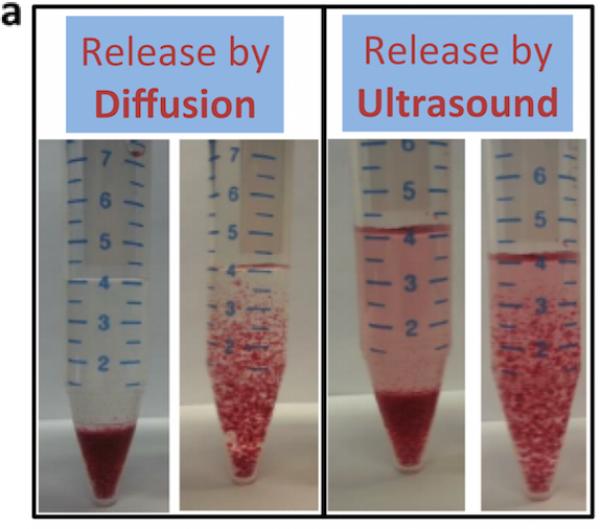



Increasing the surface-to-volume ratio increases the ultrasound stimulated release rate of the AuNPs. Alginate hydrogel microbeads loaded with PEG-AuNPs did not release significant NPs until stimulated with ultrasound (Left images = microbeads settled to bottom of container; Right images = microbeads perturbed and in suspension). (b) TEM images of media containing released nanoparticles (scalebar 250 nm). (c) Cumulative release of peg-AuNPs from alginate microbeads during a 5hr period (control = 0 mW cm−2 ; US = 9.6 mW cm−2 ; 2.5 mins per hr) following overnight storage at 37°C. (d) Cumulative release of PEG-AuNPs over 5 days of storage (‘5-day diffusion’) vs. cumulative release during a 5hr period in response to ultrasound treatment on day 5 (US = 9.6 mW cm−2; 2.5 mins per hr). ** = p<0.01; *** = p<0.001.
Studies were then performed to confirm that the microbeads were capable of holding the AuNPs for longer periods before release, while still maintaining their ability to respond to ultrasound. Incubation for 5 days led to 1- 4% of the nanoparticles being released by diffusion (Figure 2d). When these samples were treated with ultrasound after 5 days, there was approximately a 5,000-fold increase in release rate during the time of ultrasound application, and a ~10-fold increase in the cumulative amount released over the entire time.
Fabrication and testing of bioactive nanoparticles
Next, bioactive nanoparticles were fabricated using BMP-2. Gold nanoparticles can bind thiol residues found in cysteine groups, and this does not typically adversely affect the factor's bioactivity.[21, 24] The structure of BMP-2 reveals that it contains 7 cysteine groups (Figure 3). BMP-2 conjugation to gold seed (19 nm) and 30nm particles was tested (Figure S3), and 19nm seed AuNPs (zeta potential, ζ = −22.8 mV; Figure 3a) were then used for all further studies as the end hydrodynamic radius (~150 nm; ζ = −24.1 mV) more closely matched the PEG-AuNPs. These particles bound the BMP-2 at an efficiency of 49% (final concentration of 1.01μg BMP-2/mg AuNPs) and remained stable and did not aggregate when added to culture media. To analyze bioactivity, free BMP-2 and BMP-2 that had been conjugated to nanoparticles was added to cell culture medium to examine their osteogenic potential. Unconjugated BMP-2 demonstrated increased osteogenesis (as measured by Alkaline Phosphatase (ALP) activity) over DMEM (negative control) and osteogenic media (positive controls) in the range of 10 – 500ng/ml (statistically significant at BMP-2 = 500 ng ml−1; Figure 3c, d). PEG-AuNPs alone (added at an equimolar concentration as the highest BMP-AuNPs concentration tested, 2.15 nM) showed a slight – yet non-significant – increase in osteogenic activity over controls. However, when 100 ng ml−1 BMP-2 conjugated to AuNP was added to the media (2.15 nM AuNPs), measured ALP levels were equivalent to the maximum dose of free BMP-2 tested (500 ng ml−1; p = 1.0).
Figure 3.

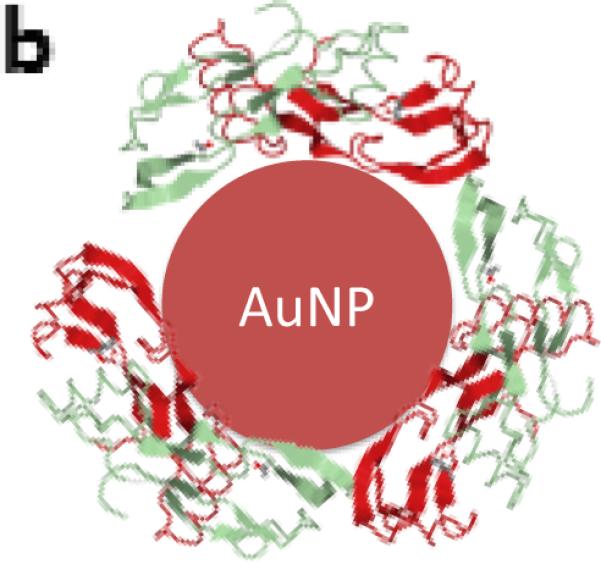


Recombinant human-bone morphogenetic protein-2 (BMP-2) can be conjugated to AuNPs using thiol groups and these BMP-2–AuNPs are biologically active. (a) BMP-2 protein and its amino acid sequence sequence, with the cysteine residues highlighted (Image from: Research Collaboratory for Structurual Bioinformatics Protein Data Bank, www.rcsb.org). (b) Thiol bonds can be used to covalently conjugate the BMP-2 to the AuNPs (final concentration following conjugation = 1.01 μg BMP-2/mg AuNPs; see methods in Supporting Information for details). (c) Unconjugated protein (10, 100, 300 ng ml−1) or AuNP-conjugated protein (10, 100 ng BMP-2 ml−1) were added to medium over mouse MSCs (D1 cells) and osteogenic activity was analyzed via alkaline phosphatase staining (blue color) on day 7. DMEM only (no protein or nanoparticles), osteo media (DMEM supplemented with l-ascorbic acid and β-glycerophosphate) and PEG-AuNPs supplemented media, were used as controls. (d) Quantification of alkaline phosphatase (ALP) staining for control groups, unconjugated BMP-2 (10, 100, 300, 500 ng ml−1) and AuNPs (PEG and BMP-2; BMP-2 concentrations of 10 and 100ng ml−1). ** = P<0.01.
Testing of on-demand released BMP-2–AuNPs for maintained bioactivity
Finally, studies were performed to examine whether BMP-2–AuNPs can be loaded into the alginate microbeads, stimulated with ultrasound and maintain their bioactivity (Figure 4). Exposure to ultrasound did not affect the hydrodynamic radius or zeta potential of unencapsulated particles (Figure 4b). Following incorporation of BMP-2–AuNPs into alginate microparticles and subsequent stimulation with ultrasound, the resulting supernatant was added to mMSCs. The released BMP-2–AuNPs led to a two-fold increase in ALP activity over osteogenic media controls (Figure 4c), confirming the bioactivity of the NPs following alginate loading and US-stimulated release. The supernatant from diffusion-released AuNPs had no impact on ALP activity, as expected (Figure 4c).
Figure 4.



BMP-2–AuNPs released from alginate gels in response to US are biologically active. (a) Schematic overview of approach: US is used to release BMP-2–AuNPs from the alginate microbeads under sterile conditions and these are subsequently added to D1 cell cultures; medium from diffusion only release is also tested. Alkaline Phosphatase activity at day 7 is used to analyze maintained bioactivity. (b) Dynamic light scattering measurements of hydrodynamic diameters of bare gold nanoparticles (‘Seed AuNPs’) following fabrication and following conjugation with BMP-2 (‘BMP-2–AuNPs, 0 US’). These particles were then treated with ultrasound (9.6 mW cm−2) for one (1 × 2.5 min) or two (2 × 2.5 min) rounds of ultrasound to test whether this stripped the BMP-2 from the AuNPs. (c) Quantification of alkaline phosphatase (ALP) staining for diffusion only and US-released (US = 9.6 mW cm−2; 2.5 mins per hr for 10hrs) AuNPs from alginate beads. ** = P<0.015.
The results of these studies demonstrate that nanoparticles physically entrapped in alginate have a low basal release rate that can be dramatically increased when triggered by ultrasound. Growth-factor conjugated AuNPs released via ultrasound maintain their bioactivity, as demonstrated by maintenance of osteogenic activity. Taken together, these results suggest the potential of this system to provide burst release of drugs from a depot on-demand, over multiple days.
A striking increase in release rate with ultrasound was achieved in this study. The baseline release rate of nanoparticles from the gels used in these studies was near-zero, leading to the ratio of US-stimulated release rate: non-US release reaching ~200-fold when stimulated at 24 hrs and ~5,000-fold after waiting 5 days. A previous demonstration of protein and small molecule release from ultrasound-responsive alginate gels, in contrast, demonstrated only ~10-fold increase in release rate during ultrasound.[2] In the previous work, the affinity-based interactions between the hydrogel and bioactive agents resulted in a much higher baseline release rate, diminishing the impact of US release. Although detailed mechanistic studies were not performed here, it is possible that the increased relative release rate at 5 days was caused by a replacement of the divalent ions crosslinking the alginate with monovalent ions from the surrounding media, or by relaxation of stresses imposed on the gels by inclusion of the nanoparticles.[25] The release rate can also be controlled by increasing gel surface-to-volume ratio, which as shown here does not affect diffusion-only release; increasing the ultrasound duration; or increasing the number of repeat cycles.[2, 14] In this study, ultrasound treatments were limited to 2.5 mins to maintain temperatures below 37 °C. These studies utilized a nanoparticle whose size could be readily controlled and whose electrostatic interactions with anionic alginate are minimized due to their negative charge and PEG coating [26]. Therapeutic nanoparticles are likely to have charge interactions with alginate that retard release, on top of the steric hindrance, and it will likely be necessary to evaluate this impact for each type of nanoparticle used in the system.
Gold nanoparticles were used for the bioactivity study due to the large volume of research on their biological effects, their clinical potential and use in clinical trials, and their ability to covalently conjugate proteins.[18, 21, 24] BMP-2 was chosen as a model drug molecule due to its clinical use, clinically demonstrated requirement for spatial and temporal control [27], and previous demonstrations of enhanced efficacy when it is released in a delayed manner.[6] Herein, BMP-2 was successfully conjugated to gold nanoparticles and this enhanced its bioactivity. This effect of nanoparticle presentation has not been demonstrated for BMP-2 previously to our knowledge, but has been shown for other factors.[19, 28] Previous investigators have attached BMP-2 molecules to much larger particles (~600 nm, e.g., [29]) and saw an increase in hydrodynamic radius that was consistent with the unconjugated molecule (~10 nm[30]). The observed larger shift in hydrodynamic radius here is consistent with other authors that have conjugated proteins to similar size AuNPs via thiol bonds (e.g., [24]) and is possibly due to biomolecule aggregation on the AuNP surface. A non-significant enhancement of PEG-AuNPs on osteogenesis was also observed. Previous authors have demonstrated the potential of bare AuNPs to stimulate osteogenic differentiation of MSCs through binding with cytoplasmic proteins.[31] However, it is unclear whether the addition of PEG to the nanoparticles in the present study inhibited this mechanism.
This work serves as a proof-of-principle study to demonstrate a novel approach for on-demand delivery of nanotherapeutics. Although the nanoparticle field has made dramatic advances in targeting and homing of nanoparticles, avoiding the reticulo-endothelial system, and extending circulation time, these still remain significant challenges for the field.[18] This work may offer an alternative approach for delivering nanotherapeutics locally in injectable microbeads and, as demonstrated herein, allow for their triggered release in a precise fashion. As the settings and materials utilized in this study are consistent with a previous report [2] that was shown to be efficacious in vitro and in vivo, it is expected this system should be translatable to in vivo applications. Furthermore, the materials and ultrasound levels used in this system have been previously explored for clinical use or are currently being used as part of clinical products. Future work will fully explore the in vivo efficacy of the system, its safety profile and a wider range of nanoparticles and bioactive agents. This platform for on-demand delivery of bioactive nanoparticles is expected to provide an exciting tool for both researchers and clinicians to further explore the importance of temporal co-ordination of factor delivery.
Supplementary Material
Acknowledgements
The authors acknowledge Steven Perrault, Wyss Institute for Biologically Inspired Engineering for nanoparticle advice. For funding we thank NIH (R01 DE019917); CJK acknowledges RCSI's Office of Research and Innovation Seed Fund Award (Grant Number GR 14-0963) and Science Foundation Ireland (SFI) under Grant Number SFI/12/RC/2278; HS acknowledges the Fulbright Program; SMK acknowledges his University of Rhode Island College of Engineering startup funds and an RI-INBRE Proposal Development Award (NIH/NIBMS 2P20GM103430).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Cathal J. Kearney, Harvard University School of Engineering and Applied Sciences and Wyss Institute for Biologically Inspired Engineering, 40 Oxford St., Cambridge, MA 02138, USA Department of Anatomy, Tissue Engineering Research Group and Advanced Materials and Bioengineering Research Center, Royal College of Surgeons in Ireland, 123 St. Stephen's Green, Dublin, Ireland.
Dr. Hadas Skaat, Harvard University School of Engineering and Applied Sciences and Wyss Institute for Biologically Inspired Engineering, 40 Oxford St., Cambridge, MA 02138, USA
Prof. Stephen M. Kennedy, Harvard University School of Engineering and Applied Sciences and Wyss Institute for Biologically Inspired Engineering, 40 Oxford St., Cambridge, MA 02138, USA Department of Electrical, Computer and Biomedical Engineering & Department of Chemical Engineering, The University of Rhode Island, 45 Upper College Rd, Kingston, RI 02881, USA.
Jennifer Hu, Harvard University School of Engineering and Applied Sciences and Wyss Institute for Biologically Inspired Engineering, 40 Oxford St., Cambridge, MA 02138, USA.
Max Darnell, Harvard University School of Engineering and Applied Sciences and Wyss Institute for Biologically Inspired Engineering, 40 Oxford St., Cambridge, MA 02138, USA.
Theresa M. Raimondo, Harvard University School of Engineering and Applied Sciences and Wyss Institute for Biologically Inspired Engineering, 40 Oxford St., Cambridge, MA 02138, USA
Prof. David J. Mooney, Harvard University School of Engineering and Applied Sciences and Wyss Institute for Biologically Inspired Engineering, 40 Oxford St., Cambridge, MA 02138, USA.
References
- 1.Kearney CJ, Mooney DJ. Nat. Mat. 2013;12:1004. doi: 10.1038/nmat3758. [DOI] [PubMed] [Google Scholar]
- 2.Huebsch N, Kearney CJ, Zhao X, Kim J, Cezar CA, Suo Z, Mooney DJ. Proc. Natl. Acad. Sci. U. S. A. 2014;111:9762. doi: 10.1073/pnas.1405469111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldman A, Majumder B, Dhawan A, Ravi S, Goldman D, Kohandel M, Majumder PK, Sengupta S. Nat. Commun. 2015;6:6139. doi: 10.1038/ncomms7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Nat. Biotechnol. 2001;19:1029. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 5.Huang X, Brazel CS. J. Control. Release. 2001;73:121. doi: 10.1016/s0168-3659(01)00248-6. [DOI] [PubMed] [Google Scholar]
- 6.Betz OB, Betz VM, Nazarian A, Egermann M, Gerstenfeld LC, Einhorn TA, Vrahas MS, Bouxsein ML, Evans CH. Gene Ther. 2007;14:1039. doi: 10.1038/sj.gt.3302956. [DOI] [PubMed] [Google Scholar]
- 7.Hoare TR, Kohane DS. Polymer. 2008;49:1993. [Google Scholar]
- 8.Ron ES, Bromberg LE. Adv. Drug Del. Rev. 1998;31:197. doi: 10.1016/s0169-409x(97)00121-x. [DOI] [PubMed] [Google Scholar]
- 9.Hoare T, Santamaria J, Goya GF, Irusta S, Lin D, Lau S, Padera R, Langer R, Kohane DS. Nano Lett. 2009;9:3651. doi: 10.1021/nl9018935. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhao X, Kim J, Cezar CA, Huebsch N, Lee K, Bouhadir K, Mooney DJ. Proc. Natl. Acad. Sci. U. S. A. 2011;108:67. doi: 10.1073/pnas.1007862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nair M, Guduru R, Liang P, Hong J, Sagar V, Khizroev S. Nat. Commun. 2013;4:1707. doi: 10.1038/ncomms2717. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez-Lorenzo C, Bromberg L, Concheiro A. Photochem. Photobiol. 2009;85:848. doi: 10.1111/j.1751-1097.2008.00530.x. [DOI] [PubMed] [Google Scholar]
- 12.Farra R, Sheppard NF, McCabe L, Neer RM, Anderson JM, Santini JT, Cima MJ, Langer R. Sci. Transl. Med. 2012;4:122ra21. doi: 10.1126/scitranslmed.3003276. [DOI] [PubMed] [Google Scholar]
- 13.Epstein-Barash H, Orbey G, Polat BE, Ewoldt RH, Feshitan J, Langer R, Borden MA, Kohane DS. Biomat. 2010;31:5208. doi: 10.1016/j.biomaterials.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kost J, Leong K, Langer R. Proc. Natl. Acad. Sci. U. S. A. 1989;86:7663. doi: 10.1073/pnas.86.20.7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitragotri S. Nat. Rev. Drug Discov. 2005;4:255. doi: 10.1038/nrd1662. [DOI] [PubMed] [Google Scholar]
- 16.Mura S, Nicolas J, Couvreur P. Nat. Mat. 2013;12:991. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 17.Boontheekul T, Kong HJ, Mooney DJ. Biomat. 2005;26:2455. doi: 10.1016/j.biomaterials.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 18.Shi J, Votruba AR, Farokhzad OC, Langer R. Nano Lett. 2010;10:3223. doi: 10.1021/nl102184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skaat H, Ziv-Polat O, Shahar A, Last D, Mardor Y, Margel S. Adv. Health. Mat. 2012;1:168. doi: 10.1002/adhm.201100056. [DOI] [PubMed] [Google Scholar]
- 20.Perrault SD, Chan WCW. J. Am. Chem. Soc. 2009;131:17042. doi: 10.1021/ja907069u. [DOI] [PubMed] [Google Scholar]
- 21.Powell AC, Paciotti GF, Libutti SK. Methods in molecular biology (Clifton, N.J.) 2010;624:375. doi: 10.1007/978-1-60761-609-2_25. [DOI] [PubMed] [Google Scholar]
- 22.Rahme K, Chen L, Hobbs RG, Morris MA, O'Driscoll C, Holmes JD. RSC Adv. 2013;3:6085. [Google Scholar]
- 23.Fusco S, Sakar MS, Kennedy S, Peters C, Bottani R, Starsich F, Mao A, Sotiriou GA, Pané S, Pratsinis SE, Mooney D, Nelson BJ. Adv. Mater. 2014;26:952. doi: 10.1002/adma.201304098. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Cao L, Shvartsman D, Silva EA, Mooney DJ. Nano Lett. 2011;11:694. doi: 10.1021/nl103812a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee KY, Mooney DJ. Prog. Polym. Sci. 2012 doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X-D, Wu D, Shen X, Liu P-X, Yang N, Zhao B, Zhang H, Sun Y-M, Zhang L-A, Fan F-Y. Int. J. Nanomed. 2011;6:2071. doi: 10.2147/IJN.S21657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carragee EJ, Hurwitz EL, Weiner BK. Spine J. 2011;11:471. doi: 10.1016/j.spinee.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Giljohann DA, Seferos DS, Prigodich AE, Patel PC, Mirkin CA. J. Am. Chem. Soc. 2009;131:2072. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao L, Wang J, Hou J, Xing W, Liu C. Biomat. 2014;35:684. doi: 10.1016/j.biomaterials.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 30.Scheufler C, Sebald W, Hülsmeyer M. J. Mol. Biol. 1999;287:103. doi: 10.1006/jmbi.1999.2590. [DOI] [PubMed] [Google Scholar]
- 31.Yi C, Liu D, Fong C-C, Zhang J, Yang M. ACS Nano. 2010;4:6439. doi: 10.1021/nn101373r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.