

Abstract
Magnesium (Mg2+) is the second most abundant cellular cation and is essential for all stages of life, from the early embryo to adult. Mg2+ deficiency causes or contributes to many human diseases, including migraine headaches, Parkinson’s disease, Alzheimer’s disease, hypotension, type 2 diabetes mellitus and cardiac arrhythmias. Although the concentration of Mg2+ in the extracellular environment can vary significantly, the total intracellular Mg2+ concentration is actively maintained within a relatively narrow range (14 – 20 mM) via tight, yet poorly understood, regulation of intracellular Mg2+ by Mg2+ transporters and Mg2+-permeant ion channels. Recent studies have continued to add to the growing number of Mg2+ transporters and ion channels involved in Mg2+ homeostasis, including TRPM6 and TRPM7, members of the transient receptor potential (TRP) ion channel family. Mutations in TRPM6, including amino acid substitutions that prevent its heterooligomerization with TRPM7, occur in the rare autosomal-recessive disease hypomagnesemia with secondary hypocalcemia (HSH). However, is the fact that genetic ablation of either gene in mice results in early embryonic lethality that has raised the question of whether these channels’ capacity to mediate Mg2+ influx plays an important role in embryonic development. Here we review what is known of the function of Mg2+ in early development and summarize recent findings regarding the function of the TRPM6 and TRPM7 ion channels during embryogenesis.
Keywords: magnesium, TRPM7, TRPM6, ion channel, gastrulation, and embryonic development
Introduction
Unlike it’s flashy divalent cousin Ca2+, Mg2+ faithfully labors without fanfare in numerous vital biological processes, ranging from metabolic regulation, to DNA and RNA synthesis, neurotransmission, and cell stress reactions (Romani, 2007). Mg2+ homeostasis regulation in mammals occurs in the intestine, kidney and bone, where the divalent cation is initially absorbed, excreted and stored in its hydroxylapatite form, respectively. The kidney is believed to be the primary site for regulation of whole body Mg2+ homeostasis, where the balance of Mg2+ reabsorption and excretion is controlled. It is well known that disruption of Mg2+ homeostasis is associated with numerous human diseases (for an excellent comprehensive review see (de Baaij et al., 2015)). Low level of Mg2+ intake has been linked to the etiology of preeclampsia, stroke, diabetes, and cardiovascular disease. Beyond these pathologies, which primarily affects adults, it has been reported that insufficient Mg2+ intake during pregnancy may increase the risk of preterm birth and low birth weight (Conradt et al., 1984, Doyle et al., 1989, Makrides and Crowther, 2001).
Given the cation’s association with fetal development and human diseases, it is very important to understand the molecular mechanisms that govern Mg2+ homeostasis. In this review, we summarize data illustrating the importance of Mg2+ homeostasis during embryonic development. We focus primarily on data from Xenopus laevis, where the most extensive and detailed analysis of the impact of Mg2+ on embryonic development has been conducted; this is presumably because Xenopus embryos can be developed ex-vivo and the concentration of ions bathing the embryo can be easily manipulated. We also discuss recent studies regarding the function of TRPM7 and TRPM6 ion channels in Mg2+ homeostasis and embryogenesis. Finally, we end our focused review by sharing our own unpublished results on the impact of Mg2+ on Xenopus development, including new observations that may be relevant to a role for this understudied cation in Parkinson’s disease. For more detailed information, we refer the reader to more comprehensive reviews on TRPM6 and TRPM7 channels as well as Mg2+ homeostasis (Komiya et al., 2014, Romani, 2011, Runnels, 2011).
Cellular roles for Mg2+ and its regulation by ion channels and transporters
Magnesium is involved in a wide variety of biological functions, including the cell cycle, ion channel regulation, ATPase activity, protein and nucleic acid synthesis, metabolism reactions, maintenance of body temperature and neurotransmission in nervous system (Romani, 2007). Mg2+ is a cofactor for many enzymes’ activities. It has been estimated that at least 600 enzymatic reactions are directly or indirectly regulated by Mg2+ (de Baaij et al., 2015). Total intracellular Mg2+ is 14 – 20 mM, making it the second most abundant cation (Grubbs and Maguire, 1987, Romani, 2011). However, unlike Ca2+, whose cytosolic concentration can vary 10–100 fold, the free concentration of Mg2+ is comparably steady, hovering between 0.4 – 1 mM in concentration. This has lead some to question whether Mg2+ can function as a second messenger like Ca2+. Nevertheless, rapid fluxes of Mg2+ into and out of cells have been detected in a number of cell types (Matsuura et al., 1993, Romani and Scarpa, 1990a, Romani and Scarpa, 1990b). Disruption one of these processes was recently shown to be responsible for a novel X-linked human immunodeficiency characterized by CD4 lymphopenia, severe chronic viral infections, and defective T-lymphocyte activation (Li et al., 2011). Mutations in the Mg2+ transporter MAGT1 abolished Mg2+ influx required for activation of phospholipase C-γ1 (PLC-γ1) and Ca2+ influx in T cells. Thus the impact of Mg2+ on cell signaling may be underappreciated. Indeed, low Mg2+ availability disrupts cell proliferation and cell differentiation in cell culture systems. Under low Mg2+ conditions, HL-60 leukemic cells accumulate in the G0/G1 phase of cell cycle and have altered granulocytic differentiation (Covacci et al., 1998). Low Mg2+ availability also inhibits cell proliferation of mammary epithelial HC11 cells by increasing oxidative stress (Wolf et al., 2009). Mg2+ deficiency has been etiologically linked to many human diseases as well (de Baaij et al., 2015, Volpe, 2013). For example, it has been reported that low Mg2+ and Ca2+ intake is associated with Parkinson’s disease (PD) and higher dietary intake of Mg2+ as well as iron and zinc may have a protective effect against PD (Miyake et al., 2011, Oyanagi et al., 2006). In addition, the highly selective Mg2+ transporter NIPA2 is mutated in patients with childhood absence epilepsy (CAE). The NIPA2 protein mutants were expressed in the cytosol, whereas the wild type protein properly localized to the cell border (Xie et al., 2014). Indeed, it is estimated that up to 60 % of all critically ill patients are Mg2+ deficient (de Baaij et al., 2015). Therefore, regulation of cellular as well as whole body Mg2+ homeostasis is critical for human health. Consequently, normal cells work aggressively to maintain a relatively narrow range of intracellular free Mg2+ concentration, regardless of the extracellular Mg2+ concentration. This responsibility largely falls to Mg2+ transporters and ion channels.
Magnesium transporters were originally identified in prokaryotes and protozoan (Quamme, 2010). In 1976, CorA was identified as a Mg2+ transporter in bacteria. CorA has two transmembrane domains and function as a Mg2+ transporter when oligomerized (Park et al., 1976). CorA can permeate Co2+, Ni2+ as well as Mg2+ and CorA is the primary transporter responsible for Mg2+ intake in most prokaryotes. In an effort to search for new CorA family members and homologues, many other Mg2+ transporters such as MgtE, Alr1, and Mrs2, have been identified in bacteria, fungi and yeast systems (Bui et al., 1999, Graschopf et al., 2001). These discoveries led to the identification of mammalian’s Mg2+ transporters, which were identified in part based on homologies to their ancestral counterparts. While some of the identified transporters exhibit relatively high specificity for Mg2+ transport, other molecules shown to play a role in Mg2+ homeostasis were surprisingly found to be relatively non-selective (see (de Baaij et al., 2015) for a review). The TRPM7 ion channel, which has been shown to play a vital role in both cellular and whole animal Mg2+ homeostasis, is a good example (Ryazanova et al., 2010, Schmitz et al., 2003). TRPM7 exhibits high permeation of Mg2+, but is also permeable to other essential divalent cations, including Ca2+ and Zn2+ (Monteilh-Zoller et al., 2003). Likewise, TRPM6, a homologue of TRPM7 that is mutated in the rare autosomal disorder familial hypomagnesemia with secondary hypocalcemia (HSH), is permeable to a similar range of divalent cations as TRPM7 (Li et al., 2006, Voets et al., 2004). TRPM7 and TRPM6 are members of transient receptor potential (TRP) superfamily, which comprises a large number of cation-permeable ion channels, many of which execute critical functions in sensory systems (Clapham, 2003, Nadler et al., 2001, Runnels et al., 2001). Among ion channels, TRPM7 and TRPM6 are quite unique because they are bifunctional molecules (“chanzymes”) with functional kinases domains attached to COOH-termini (Figure 1). TRPM7 appears to be ubiquitously expressed, whereas the expression of TRPM6 is more restricted, showing highest expression in the intestine and the distal convoluted tubule (DCT) of the kidney (Schlingmann et al., 2002, Walder et al., 2002). The DCT is the final site for Mg2+ reabsorption before urinary excretion and functions to balance the body’s Mg2+ content. TRPM6 is posited to play a critical role in this process. A positional candidate gene approach identified TRPM6 as the gene mutated in familial HSH (Schlingmann et al., 2002, Walder et al., 2002). Individuals affected by HSH are characterized by low serum Mg2+ and Ca2+. Patients exhibit neurologic symptoms such as seizures and muscle spasms (Shalev et al., 1998). Life-long supplementation of Mg2+ can be used to treat HSH. Thus far, however, there has been no report of a mutation in the TRPM7 gene for a HSH patient. While a majority of the mutations in individuals affected with HSH are either nonsense or frameshift mutations in TRPM6 that are easily compatible with a loss-of-function phenotype, one missense mutation entails the exchange of a highly conserved serine for a leucine at amino acid position 141 (S141L), which disrupts the ability of TRPM6 to form heterooligomers with TRPM7 (Schlingmann et al., 2002). In two sets of studies, TRPM6 was reported to be dependent upon TRPM7 for cell surface expression (Chubanov et al., 2004, Schmitz et al., 2005). Full-length variants of TRPM6 failed to form functional channel complexes when heterologously expressed in HEK-293 cells and Xenopus oocytes (Chubanov et al., 2004). This data suggests that TRPM7 may also participate in whole body Mg2+ homeostasis, either by itself, or by forming heterooligomeric channels with TRPM6. In support of such a role, heterozygous mice engineered to lack TRPM7’s kinase domain (TRPM7ΔKINASE) exhibited a mild hypomanesaemia phenotype, which was most likely due to diminished TRPM7 channel activity resulting from deletion of the kinase domain. The heterozygous TRPM7ΔKINASE mutant mice exhibited high Mg2+ excretion, altered Mg2+ homeostasis and high sensitivity to low-magnesium diet (Ryazanova et al., 2010). A more severe phenotype was observed for homozygous TRPM7ΔKINASE null embryos, which were able to initiate gastrulation and mesoderm formation, but died between day 7.5 and 8.5 of embryogenesis. Thus, studies in mice have elaborated these channels role in whole body magnesium homeostasis, but have also pointed to additional critical functions during early embryogenesis.
Fig. 1. Schematic model of the membrane topology of TRPM7 and TRPM6.
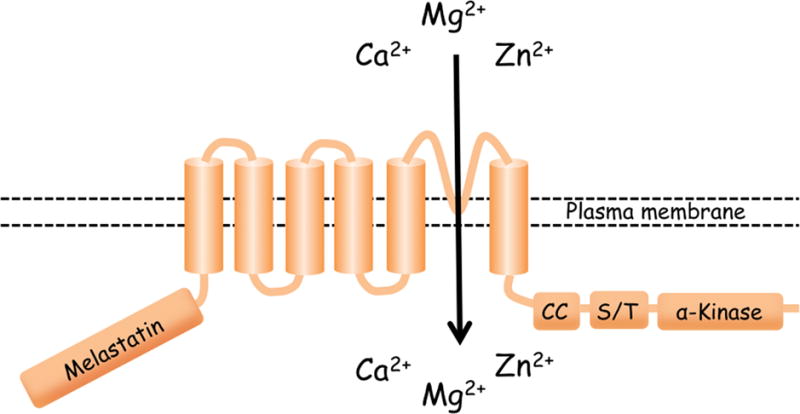
TRPM7 and TRPM6 have 6 transmembrane spanning domains and a pore-loop that is localized between fifth and sixth transmembrane domains. TRPM7 and TRPM6 possess coiled-coil (CC), Serine/Threonine-rich (S/T), and α-kinase domains in the intracellular COOH-terminus region.
TRPM7, TRPM6 and embryogenesis
The requirement of TRPM7 and TRPM6 for embryogenesis has been demonstrated by ablation of their genes in mouse models. In the mouse embryo, expression of TRPM7 is dramatically increased from embryonic day 10.5 (E10.5) to E11.5 and global expression was observed through E14.5 (Jin et al., 2008). No homozygous TRPM7 mutants were born, dying at E7.5, the period when gastrulation cell movements commence. During mouse embryogenesis, the primitive streak is formed around E6.5, after which cells start moving dynamically to establish the anterior-posterior axis. From E8.5 to E15 the neural plate is formed and the neural tube closes. Jin and colleagues also generated conditional knockout mice of TRPM7 using a tamoxifen-inducible and multiple tissue specific Cre recombinase lines (Jin et al., 2012). Tamoxifen-dependent deletion of TRPM7 at E7.5 to E8.5 caused embryonic lethality within 48–72 hours, while the depletion at E14.5 did not cause embryonic lethality, with mutant mice developing normally. Taken together, results from TRPM7 mutant mice demonstrate a temporal requirement of TRPM7 during early gastrulation in mouse. In addition, Jin et al. demonstrated that disruption of TRPM7 in induced pluripotent stem (iPS) cells induced apoptosis, indicating a contribution of TRPM7 to stem cell survival (Jin et al., 2012). A similar dependence on TRPM7 was observed for the survival of bone marrow derived mesenchymal stem cells and human dental pulp stem cells (Cheng et al., 2010, Cui et al., 2014), but not neural stem cells (Jin et al., 2012). This may be because not all cells solely depend upon TRPM7 for cellular magnesium homeostasis. Nevertheless, an indispensable function for TRPM7 in certain stem cell populations may impact developmental processes dependent upon cell differentiation. Later in development, TRPM7 has also been demonstrated in mice to have critical functions during nephrogenesis, the development of neural-crest-derived pigment cells, and myocardial proliferation during early cardiogenesis (Jin et al., 2012, Sah et al., 2013). Additional studies in zebrafish have also highlighted roles for TRPM7 in pigmentation, but have uncovered additional developmental roles in other organs’ development, including the pancreas (McNeill et al., 2007, Yee et al., 2011). Magnesium’s contribution to TRPM7 function later in development remains poorly understood.
Surprisingly, knockout of TRPM6 from mice is also embryonically lethal (Walder et al., 2002, Woudenberg-Vrenken et al., 2011). In experiments reported by Walder and colleagues, homozygous TRPM6 knockout embryos died by E12.5 and exhibited neural tube closure defects (Walder et al., 2002). The expression pattern of TRPM6 during embryogenesis demonstrated a significant increase at E10, suggesting TRPM6 is also temporally required for early development. Heterozygous TRPM6 knockout mice exhibited mild hypomagnesemia (Walder et al., 2002, Woudenberg-Vrenken et al., 2011). Interestingly, in one of the studies dams fed with a high Mg2+ diet slightly suppressed the embryonic lethality caused by knockout of TRPM6, suggesting that embryonic lethality may in part be due to Mg2+ deficiency. However, in the second study by Woudenberg-Vreken et al., a high Mg2+ diet failed to rescue the lethality caused by homozygous deletion of TRPM6 (Woudenberg-Vrenken et al., 2011). Studies from zebrafish also support a role for Mg2+ during early embryonic development. Zhou and Clapham have demonstrated that knockdown of the MagT1 and TUSC3 Mg2+ transporters in zebrafish embryos causes early developmental arrest, with embryos exhibiting an apparent defect in brain and eye development (Zhou and Clapham, 2009). Supplementation of Mg2+ in the growth media partially rescued the embryonic arrest caused by depletion of MagT1, demonstrating the importance of Mg2+ transporters and Mg2+ during embryogenesis. In a more recent study, mutations in the gene encoding for cyclin M2 (CNNM2) were demonstrated to be causative for mental retardation and seizures in patients with hypomagnesemia (Arjona et al., 2014). In patients with a recessive mode of inheritance, the intellectual disability is more severe and is accompanied by motor defects and brain malformations. In zebrafish, CNNM2 has two orthologues, cnnm2a and cnnm2b. Consistent with the human pathology, knockdown of cnnm2a in zebrafish resulted in reduced total magnesium content and in morphological phenotypes characterized by enlarged pericardial cavities and notochord defects. At 25 hour post-fertilization (hpf) maldevelopment of the midbrain hindbrain boundary (MHB) was observed. Cnnm2b morphants also had lower total magnesium and were characterized by enlarged pericardial cavities, kidney cysts, accumulation of cerebrospinal fluid in the cerebrum, and at 25 hpf, also exhibited defects in development of the MHB. These studies highlight a particularly important role for CNNM2, and potentially for magnesium, in heart, kidney and especially brain development.
TRPM7 function and magnesium deficiency in the Xenopus embryo
Xenopus laevis embryos constitute a classic animal model to investigate early developmental processes. Many of the signaling molecules that regulate early embryogenesis were originally identified in Xenopus. It is well known that key factors of early development, such as BMP, Wnt and FGF signaling molecules, are functionally conserved between Xenopus and mammals. Since Xenopus eggs can be externally fertilized, it is easy to observe each step of embryogenesis. This model also generates a relatively large embryo, which allows for targeted microinjection and microsurgery. Microinjection of morpholino antisense oligonucleotides (MOs), which inhibit protein translation from their mRNA targets, produces phenotypes that are typically milder than those produced by homozygous gene deletion. Because of the early embryonic lethality of TRPM7 and TRPM6 knockout mice, a moderate knockdown of the ion channels facilitates investigation of the developmental processes they are affecting. In addition, Xenopus embryos as well as dissected explants can survive in a simple buffer, allowing us to easily manipulate the ionic composition of the culture buffer and to examine the significance of different ions during early embryogenesis.
Previously, we have demonstrated a crucial role of TRPM7 in gastrulation cell movements during Xenopus embryogenesis (Liu et al., 2011). While Xenopus TRPM7 expression is very consistent throughout embryogenesis, in situ hybridization analysis showed specific TRPM7 expression in dorsal mesoderm and ectoderm during the gastrula and in neural plate during neurulation. Downregulation of TRPM7 by MO injection induced severe gastrulation defects, which were characterized by a short and curved axis and widely opened neural tube. Interestingly, co-injection of TRPM6 or the SLC41A2 Mg2+ transporter with the TRPM7 MO suppressed the MO-induced gastrulation defects. Supplementation of Mg2+, but not Ca2+, in the culture buffer was similarly effective. These results indicate Mg2+ intake through TRPM7 is required for gastrulation in the Xenopus embryo. During gastrulation, neuroectoderm and dorsal mesoderm undergo dynamic morphological changes. Polarized cells move toward the midline and intercalate mediolateraly. Afterwards the ball-shape embryo narrows and lengthens its head-tail axis. This coordinated movement of cells establishes anterior-posterior axis and is referred to as “convergent extension movements” (Wallingford et al., 2002). It is well know that non-canonical Wnt pathway plays a pivotal role in convergent extension movements (Wallingford and Habas, 2005). We have demonstrated that knockdown of TRPM7 strongly inhibits convergent extension movements and disrupts cell polarization (Liu et al., 2011). Importantly, TRPM7 MO-induced suppression of cell movements can be rescued by the supplementation of Mg2+ in culturing buffer, indicating maintaining Mg2+ concentration in embryo is critical for cell movements during early embryogenesis. Although molecular mechanism is not fully understood, our co-injection studies suggest that the TRPM7 signaling merges with non-canonical Wnt pathway. Overall, our experiments with Xenopus embryos uncovered the importance of TRPM7 as well as Mg2+ for cell movements during gastrulation. Since expression of a Mg2+ transporter and Mg2+ supplementation could rescue the defects induced by the TRPM7 MO, maintenance of cellular Mg2+ homeostasis is likely crucial for gastrulation.
In 1977 Miller and Landesman performed the first comprehensive investigation of the significance of Mg2+ during Xenopus embryogenesis (Miller and Landesman, 1977). Typically, Xenopus embryos are cultured in a simple buffer such as 0.1 × MMR or 0.1 × MBS, solutions that contains 0.1 μM MgSO4. 0.1 × MMR contains (in mM) 0.5 HEPES (pH 7.8), 10 NaCl, 0.2 KCl, 0.2 CaCl2 and 0.1 MgSO4. 0.1 × MBS contains (in mM) 0.5 HEPES (pH 7.8), 8.8 NaCl, 0.1 KCl, 0.07 CaCl2, 0.1 MgSO4, 0.25 NaHCO3. These buffers also contain sodium, potassium, and calcium ions, which are also essential for embryonic development since Xenopus embryos cannot survive in distilled water alone. In their experiments, the authors determined the impact of Mg2+ on embryogenesis by varying the concentration of Mg2+ in the culture buffer from 100 nM to 10 mM. When the Mg2+ concentration was lower than 10 μM, embryos exhibited reduced melanophores at stage 35–36. By stage 40 the embryos showed shorter tail expansion, abnormal coiling of the gut, slow head enlargement, restricted heart growth, and edema. In addition, abnormal somite development was observed, which may be related to paralysis of the Mg2+ deficient embryos. In control embryos, the total Mg2+ content began to increase after hatching. By comparison, embryos cultured in Mg2+ deficient buffer maintained a very low level of total Mg2+ throughout development. Their results suggested that proper Mg2+ uptake is a critical factor during early developmental processes in Xenopus.
The Miller and Landesman article has very detailed drawings of the embryonic phenotypes caused by Mg2+ deficiency, but to the best of our knowledge there are no photographs of the embryos from their experiments. To update the information on Mg2+ deficiency during Xenopus embryogenesis, we conducted similar experiments to those of Miller and Landesman by examining the effect of Mg2+ depletion from culture medium on the developmental processes of Xenopus laevis (Figure 2A). We observed the morphology of Xenopus embryos cultured in regular MMR solution or Holtfreter’s buffer with or without 0.1 mM MgSO4. It should be noted that our buffer does not contain the chelating agent, sodium pyrophosphate, since Miller and Landesman reported that sodium pyrophosphate induces cell dissociation and inhibits gastrulation.
Fig. 2. Magnesium depletion causes developmental abnormalities in Xenopus laevis embryos.
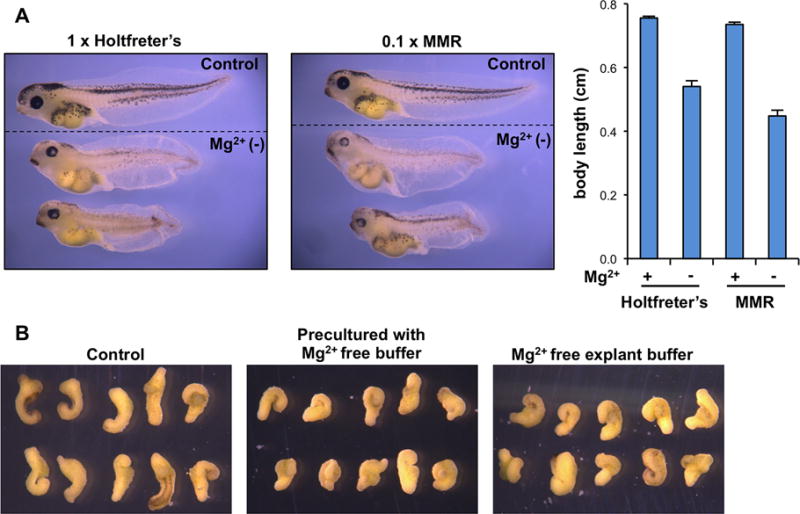
(A) Mg2+ deficient embryos showed pigmentation defects and a shortened axis. Top embryos in each panel are wild type embryos reared in normal buffer. Right panel shows average of body length at stage 42. (B) Mg2+ depletion suppressed convergent extension movements. At stage 10.5, dorsal marginal zone was dissected and cultured in buffer with or without Mg2+ until siblings reach stage 22–23.
Consistent with their earlier findings, early developmental processes such as cleavage, early gastrulation, and neurulation were not significantly affected by Mg2+ depletion in our experiments (data not shown). At the tadpole stage, embryos cultured in Mg2+ deficient media exhibited lighter pigmentation in eyes and body than control embryos reared in normal buffer (Figure 2A). All embryos cultured in Mg2+ deficient media did not elongate their axis properly: the length of anterior-posterior axis was significantly shorter than control embryos. In addition, embryos cultured in Mg2+ deficient media had spinal cords that were not straight, with some of embryos exhibiting a mildly kinked axis. Many of the embryos cultured in Mg2+ deficient media had a slightly smaller head size. Although we did not perform histological exams, in some of embryos the gut appeared abnormal as reported by Miller and Landesman. We did not observe severe defects during early gastrulation and the reason for this is not clear. The concentration of Mg2+ from egg to the stage 9 embryo, just before gastrulation begins, does not vary much from 14 mM (Slack et al., 1973). The rate at which the embryos lose Mg2+ in Mg2+ deficient media is not known and could be too slow to produce a severe phenotype at earlier stages in development. More research on the regulation of Mg2+ homeostasis in the developing embryo is required to address this question.
Nevertheless, given the fact that Mg2+ supplementation can rescue the convergent extension defect induced by TRPM7 depletion suggests that Mg2+ influences cell movements during early embryogenesis. In addition to the result from Xenopus embryos, Su et al. have demonstrated the essential role of TRPM7 and Mg2+ in cytoskeletal arrangements and polarized cell movements in fibroblasts (Su et al., 2011). Knockdown of TRPM7 by RNAi inhibited the activity of Rho, Rac, and Cdc42, which interfered with the establishment of cell polarity and lamellipodia formation in a cellular wound assay. Similar to what was observed in Xenopus experiments, these cellular defects could be rescued by expression of the Mg2+ transporter SLC41A2. These results support a role for Mg2+ as well as for TRPM7 in controlling polarized cell movements. To directly determine whether Mg2+ influences gastrulation cell movements we investigated the effect of Mg2+ depletion on convergent extension using a Keller explant assay. After fertilization, embryos were grown in regular MMR or Mg2+-free MMR until the early gastrula stage, after which the dorsal marginal zone was dissected. The isolated explants were then cultured in buffer containing Mg2+ or Mg2+-free buffer. For the control condition (with Mg2+), dorsal tissue explants underwent convergent extension movements and became well elongated. The explants that were pre-cultured with Mg2+-free MMR also elongated, but the length was shorter than the controls, indicating that elongation was impaired (Figure 2B). These results suggest that the short axis phenotype in embryos cultured in Mg2+ deficient media could be explained by mild inhibition of convergent extension cell movements.
The results from Miller and Landesman as well as our own results indicate a vital contribution of Mg2+ to pigmentation development. Body pigmentation develops from the presence of melanin in melanocytes (the melanophore is the non-mammalian vertebrate counterpart of the mammalian melanocyte) and retinal pigment epithelium. Melanin is produced by specialized lysosomal-related organelles called melanosomes. Interestingly, it has been reported that the TRPM7, TRPM1 and TRPA1 ion channels are present in melanocytes and are involved in the regulation of melanin synthesis (Bellono and Oancea, 2014). As mentioned earlier, TRPM7 mutant zebrafish exhibit pigmentation defects as a result of having fewer melanophores (McNeill et al., 2007). The loss of pigmentation appears to be caused by apoptotic and necrotic cell death of melanoblasts, precursor cells of melanophores, as well as melanophores. In melanophores of TRPM7 mutant zebrafish, many irregular shaped melanosomes were observed. In the surrounding tissue, melanosomes were also seen, suggesting that the plasma membrane of melanophore cells ruptured and caused necrotic cell death. Interestingly, inhibition of melanin synthesis by a tyrosinase inhibitor can rescue melanophore cell death in the TRPM7 mutant zebrafish (McNeill et al., 2007). These results indicate that abnormal melanin synthesis is related to the death of melanophores of TRPM7 mutant fish. In addition, Mg2+ supplementation was also partially successful in suppressing the death of melanophores. Melanosomes contain a high concentration of metals, including iron, zinc, copper, calcium and magnesium, indicating that the organelle may also function as an intracellular storage site for these metals (Hong and Simon, 2007). Although the molecular mechanisms remain unclear, the data suggests that Mg2+ accumulation into melanosomes may contribute to melanogenesis. Mg2+ may be affecting melanin synthesis by binding to enzymes or intermediates of melanin synthesis that contribute to pigmentation development. Thus, loss of TRPM7 or embryonic development in a low Mg2+ environment causes abnormal melanin synthesis that somehow contributes to melanocyte cell death. This could explain for why melanin-positive cells may be more sensitive to loss of TRPM7 and to Mg2+ deficiency than other cell types.
Magnesium homeostasis and neurodegenerative diseases
In addition to defects of cell movement and pigmentation, we observed that embryos grown in Mg2+-free medium did not readily move at the tadpole stage. Control tadpoles swam actively in the culture dish, whereas Mg2+ deficient tadpoles exhibited a paralysis phenotype (Supplementary Movie 1). When touched with a paintbrush, Mg2+ deficient tadpoles were often unresponsive to touch stimulation. In comparison to control tadpoles that swam away immediately when touched, some Mg2+ deficient tadpoles twitched and swam away abnormally slow. This touch response defect was of our particular interest because TRPM7 mutant zebrafish lines have been shown to also exhibit movement defects (Decker et al., 2014, Low et al., 2011). In wild type zebrafish, a light-to-dark transition stimulates swimming behavior. Decker and colleagues demonstrated that the swimming distance of TRPM7 mutant fishes under these conditions was shorter than wild type. Interestingly, the paralysis phenotype of TRPM7 mutant zebrafish is reported to be due to abnormal differentiation of dopaminergic neurons. Decker et al. demonstrated that in the brain of TRPM7 mutant zebrafish the number of cells expressing tyrosine hydroxylase (an enzyme involved in dopamine synthesis) was significantly lower than in wild type fish. Interestingly, application of levodopa (L-dopa), a precursor of dopamine, could rescue the motility defect of TRPM7 mutants. Their results suggested that loss of function mutations in the TRPM7 gene in zebrafish is caused by abnormal differentiation of dopaminergic neurons, which ultimately resulted in a motility defect. However, mutation in TRPM7 has also shown to be responsible for a touch-unresponsive zebrafish mutant. This phenotype could be alleviated by forcing the expression of TRPM7 in primary sensory neurons, suggesting that TRPM7 is required transiently for function or differentiation of embryonic primary sensory neurons (Low et al., 2011). A defect in a touch-evoked escape behavior was also observed in cnnm2a and cnnm2b zebrafish morphants, which was speculated to be caused to lack of excitation of sensory neurons via TRPM7 and/or to defects in early brain development (Arjona et al., 2014). Given the similarity of the phenotypes between Mg2+ deficient embryos and the cnnm2 and TRPM7 mutant fishes, the movement defect in Mg2+ deficient Xenopus tadpoles could be caused by a defect in sensory neurons and/or dopaminergic neurons. This hypothesis remains to be explored.
Loss of dopaminergic neurons is a hallmark of Parkinson’s disease (PD). Interestingly, these dopaminergic neurons are neuromelanin-positive. Given the fact that both TRPM7 and Mg2+ contribute to melanophore cell development as well as to the motility of zebrafish and Xenopus embryos, it is tempting to speculate that Mg2+ deficiency may be related to etiology of Parkinson’s disease. In fact, epidemiological evidence and animal studies support a link between Mg2+ deficiency and loss of dopaminergic neurons in PD. In Guam and Kii peninsula in Japan, where extremely low concentrations of Mg2+ and Ca2+ in drinking water have been reported, individuals have a higher risk of developing parkinsonism-dementia complex (PDC) and amyotrophic lateral sclerosis (ALS). Parkinson’s patients have also been found to have low Mg2+ concentrations in cortex, white matter, basal ganglia, and brain stem (Yasui et al., 1992). Oyanagi et al. have attempted to replicate the condition of low Mg2+ and Ca2+ intake in rats. Rats were fed varying concentrations of Mg2+ and Ca2+ in their diet for over a generation; a significant loss of dopaminergic neurons occurred in the substantia nigra of rats fed a low Mg2+ diet (Oyanagi et al., 2006). Furthermore, it has been shown that Mg2+ supplementation of the culture medium for an in vitro PD model prevented loss of dopaminergic neurons induced by the parkinsonian agent (1-methyl-4-phenylpyridinium, MMP+) (Hashimoto et al., 2008). Another group investigated the effect of low Mg2+ and Ca2+ diet on PD using mice. Mice fed low Mg2+ and Ca2+ for 3–6 weeks developed akinesia and rigidity concomitant with a decrease in the number of their dopaminergic neurons (Taniguchi et al., 2013). Interestingly, a polymorphism in the TRPM7 gene (T1482I) has been associated with a subset of patients with Guamanian amyotrophic lateral sclerosis (ALS-G) or parkinsonism dementia (PD-G). The T1482I mutation may render the TRPM7 channel more sensitive to inhibition by intracellular Mg2+ (Hermosura et al., 2005), although this finding has been disputed (Demeuse et al., 2006). In addition, a rare gain-of-function mutation in the Mg2+ transporter gene SLC41A1, which was shown to lower intracellular Mg2+ when expressed in HEK-293 cells, is putatively linked to PD (Kolisek et al., 2013). Moreover, SLC41A1 is located on the PARK16 locus that is associated with Parkinson’s disease. Taken together these studies suggest that an appropriate amount of Mg2+ is required for survival, differentiation and/or function of dopaminergic neurons and that Mg2+ deficiency may be related to the etiology of PD. Since, TRPM7 appears to be involved in the function of dopaminergic neurons, magnesium’s role in this devastating disease warrants further investigation.
Conclusion
Magnesium is an abundant and essential divalent cation in the body. Not surprisingly, disruption of Mg2+ homeostasis has the potential to contribute to birth defects as well as to many other human diseases. Knockout mouse studies have uncovered indispensable roles for TRPM7 and TRPM6 during early development. However, experiments in zebrafish and Xenopus embryos have given insight into the role that Mg2+ may be playing not only in gastrulation, but in body pigmentation, brain development and body movement as well. Excitingly, these observations may be relevant to the etiology of many human diseases, including neural tube closure defects and neurological disorders such as epilepsy and Parkinson’s disease. The mechanisms by which Mg2+ exerts its effects during development and disease are still poorly understood. This is in part due to the fact that Mg2+ functions as a cofactor in so many cellular processes. In addition, Mg2+ could be indirectly affecting the homeostasis of Ca2+ or other metal ions. Gain-of-function and loss-of-function studies, such as those that have been conducted in Xenopus and zebrafish, have the potential to uncover the mechanisms by which Mg2+ exerts its in vivo effects. Research into the function of Mg2+ will give much needed insight into approaches that may prevent or limit the devastating effects of the diseases caused or made worse by dysregulation of this vital divalent cation.
Supplementary Material
Embryos reared in Mg2+ deficient buffer until tadpole stage displayed movement defects compared to wild type embryos reared in normal buffer. Control tadpoles swam away immediately when touched with a paintbrush. In contrast, Mg2+ deficient tadpoles did not readily respond to touch: some tadpoles swam away abnormally slow whereas others did not respond at all.
Acknowledgments
This work was supported by the generous support of the National Institutes of Health, NIGMS (GM080753) to LWR. We are also grateful to Yosh Beier for his help in German translation of original articles we consulted in preparation of this review.
References
- ARJONA FJ, DE BAAIJ JH, SCHLINGMANN KP, LAMERIS AL, VAN WIJK E, FLIK G, REGELE S, KORENKE GC, NEOPHYTOU B, RUST S, et al. CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS Genet. 2014;10:e1004267. doi: 10.1371/journal.pgen.1004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELLONO NW, OANCEA EV. Ion transport in pigmentation. Arch Biochem Biophys. 2014;563C:35–41. doi: 10.1016/j.abb.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUI DM, GREGAN J, JAROSCH E, RAGNINI A, SCHWEYEN RJ. The bacterial magnesium transporter CorA can functionally substitute for its putative homologue Mrs2p in the yeast inner mitochondrial membrane. J Biol Chem. 1999;274:20438–20443. doi: 10.1074/jbc.274.29.20438. [DOI] [PubMed] [Google Scholar]
- CHENG H, FENG JM, FIGUEIREDO ML, ZHANG H, NELSON PL, MARIGO V, BECK A. Transient receptor potential melastatin type 7 channel is critical for the survival of bone marrow derived mesenchymal stem cells. Stem Cells Dev. 2010;19:1393–1403. doi: 10.1089/scd.2009.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUBANOV V, WALDEGGER S, MEDEROS Y SCHNITZLER M, VITZTHUM H, SASSEN MC, SEYBERTH HW, KONRAD M, GUDERMANN T. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A. 2004;101:2894–2899. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLAPHAM DE. TRP channels as cellular sensors. Nature. 2003;426:517–24. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- CONRADT A, WEIDINGER H, ALGAYER H. [Reduced frequency of gestoses in beta-mimetic treated risk pregnancies with added magnesium therapy] Geburtshilfe Frauenheilkd. 1984;44:118–123. doi: 10.1055/s-2008-1036865. [DOI] [PubMed] [Google Scholar]
- COVACCI V, BRUZZESE N, SGAMBATO A, DI FRANCESCO A, RUSSO MA, WOLF FI, CITTADINI A. Magnesium restriction induces granulocytic differentiation and expression of p27Kip1 in human leukemic HL-60 cells. J Cell Biochem. 1998;70:313–322. doi: 10.1002/(sici)1097-4644(19980901)70:3<313::aid-jcb4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- CUI L, XU SM, MA DD, WU BL. The effect of TRPM7 suppression on the proliferation, migration and osteogenic differentiation of human dental pulp stem cells. Int Endod J. 2014;47:583–593. doi: 10.1111/iej.12193. [DOI] [PubMed] [Google Scholar]
- DE BAAIJ JH, HOENDEROP JG, BINDELS RJ. Magnesium in man: implications for health and disease. Physiol Rev. 2015;95:1–46. doi: 10.1152/physrev.00012.2014. [DOI] [PubMed] [Google Scholar]
- DECKER AR, MCNEILL MS, LAMBERT AM, OVERTON JD, CHEN YC, LORCA RA, JOHNSON NA, BROCKERHOFF SE, MOHAPATRA DP, MACARTHUR H, et al. Abnormal differentiation of dopaminergic neurons in zebrafish trpm7 mutant larvae impairs development of the motor pattern. Dev Biol. 2014;386:428–439. doi: 10.1016/j.ydbio.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEMEUSE P, PENNER R, FLEIG A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol. 2006;127:421–434. doi: 10.1085/jgp.200509410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOYLE W, CRAWFORD MA, WYNN AH, WYNN SW. Maternal magnesium intake and pregnancy outcome. Magnes Res. 1989;2:205–210. [PubMed] [Google Scholar]
- GRASCHOPF A, STADLER JA, HOELLERER MK, EDER S, SIEGHARDT M, KOHLWEIN SD, SCHWEYEN RJ. The yeast plasma membrane protein Alr1 controls Mg2+ homeostasis and is subject to Mg2+-dependent control of its synthesis and degradation. J Biol Chem. 2001;276:16216–16222. doi: 10.1074/jbc.M101504200. [DOI] [PubMed] [Google Scholar]
- GRUBBS RD, MAGUIRE ME. Magnesium as a regulatory cation: criteria and evaluation. Magnesium. 1987;6:113–127. [PubMed] [Google Scholar]
- HASHIMOTO T, NISHI K, NAGASAO J, TSUJI S, OYANAGI K. Magnesium exerts both preventive and ameliorating effects in an in vitro rat Parkinson disease model involving 1-methyl-4-phenylpyridinium (MPP+) toxicity in dopaminergic neurons. Brain Res. 2008;1197:143–151. doi: 10.1016/j.brainres.2007.12.033. [DOI] [PubMed] [Google Scholar]
- HERMOSURA MC, NAYAKANTI H, DOROVKOV MV, CALDERON FR, RYAZANOV AG, HAYMER DS, GARRUTO RM. A TRPM7 variant shows altered sensitivity to magnesium that may contribute to the pathogenesis of two Guamanian neurodegenerative disorders. Proc Natl Acad Sci U S A. 2005;102:11510–11515. doi: 10.1073/pnas.0505149102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONG L, SIMON JD. Current understanding of the binding sites, capacity, affinity, and biological significance of metals in melanin. J Phys Chem B. 2007;111:7938–7947. doi: 10.1021/jp071439h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIN J, DESAI BN, NAVARRO B, DONOVAN A, ANDREWS NC, CLAPHAM DE. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science. 2008;322:756–760. doi: 10.1126/science.1163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIN J, WU LJ, JUN J, CHENG X, XU H, ANDREWS NC, CLAPHAM DE. The channel kinase, TRPM7, is required for early embryonic development. Proc Natl Acad Sci U S A. 2012;109:E225–233. doi: 10.1073/pnas.1120033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLISEK M, SPONDER G, MASTROTOTARO L, SMORODCHENKO A, LAUNAY P, VORMANN J, SCHWEIGEL-RONTGEN M. Substitution p.A350V in Na(+)/Mg(2)(+) exchanger SLC41A1, potentially associated with Parkinson’s disease, is a gain-of-function mutation. PLoS One. 2013;8:e71096. doi: 10.1371/journal.pone.0071096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOMIYA Y, SU LT, CHEN HC, HABAS R, RUNNELS LW. Magnesium and embryonic development. Magnes Res. 2014;27:1–8. doi: 10.1684/mrh.2014.0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI FY, CHAIGNE-DELALANDE B, KANELLOPOULOU C, DAVIS JC, MATTHEWS HF, DOUEK DC, COHEN JI, UZEL G, SU HC, LENARDO MJ. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature. 2011;475:471–476. doi: 10.1038/nature10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI M, JIANG J, YUE L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol. 2006;127:525–37. doi: 10.1085/jgp.200609502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU W, SU LT, KHADKA DK, MEZZACAPPA C, KOMIYA Y, SATO A, HABAS R, RUNNELS LW. TRPM7 regulates gastrulation during vertebrate embryogenesis. Dev Biol. 2011;350:348–357. doi: 10.1016/j.ydbio.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOW SE, AMBURGEY K, HORSTICK E, LINSLEY J, SPRAGUE SM, CUI WW, ZHOU W, HIRATA H, SAINT-AMANT L, HUME RI, et al. TRPM7 is required within zebrafish sensory neurons for the activation of touch-evoked escape behaviors. J Neurosci. 2011;31:11633–11644. doi: 10.1523/JNEUROSCI.4950-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAKRIDES M, CROWTHER CA. Magnesium supplementation in pregnancy. Cochrane Database Syst Rev. 2001:CD000937. doi: 10.1002/14651858.CD000937. [DOI] [PubMed] [Google Scholar]
- MATSUURA T, KANAYAMA Y, INOUE T, TAKEDA T, MORISHIMA I. cAMP-induced changes of intracellular free Mg2+ levels in human erythrocytes. Biochim Biophys Acta. 1993;1220:31–36. doi: 10.1016/0167-4889(93)90093-5. [DOI] [PubMed] [Google Scholar]
- MCNEILL MS, PAULSEN J, BONDE G, BURNIGHT E, HSU MY, CORNELL RA. Cell death of melanophores in zebrafish trpm7 mutant embryos depends on melanin synthesis. J Invest Dermatol. 2007;127:2020–2030. doi: 10.1038/sj.jid.5700710. [DOI] [PubMed] [Google Scholar]
- MILLER JC, LANDESMAN R. Magnesium deficiency in embryos of Xenopus laevis. J Embryol Exp Morphol. 1977;39:97–113. [PubMed] [Google Scholar]
- MIYAKE Y, TANAKA K, FUKUSHIMA W, SASAKI S, KIYOHARA C, TSUBOI Y, YAMADA T, OEDA T, MIKI T, KAWAMURA N, et al. Dietary intake of metals and risk of Parkinson’s disease: a case-control study in Japan. J Neurol Sci. 2011;306:98–102. doi: 10.1016/j.jns.2011.03.035. [DOI] [PubMed] [Google Scholar]
- MONTEILH-ZOLLER MK, HERMOSURA MC, NADLER MJ, SCHARENBERG AM, PENNER R, FLEIG A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol. 2003;121:49–60. doi: 10.1085/jgp.20028740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NADLER MJ, HERMOSURA MC, INABE K, PERRAUD AL, ZHU Q, STOKES AJ, KUROSAKI T, KINET JP, PENNER R, SCHARENBERG AM, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- OYANAGI K, KAWAKAMI E, KIKUCHI-HORIE K, OHARA K, OGATA K, TAKAHAMA S, WADA M, KIHIRA T, YASUI M. Magnesium deficiency over generations in rats with special references to the pathogenesis of the Parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Neuropathology. 2006;26:115–128. doi: 10.1111/j.1440-1789.2006.00672.x. [DOI] [PubMed] [Google Scholar]
- PARK MH, WONG BB, LUSK JE. Mutants in three genes affecting transport of magnesium in Escherichia coli: genetics and physiology. J Bacteriol. 1976;126:1096–1103. doi: 10.1128/jb.126.3.1096-1103.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUAMME GA. Molecular identification of ancient and modern mammalian magnesium transporters. Am J Physiol Cell Physiol. 2010;298:C407–429. doi: 10.1152/ajpcell.00124.2009. [DOI] [PubMed] [Google Scholar]
- ROMANI A. Regulation of magnesium homeostasis and transport in mammalian cells. Arch Biochem Biophys. 2007;458:90–102. doi: 10.1016/j.abb.2006.07.012. [DOI] [PubMed] [Google Scholar]
- ROMANI A, SCARPA A. Hormonal control of Mg2+ transport in the heart. Nature. 1990a;346:841–844. doi: 10.1038/346841a0. [DOI] [PubMed] [Google Scholar]
- ROMANI A, SCARPA A. Norepinephrine evokes a marked Mg2+ efflux from liver cells. FEBS Lett. 1990b;269:37–40. doi: 10.1016/0014-5793(90)81113-3. [DOI] [PubMed] [Google Scholar]
- ROMANI AM. Cellular magnesium homeostasis. Arch Biochem Biophys. 2011;512:1–23. doi: 10.1016/j.abb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUNNELS LW. TRPM6 and TRPM7: A Mul-TRP-PLIK-cation of channel functions. Curr Pharm Biotechnol. 2011;12:42–53. doi: 10.2174/138920111793937880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUNNELS LW, YUE L, CLAPHAM DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–1047. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- RYAZANOVA LV, RONDON LJ, ZIERLER S, HU Z, GALLI J, YAMAGUCHI TP, MAZUR A, FLEIG A, RYAZANOV AG. TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat Commun. 2010;1:109. doi: 10.1038/ncomms1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAH R, MESIRCA P, MASON X, GIBSON W, BATES-WITHERS C, VAN DEN BOOGERT M, CHAUDHURI D, PU WT, MANGONI ME, CLAPHAM DE. Timing of myocardial trpm7 deletion during cardiogenesis variably disrupts adult ventricular function, conduction, and repolarization. Circulation. 2013;128:101–114. doi: 10.1161/CIRCULATIONAHA.112.000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLINGMANN KP, WEBER S, PETERS M, NIEMANN NEJSUM L, VITZTHUM H, KLINGEL K, KRATZ M, HADDAD E, RISTOFF E, DINOUR D, et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31:166–170. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- SCHMITZ C, DOROVKOV MV, ZHAO X, DAVENPORT BJ, RYAZANOV AG, PERRAUD AL. The channel kinases TRPM6 and TRPM7 are functionally nonredundant. J Biol Chem. 2005;280:37763–37771. doi: 10.1074/jbc.M509175200. [DOI] [PubMed] [Google Scholar]
- SCHMITZ C, PERRAUD AL, JOHNSON CO, INABE K, SMITH MK, PENNER R, KUROSAKI T, FLEIG A, SCHARENBERG AM. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200. doi: 10.1016/s0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- SHALEV H, PHILLIP M, GALIL A, CARMI R, LANDAU D. Clinical presentation and outcome in primary familial hypomagnesaemia. Arch Dis Child. 1998;78:127–130. doi: 10.1136/adc.78.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLACK C, WARNER AE, WARREN RL. The distribution of sodium and potassium in amphibian embryos during early development. J Physiol. 1973;232:297–312. doi: 10.1113/jphysiol.1973.sp010271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SU LT, LIU W, CHEN HC, GONZALEZ-PAGAN O, HABAS R, RUNNELS LW. TRPM7 regulates polarized cell movements. Biochem J. 2011;434:513–521. doi: 10.1042/BJ20101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANIGUCHI R, NAKAGAWASAI O, TAN-NO K, YAMADERA F, NEMOTO W, SATO S, YAOITA F, TADANO T. Combined low calcium and lack magnesium is a risk factor for motor deficit in mice. Biosci Biotechnol Biochem. 2013;77:266–270. doi: 10.1271/bbb.120671. [DOI] [PubMed] [Google Scholar]
- VOETS T, NILIUS B, HOEFS S, VAN DER KEMP AW, DROOGMANS G, BINDELS RJ, HOENDEROP JG. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem. 2004;279:19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- VOLPE SL. Magnesium in disease prevention and overall health. Adv Nutr. 2013;4:378S–383S. doi: 10.3945/an.112.003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALDER RY, LANDAU D, MEYER P, SHALEV H, TSOLIA M, BOROCHOWITZ Z, BOETTGER MB, BECK GE, ENGLEHARDT RK, CARMI R, et al. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet. 2002;31:171–174. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- WALLINGFORD JB, FRASER SE, HARLAND RM. Convergent extension: the molecular control of polarized cell movement during embryonic development. Dev Cell. 2002;2:695–706. doi: 10.1016/s1534-5807(02)00197-1. [DOI] [PubMed] [Google Scholar]
- WALLINGFORD JB, HABAS R. The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development. 2005;132:4421–4436. doi: 10.1242/dev.02068. [DOI] [PubMed] [Google Scholar]
- WOLF FI, TRAPANI V, SIMONACCI M, BONINSEGNA A, MAZUR A, MAIER JA. Magnesium deficiency affects mammary epithelial cell proliferation: involvement of oxidative stress. Nutr Cancer. 2009;61:131–136. doi: 10.1080/01635580802376360. [DOI] [PubMed] [Google Scholar]
- WOUDENBERG-VRENKEN TE, SUKINTA A, VAN DER KEMP AW, BINDELS RJ, HOENDEROP JG. Transient receptor potential melastatin 6 knockout mice are lethal whereas heterozygous deletion results in mild hypomagnesemia. Nephron Physiol. 2011;117:p11–19. doi: 10.1159/000320580. [DOI] [PubMed] [Google Scholar]
- XIE H, ZHANG Y, ZHANG P, WANG J, WU Y, WU X, NETOFF T, JIANG Y. Functional Study of NIPA2 Mutations Identified from the Patients with Childhood Absence Epilepsy. PLoS One. 2014;9:e109749. doi: 10.1371/journal.pone.0109749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YASUI M, KIHIRA T, OTA K. Calcium, magnesium and aluminum concentrations in Parkinson’s disease. Neurotoxicology. 1992;13:593–600. [PubMed] [Google Scholar]
- YEE NS, ZHOU W, LIANG IC. Transient receptor potential ion channel Trpm7 regulates exocrine pancreatic epithelial proliferation by Mg2+-sensitive Socs3a signaling in development and cancer. Dis Model Mech. 2011;4:240–254. doi: 10.1242/dmm.004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU H, CLAPHAM DE. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc Natl Acad Sci U S A. 2009;106:15750–15755. doi: 10.1073/pnas.0908332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Embryos reared in Mg2+ deficient buffer until tadpole stage displayed movement defects compared to wild type embryos reared in normal buffer. Control tadpoles swam away immediately when touched with a paintbrush. In contrast, Mg2+ deficient tadpoles did not readily respond to touch: some tadpoles swam away abnormally slow whereas others did not respond at all.