

Abstract
Concentrations of the chemokine fractalkine (FKN) are increased in patients with chronic heart failure, and our previous studies show that aged mice lacking the prostaglandin E2 EP4 receptor subtype (EP4-KO) have increased cardiac FKN, with a phenotype of dilated cardiomyopathy. However, how FKN participates in the pathogenesis of heart failure has rarely been studied. We hypothesized that FKN contributes to the pathogenesis of heart failure and that anti-FKN treatment prevents heart failure induced by myocardial infarction (MI) more effectively in EP4-KO mice. Male EP4-KO mice and wild-type littermates underwent sham or MI surgery and were treated with an anti-FKN antibody or control IgG. At 2 weeks post-MI, echocardiography was performed and hearts were excised for determination of infarct size, immunohistochemistry and Western blot of signalling molecules. Given that FKN protein levels in the left ventricle were increased to a similar extent in both strains after MI and that anti-FKN treatment improved survival and cardiac function in both strains, we subsequently used only wild-type mice to examine the mechanisms whereby anti-FKN is cardioprotective. Myocyte cross-sectional area and interstitial collagen fraction were reduced after anti-FKN treatment, as were macrophage migration and gelatinase activity. Activation of ERK1/2 and p38 MAPK were reduced after neutralization of FKN. In vitro, FKN increased fibroblast proliferation. In conclusion, increased FKN contributes to heart failure after MI. This effect is not exacerbated in EP4-KO mice, suggesting that there is no link between FKN and lack of EP4. Overall, inhibition of FKN may be important to preserve cardiac function post-MI.
Introduction
Fractalkine (FKN), also known as CX3CL1, is the only member of the CX3C chemokine family, characterized by the presence of three amino acid residues (X3) localized between two cysteine residues, thus forming a disulfide bond, a CX3C motif, and also a transmembrane domain. The full-length fractalkine protein consists of 397 amino acids encoded by the CX3CL1 gene mapped on chromosome 16 in humans (Pan et al. 1997; Bazan et al. 1997) and of 395 amino acids encoded by the neurotactin gene mapped on chromosome 11 in mice (Rossi et al. 1998). Fractalkine is a unique dual-function chemokine that exists in two forms; a soluble form, which acts as a chemoattractant, and a membrane-bound form acting as an adhesion molecule (Umehara et al. 2004). Fractalkine, its receptor CX3CR1 and monocyte chemoattractant protein 1 have been identified as chemokines and receptors that have an important role in the migration and recruitment of monocytes during the pathogenesis of several inflammatory diseases, including atherosclerosis (Zhou et al. 2012). Fractalkine was recently identified as an independent key factor in the pathogenesis of plaque vulnerability and subsequent plaque rupture (Li et al. 2012). Fractalkine has also been associated with cardiac injury. Patients with acute myocardial infarction (MI) had significantly elevated levels 3 and 12 h after percutaneous coronary intervention compared with patients who had stable angina pain (Njerve et al. 2014). Thus, FKN has a very important role in the pathology of coronary heart disease.
Myocardial infarction is one of the most frequent causes of death. More than 20% of cardiovascular deaths are caused by coronary heart disease (Go et al. 2013). Furthermore, even though many patients survive an acute MI, most of them inevitably suffer from heart failure (HF; Fox et al. 2001), which is likely to result from left ventricular (LV) remodelling, leading to functional decompensation and then HF (Sutton & Sharpe, 2000). Previous studies have confirmed that ischaemia-induced myocardial fibrosis, inflammation and apoptosis are essential factors in the process of cardiac remodelling after MI (Porter & Turner, 2009). There are few studies that have examined the effects of FKN on models of cardiac injury. Xuan et al. (2011) showed that inhibition of FKN reduces cardiac remodelling after transaortic constriction or MI. However, their study did not examine the effects of FKN neutralization on macrophage migration, matrix metalloproteinase activity, myocyte apoptosis or fibroblast proliferation, and a neutralizing antibody was given after the initial wave of inflammation. Also, our study differs from theirs in that we examined the effect of anti-FKN in a cardiac myocyte-selective model of mice lacking the prostaglandin E2 EP4 receptor subtype (EP4-KO), because our earlier studies had shown that older EP4-KO mice had elevated cardiac concentrations of this chemokine. Thus, the mechanism whereby anti-FKN affects the infarcted heart is still not established.
In a previous study from our laboratory, FKN was increased in left ventricular samples from old male EP4-KO mice that displayed a phenotype of dilated cardiomyopathy with reduced ejection fraction (Harding et al. 2010), indicating that prostaglandin E2 acting on its EP4 receptor protects the heart from ischaemic injury. Thus, in the present study we hypothesized that FKN contributes to heart failure and that treatment with anti-FKN prevents heart failure induced by MI. Furthermore, we hypothesized that FKN neutralization would be more effective in EP4-KO mice.
Methods
Ethical approval
All animal experiments were approved by the Henry Ford Health System Institutional Animal Care and Use Committee in accordance with Federal guidelines.
Generation and genotyping of EP4-KO mice
Generation of cardiac myocyte-specific EP4-KO by a Cre-mediated process has been described previously. Genotyping and breeding details have also been described previously (Harding et al. 2010). In our initial experiments, we used both wild-type (WT) and EP4-KO mice. Importantly, these mice are on a mixed genetic background and differ only by the presence or absence of Cre. As our initial experiments in this study showed no difference between strains in their response to anti-FKN, subsequent experiments used only WT mice.
Culture of adult mouse cardiac fibroblasts
Adult mouse cardiac fibroblasts were obtained using a modification of the technique designed to harvest adult mouse cardiac myocytes. Briefly, mice were anaesthetized by inhalation of isoflurane (3% isoflurane with 1 l min−1 oxygen), their hearts were removed and a cannula inserted into the aorta for retrograde perfusion. Mouse hearts were then digested as described previously by us (Harding et al. 2010), and the supernatant obtained after digestion was removed. This supernatant was centrifuged at 100g for 1 min (4°C), and the resulting supernatant, which contains the fibroblasts, was centrifuged at 145g and the pellet resuspended in 5 ml of Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum. Cells were plated on a 60 mm dish and after 2–3 h, the medium was gently removed and fresh medium added. Cells were allowed to grow and typically passaged at a ratio of 1:3. All experiments on cardiac fibroblast proliferation were performed using hearts from 10- to 12-week-old male C57BL/6 mice, and cells were used at passage 3.
Fractalkine enzyme-linked immunosorbent assay (ELISA)
Fractalkine was measured in left ventricular homogenates using a quantitative sandwich enzyme immunoassay (Mouse CX3CL1/Fractalkine Quantikine ELISA, catalogue no. MCX310; R&D Systems Minneapolis, MN, USA). Left ventricular homogenates were centrifuged at 10,000g for 10 min at 4°C. After centrifugation, 100 μl of supernatant was subjected to ELISA according to the manufacturer’s instructions. Samples were measured in duplicate and fractalkine concentrations normalized to total tissue protein, respectively, which was determined in lysates according to the method of Lowry.
Myocardial infarction model and treatment with fractalkine neutralizing antibody (anti-FKN)
Male mice 10–12 weeks old were anaesthetized with pentobarbital (45 mg kg−1) (Akorn, Inc., Lake Forest, IL. USA), intubated, and ventilated with a small rodent ventilator (Harvard Apparatus, Holliston, MA, USA) at a rate of 90 cycles min−1 with a tidal volume of 0.5 ml and a positive end-expiratory pressure of 2 cmH2O. A left side thoracotomy was performed, and the pericardium was incised. Myocardial infarction was then induced through permanent ligation of the left anterior descending coronary artery with an 8–0 silk suture proximal to its bifurcation from the main stem. Sham surgery was performed as described above except that the suture was not tied around the artery. The chest incision was subsequently closed with a 4–0 silk suture. Mice were then allowed to recover in a temperature-controlled environment. After surgery, mice were given a dose of buprenorphine (0.05 mg kg−1 s.c.) every 12 h for 2 days.
We studied the following groups of mice for each strain (the numbers for each group are given in parentheses): WT sham + IgG (8), WT sham + anti-FKN (6), WT MI + IgG (26), WT MI + anti-FKN (25), EP4-KO sham + IgG (7), EP4-KO sham + anti-FKN (5), EP4-KO MI + IgG (29) and EP4-KO MI + anti-FKN (28), for a total of eight different groups. Treatment was started on the day of surgery and continued for 2 weeks. We administered 40 μg kg−1 day−1 i.p. of a function-blocking anti-FKN antibody that was raised against the chemotactic domain of FKN (purified rabbit anti-mouse fractalkine, TP233; Torrey Pines Biolabs, Houston, TX, USA) or rabbit IgG (Protein Mods, Madison, WI, USA) as described by Xuan et al. (2011) and Robinson et al. (2000).
Survival
Survival analysis was performed on all mice. During the study period, the cages were inspected daily for dead animals. All dead mice were examined for the presence of MI as well as cardiac rupture.
Echocardiography
Two weeks after MI, cardiac function was measured with a Doppler echocardiographic system equipped with a 15 MHz linear array transducer (Acuson c256, Mountain View, CA, USA). All studies were performed on conscious mice, which is a standard procedure in our laboratory, and as we have previously described (Yang et al. 1999; Harding et al. 2010, 2011). The following parameters were obtained: (i) LV chamber dimensions and wall thickness, where LVDd is diastolic LV dimension, LVDs is systolic LV dimension and PWTd is diastolic posterior wall thickness; and (i) ejection fraction (LVEF), which is equivalent to [(LVAd – LVAs)/LVAd] × 100, where LVAd is LV diastolic area and LVAs is LV systolic area.
Histological assessment of interstitial collagen fraction (ICF), myocyte cross-sectional area (MCSA), infarct size (IS) of the infarct area
At the end of the experimental period, mice were anaesthetized with sodium pentobarbital (100 mg kg−1 i.p.) and the heart was injected with 15% potassium chloride, leading to its arrest in diastole. The heart was removed, washed in ice-cold PBS, and the atria and right ventricle were dissected. Mouse left ventricles were harvested and sectioned transversely into four slices from apex to base. Three sections (sections A, B and C) were frozen in isopentane and stored at −80°C for determination of ICF, MCSA and IS, as previously described (Qian et al. 2008). Measurements were made using MicroSuite Biological Imaging Software (Melville, NY, USA).
Immunohistochemical staining of fractalkine and macrophages
Frozen sections of left ventricle were used to detect both FKN and macrophage infiltration. Tissue sections (5 μm thick) were allowed to reach room temperature and were then fixed in ice-cold acetone for 10 min. Sections were incubated with 3% hydrogen peroxide to quench endogenous peroxidase, rinsed, and then incubated with 1% bovine serum albumin for 10 min before incubation with primary antibodies against FKN at 1:200 dilution (Cx3Cl1, catalogue no. ab25088; Abcam Cambridge, MA. USA) or the macrophage marker CD68 at 1:200 dilution (CD68, catalogue no. MCA1957; AbD Serotec, Raleigh, NC. USA) at 4°C overnight. Sections were rinsed and then incubated for 40 min with a biotinylated rabbit anti-rat antibody, rinsed, and then incubated with avidin–biotin complex reagent for 40 min. Bound antibodies were visualized following incubation with AEC (3-amino-9-ethylcarbazole) single solution (Invitrogen, Carsbad, CA, USA) for 3–5 min. Sections of experimental tissue were incubated with isotype-matched normal antibody as a negative control. Sections were counterstained with Mayer’s Haematoxylin and dehydrated for microscopic examination. Images were digitally quantified using the ImageJ system (version 1.42; National Institutes of Health, Bethesda, MD, USA) with CD68 positivity assessed in the remote zone and expressed as the number of macrophages per square millimetre.
Effect of fractalkine on fibroblast proliferation
The effect of FKN on proliferation of adult mouse fibroblasts (AVFs) was determined using the TACS MTT cell proliferation kit (Trevigen Inc., Gaithersburg, MD, USA). Briefly, AVFs were cultured until passage 2, and at ~90% confluence were seeded onto a 96-well plate at a density of 20,000 cells per well. They were allowed to attach overnight and were then serum starved for 24 h. Cells were treated with FKN (0–200 ng ml−1) for 24 h in fresh media without serum and were then processed according to the manufacturer’s instructions. Absorbance was measured at 550 nm using a plate reader, and the average reading of wells without cells (blanks) was subtracted from each measurement. All samples were assayed in triplicate.
TUNEL staining for apoptosis
Serial 5-μm-thick sections of left ventricles were cut onto slides. After storage at −80°C, frozen sections were allowed to come to room temperature for 5 min, and then apoptosis-related DNA fragmentation was determined by the terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay using a TUNEL Apoptosis Detection Kit (Millipore, Temecula, CA, USA) following the manufacturer’s instructions. Specimens were examined and photographed with a Nikon eclipse E600 light microscope. Images were captured with the NIS-Elements BR imaging software. On TUNEL-stained sections, apoptotic cells in the border zone were quantified by counting the number of cells in randomly selected fields of a total of five sections. The apoptotic index was expressed as the number of apoptotic cells per field.
Protein extraction and Western blot analysis
Protein was isolated from samples of left ventricle after homogenization in a lysis buffer containing 150 mM NaCl, 50 mM Tris–Cl (pH 7.5), 0.5% deoxycholate, 0.1% SDS and 1% nonidet P-40 with phosphatase and protease inhibitor tablets (PhosSTOP and Complete Mini, EDTA-free protease inhibitor cocktail tablets; Roche Diagnostics, Indianapolis, IN. USA). After homogenization, samples were centrifuged at 3000g for 10 min (4°C), and the supernatants were removed for Western blot analysis. The protein concentration was determined with the Coomassie protein assay kit using bovine serum albumin as the standard. Aliquots of samples (50 μg protein) were subjected to SDS-PAGE and electrotransferred to a polyvinyl difluoride membrane at 36 V overnight at 4°C. The membrane was incubated in 5% non-fat milk in PBS containing 0.1% Tween 20 (PBS-T) for 1 h at room temperature and then overnight at 4°C in the same buffer containing primary antibodies to phospho-p38 mitogen-activated protein kinase (p-p38 MAPK, catalogue no. 4511S; p38 MAPK, catalogue no. 9212S; Cell Signaling, Danvers, MA. USA) or phospho-extracellular regulated signal kinase 1/2 (p-ERK1/2, catalogue no. 9101 L; ERK1/2, catalogue no. 9102; Cell Signaling). The membrane was washed with PBS-T and incubated with horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h. After being washed, the membrane was developed with SuperSignal West Pico Chemiluminescent reagent (Thermoscientific, Rockford, IL. USA) at room temperature. The signal was detected through an exposure to Fuji RX film and analysed by scanning densitometry. The target protein was normalized to the loading control and expressed as the fold increase versus control.
In situ zymography
In situ zymography was performed on frozen sections as previously described (Tsujita et al. 2007). Fluorescein-conjugated, dye-quenched gelatin from pig skin (DQ™-gelatin) was obtained from Molecular Probes (Invitrogen, Basel, Switzerland). A 1 mg ml−1 stock solution of DQ-gelatin was prepared in gelatinase reaction buffer (150 mM NaCl, 5 mM CaCl2, 0.2 mM NaN3 and 50 mM Tris–HCl, pH 7.6) and stored at 4°C. The working solution for in situ zymography was made by directly diluting DQ-gelatin stock solution in reaction buffer to a final concentration of 20 μg ml−1. Unfixed cryosections were thawed, rounded with a wax pen, overlaid with 25 μl DQ-gelatin working solution and incubated at 37°C in an humidified dark chamber for 5 h, and pictures were immediately taken under the microscope. Control sections were incubated in the presence of 20 mM EDTA, which inhibits gelatinase activity. Digital images showing the infarction borders were taken under the ×20 objective, and a threshold was set using ImageJ software that encompassed only the bright green fluorescence indicative of gelatinase activity. The percentage area of gelatinase activity was calculated for each animal per field and the mean calculated for each group.
Statistical analyses
Results are shown as means ± SEM. Comparisons among groups were analysed using one-way ANOVA followed by Hochberg’s post hoc test. Comparisons between two independent groups were analysed using Student’s unpaired t test when variances were equal, and a Satterwaite correction was used when variances were different. A log rank test was used to compare survival distributions. Values of P < 0.05 were considered to be statistically significant.
Results
Fractalkine expression in mouse heart
Fractalkine protein levels were increased in the hearts of mice failing because of induced MI. The results of immunohistochemistry clearly showed increased expression of FKN protein in infarcted hearts (Fig. 1). Suprisingly, quantitative analysis using ELISA on samples of homogenized left ventricle showed that FKN protein was not different between sham WT and sham EP4-KO. Furthermore, FKN protein increased in both strains after MI, with no statistical difference between strains (bar graph in Fig. 1).
Figure 1. Effect of myocardial infarction (MI) on fractalkine (FKN) expression in the heart.

A, representative immunohistochemistry staining of FKN. Scale bar is 100 μm. B, quantitative analysis of FKN in left ventricle samples by enzyme-linked immunosorbent assay. Data are presented as nanograms of FKN per milligram of protein. n = 3 per group. ***P ≤ 0.005 versus respective sham-operated control mice. Abbreviations: EP4-KO, cardiac myocyte-selective deficiency of prostaglandin E2 EP4 receptor subtype; and WT, wild-type.
Anti-fractalkine treatment improves survival after MI
After MI, we observed ~60% mortality, which did not differ between strains (Fig. 2), with the majority of mice dying within the first 7 days, due to cardiac rupture. Treatment with anti-FKN improved the survival rate in both strains. In WT mice treated with anti-FKN, 16 mice from a total of 25 mice survived (64% survival rate), whereas only 11 mice from a total of 26 mice given control IgG survived (42% survival rate, P < 0.05; Fig. 2 left panel). Similar effects were noted in EP4-KO mice, where 19 of 28 mice treated with anti-FKN survived (68% survival), whereas only 12 of 29 given IgG survived (41% survival, P < 0.05; Fig. 2 right panel).
Figure 2. Kaplan–Meier curves demonstrating increased survival after MI in both wild-type (WT; left panel) and prostaglandin E2 EP4 receptor subtype-deficient (EP4-KO) mice (right panel) treated with anti-FKN compared with IgG-treated control mice.

Differences in survival between MI mice treated with anti-FKN versus IgG-treated control MI mice were tested with the log-rank test. P < 0.05 for both strains. The number of mice in each group is given as (number surviving/total number of mice) after the group description.
Anti-fractalkine treatment improves cardiac function after MI
All echocardiography data for WT mice are presented in Table 1, and representative echocardiography data are shown in Fig. 3A, with quantitative data for ejection fraction (LVEF) and left ventricular dimension at systole (LVDs) depicted in Fig. 3B and C, respectively. As expected, 2 weeks post-MI there were significant decreases in LVEF accompanied by increases in LVDs for both strains. Anti-fractalkine treatment increased LVEF in both EP4-KO and WT mice after MI (Fig. 3B), suggesting improved cardiac function after FKN neutralization. Likewise, the increase in left ventricular dimension after MI was significantly reduced by anti-FKN treatment in WT mice (from 4.3 ± 0.44 to 2.48± 0.44 mm for systole and from 5.03 ± 0.31 to 3.74 ± 0.35 mm for diastole, P < 0.01).
Table 1.
Echocardiography data of wild-type mice treated with anti-fractalkine
| Parameter | Sham + IgG (n = 8) | Sham + anti-FKN (n = 6) | MI + IgG (n = 7) | MI + anti-FKN (n = 7) |
|---|---|---|---|---|
| HR (beats min−1) | 691 ± 21 | 712 ± 14 | 708 ± 8 | 675 ± 29 |
| BW (g) | 30.9 ± 1.7 | 27.8 ± 0.8 | 29.7 ± 0.7 | 31.3 ± 0.8 |
| LVEF (%) | 80.40 ± 2.8 | 79.40 ± 3.9 | 37.44 ± 4.3** | 57.23 ± 6.9†† |
| LVDd (mm) | 2.75 ± 0.09 | 2.80 ± 0.12 | 5.03 ± 0.31** | 3.74 ± 0.35†† |
| LVDs (mm) | 1.26 ± 0.09 | 1.10 ± 0.11 | 4.30 ± 0.44*** | 2.48 ± 0.44† |
| PWTd (mm) | 0.99 ± 0.08 | 0.90 ± 0.04 | 0.76 ± 0.04* | 0.95 ± 0.07 |
| LVAs (mm2) | 0.90 ± 0.16 | 0.90 ± 0.22 | 9.68 ± 1.97** | 4.27 ± 1.18† |
| LVAd (mm2) | 4.64 ± 0.59 | 4.1 ± 0.25 | 14.66 ± 2.44** | 8.75 ± 1.51 |
| SF (%) | 54.17 ± 2.70 | 59.1 ± 4.18 | 19.04 ± 4.74*** | 33.66 ± 6.29 |
Abbreviations: BW, body weight; FKN, fractalkine; HR, heart rate; LVAd, left ventricular area at diastole; LVAs, left ventricular area at systole; LVDd, left ventricular dimension at diastole; LVDs, left ventricular dimension at systole; LVEF, left ventricular ejection fraction; MI, myocardial infarction; PWTd, posterior wall thickness at diastole; and SF, shortening fraction.
P < 0.05,
P < 0.01 and
P < 0.005 versus sham MI;
P < 0.05 and
P < 0.01 versus MI + IgG.
Figure 3. Effect of anti-FKN treatment on left ventricle ejection fraction (LVEF) and systolic dimension (LVDs) in WT and EP4-KO mice 2 weeks post-MI.

A, representative M-mode echocardiograms. B, quantitative analysis of LVEF in WT and EP4-KO mice. C, quantitative analysis of LVDs in WT mice. n = 5–9 per group. ***P < 0.005 versus IgG-treated sham-operated mice; ††P < 0.01 and †††P < 0.005 versus IgG-treated MI mice.
Thus, contrary to our original hypothesis, anti-FKN treatment was equally effective in both strains; therefore, our subsequent studies examined the mechanisms responsible for this protective effect in WT mice only.
Anti-fractalkine treatment reduces infarct size and prevents cardiac hypertrophy and remodelling after MI
Trichrome staining was used to detect the size of the infarcted area 2 weeks post-MI. Infarct size was significantly reduced after treatment with anti-FKN (Fig. 4).
Figure 4. Effect of anti-FKN treatment on infarct size in WT mice 2 weeks post-MI.
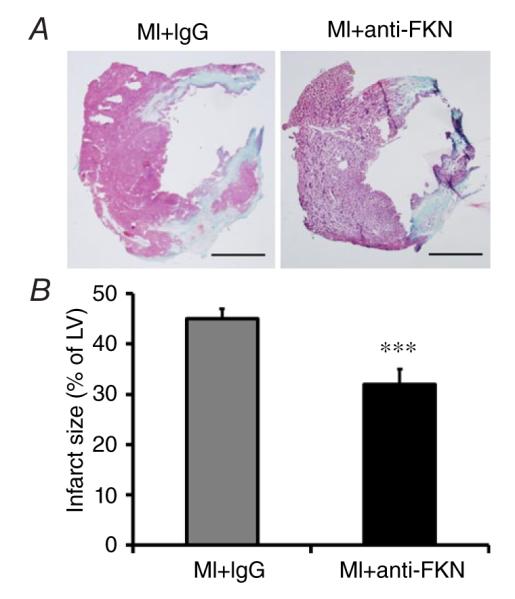
A, representative trichrome staining, showing infarcted portion of the LV. Scale bar is 500 μm. B, quantitative analysis of infarct size. n = 6–9 per group. ***P < 0.005 versus MI group treated with IgG.
As expected, both MCSA and ICF were increased following MI, and Fig. 5B demonstrates that both parameters were reduced by anti-FKN treatment.
Figure 5. Effect of anti-FKN treatment on myocyte cross-sectional area (MCSA) and interstitial collagen fraction (ICF) in WT mice 2 weeks post-MI.

A, representative images of myocyte size and interstitial collage deposition. Scale bar is 100 μm. Quantitative analysis of MCSA (B) and ICF (C). n = 5–9 per group. ***P < 0.005 versus IgG-treated sham-operated mice; ††P < 0.01 and †††P < 0.005 versus MI group treated with IgG.
Anti-fractalkine treatment reduces macrophage infiltration
To investigate the underlying mechanism(s) contributing to the protection afforded by anti-FKN treatment, we analysed macrophage infiltration in LV sections taken 14 days after MI. As expected, immunohistochemistry demonstrated a significantly increased number of macrophages (CD68 positive) in mice subject to MI compared with sham-operated mice. Figure 6 shows that anti-FKN treatment decreased macrophage infiltration in the remote zone post-MI, suggesting that anti-FKN treatment reduces macrophage activation and inflammation after MI.
Figure 6. Effect of anti-FKN treatment on macrophage infiltration in WT mice 2 weeks post-MI.

A, representative immunohistochemical staining showing infiltrated macrophages (CD68+ cells, purple colour) in the left ventricle. Scale bar is 100 μm. B, quantitative analysis of macrophage infiltration. n = 4–9 per group. ***P < 0.005 versus IgG-treated sham-operated mice; †P < 0.05 versus MI group treated with IgG.
Anti-fractalkine treatment inhibits apoptosis post-MI
At 14 days after MI, apoptosis levels in the myocardium were determined by TUNEL staining. As shown in Fig. 7, the apoptotic index (number of TUNEL-positive cells per slide) in the MI group was significantly higher than in the sham-operated group, and anti-FKN treatment reduced the number of apoptotic cells post-MI.
Figure 7. Effect of anti-FKN treatment on apoptosis in WT mice 2 weeks post-MI.

A, representative images of TUNEL staining for apoptosis (bright green spots) in the left ventricle. Scale bar is 200 μm. B, quantitative analysis of apoptotic index. n = 3 per group. ***P < 0.005 versus IgG-treated sham-operated mice; ††P < 0.01 versus MI group treated with IgG.
Anti-fractalkine treatment reduces gelatinase activity
Figure 8 shows representative in situ gelatin zymography of the LV from sham + IgG, MI + IgG and MI + anti-FKN mice. There was minimal matrix metalloproteinase activity in the sham group. Fourteen days after MI, we observed markedly enhanced gelatinase in the LV with significant attenuation of matrix metalloproteinase activity following anti-FKN treatment. The gelatinolytic area in sham-operated mice was 1.32% and was increased to 43.4% in mice subjected to MI. Treatment with the FKN neutralizing antibody reduced the gelatinolytic area to 19.3% (P < 0.05 compared with the MI group).
Figure 8. In situ zymography, showing gelatinase activity (green fluorescence) in the left ventricle.

The MI + EDTA slide is a control indicating suppression of the fluorescence (inhibiting gelatinase activity) with EDTA. Scale bar is 200 μm. Images are representative of three different animals. Bar graph shows quantification of gelatinase activity expressed as the percentage area after threshold with n = 3 mice.
Effect of FKN on proliferation of cultured mouse ventricular fibroblasts
Given that treatment with anti-FKN reduced interstitial collagen fraction, we determined the effect of FKN on proliferation of mouse cardiac fibroblasts in vitro. As shown in Fig. 9, treatment with FKN for 24 h significantly increased fibroblast proliferation at concentrations ≥50 ng ml−1. Although the lower concentrations of 5 and 10 ng ml−1 also increased proliferation, these changes did not achieve statistical significance (P = 0.09 and P = 0.06, respectively).
Figure 9. Effect of FKN on adult cardiac fibroblast proliferation.

Adult cardiac fibroblasts at passage 3 were treated with the stated concentrations of FKN for 24 h in serum-free media, and proliferation was assessed by the MTT assay with absorbance measured at 550 nm. Histogram shows FKN concentration in nanograms per millilitre on the x-axis, and the y-axis is percentage of control, with the absorbance values in vehicle-treated cells set at 100%. n = 5 separate fibroblast preparations. *P < 0.05 and ***P < 0.005 compared with vehicle control.
Anti-fractalkine treatment reduces phosphorylation of p38 MAPK and ERK1/2 MAPK after MI
Consistent with previously reported data, Fig. 10 shows that both phospho-p38 and phospho-ERK1/2 are increased in the left ventricle following MI. Treatment with anti-FKN reduced the increases noted after MI, whereas anti-FKN had no effect on these MAPKs in sham-operated animals.
Figure 10. Samples of left ventricle were examined for p38 mitogen-activated protein kinase (MAPK; left panels) and extracellular signal-related kinase 1/2 (ERK1/2; right panels) by Western blotting.

Representative Western blots for WT mice are shown in the upper part of the figure and quantitative data are shown in the bar graphs below. Phosphorylated p38 and ERK1/2 and are corrected to total p38 and ERK1/2, respectively. *P < 0.05 and **P < 0.01 versus IgG-treated sham-operated mice; †P < 0.05 versus MI mice treated with IgG. n = 3 for each group.
Discussion
This study demonstrates that anti-FKN therapy significantly retards remodelling and HF in both WT and EP4-KO mice after myocardial infarction. The key findings in this study are as follows: (i) anti-FKN therapy improved survival after permanent occlusion of the left anterior descending coronary artery in comparison to treatment with control IgG; (ii) anti-FKN therapy led to a significant reduction in infarct size after MI and remarkable improvements in cardiac remodelling and function during HF; (iii) anti-FKN therapy attenuated macrophage infiltration into heart tissue during MI and inhibited myocyte apoptosis, and in vitro tests also showed that FKN increased the proliferation of adult mouse cardiac fibroblasts; and (iv) anti-FKN-mediated cardioprotection was associated with a decrease in gelatinase activity and phosphorylation of p38 MAPK and ERK1/2. These data provide additional insights into the pleiotropic effects of FKN in cardiovascular disease and its therapeutic role in ischaemia-induced HF.
Our previous study reported that FKN expression and production were increased in the hearts of aged (28- to 32-week-old) male EP4-KO mice (Taube et al. 2013), whereas our present study used younger mice subjected to MI and showed that FKN expression and production was increased in both WT and EP4-KO mice. Cardiac function data showed that the effects of FKN neutralization were similar in both strains, so we concluded that the presence or absence of the EP4 receptor does not affect responses to anti-FKN and we performed subsequent experiments using only the one strain. These results are consistent with our data showing that neither prostaglandin E2 nor an EP4 agonist regulates FKN from cardiac myocytes (Taube et al. 2013).
In clinical investigations, expression of CX3CL1 (FKN) in endomyocardial biopsy was significantly increased in enterovirus-positive and virus-negative patients with myocarditis (Escher et al. 2011). Circulating levels of CX3CL1 were also increased in patients with chronic HF in accordance with disease severity [1.6-fold in New York Heart Association (NYHA) grade II, 2.2-fold in NYHA grade III and 2.9-fold in NYHA grade IV], with CX3CL1 protein production increased in both cardiomyocytes and fibrous tissue. However, in addition to FKN being a marker for heart failure, in vitro experiments showed that the addition of FKN to neonatal cardiomyocytes could induce the expression of markers of cardiac hypertrophy and protein phosphatases. Taken together with recent data from our laboratory showing that FKN can reduce the magnitude and speed of contraction in adult mouse myocytes, the data suggest that FKN may have a pathophysiological role in heart failure and that its inhibition or neutralization may be an effective therapeutic strategy (Husberg et al. 2008).
In our study, we found that the majority of mice died within 1 week following MI surgery, primarily due to cardiac rupture, and anti-FKN treatment remarkably reduced mortality. To elucidate the mechanism, we investigated the change of inflammation and apoptosis after MI. Inflammation is a crucial factor in the post-MI healing process. An excessive inflammatory response in the infarcted myocardium is related to adverse cardiac events, including cardiac rupture, and anti-inflammation therapy can protect from cardiac rupture (Anzai, 2013). Another important factor determining survival is apoptosis. Myocardial remodelling following MI is emerging as a key cause of chronic heart failure. Following MI, both the infarcted region and the remote non-infarcted zone undergo cardiac remodelling as a part of the wound-healing response. The LV healing response can be divided into two overlapping phases. The first step is ischaemia-induced myocyte necrosis, as cardiomyocyte death triggers the rapid activation of the complement system. Next, the viable cardiomyocytes undergo compensatory hypertrophy, which helps to maintain cardiac output (Stefanon et al. 2013; Shinde & Frangogiannis, 2014). At the same time, fibroblasts proliferate and release fibrosis-promoting cytokines that contribute positively to scar formation, with the net result being increased extracellular matrix synthesis and deposition (Shinde & Frangogiannis, 2014). Myocyte cross-sectional area and ICF, indices of cardiac hypertrophy and fibrosis, were both examined in the present study. Anti-FKN treatment reduced both the increased MCSA and ICF after MI, accompanied by an improvement in cardiac function. Subsequent in vitro experiments showed that FKN increased adult mouse cardiac fibroblast proliferation; a novel finding that supports the in vivo data showing that anti-FKN reduces the deposition of interstitial collagen. As expected, apoptosis was increased after MI, and this effect was reduced by anti-FKN treatment. Moreover, we also observed decreased macrophage migration and gelatinase activity following anti-FKN treatment, which would be expected to limit cardiac remodelling.
In the present study, we began to elucidate how neutralization of FKN exerts protective effects. Many signalling pathways have previously been implicated in the pathology of MI. Both ERK1/2 and p38 MAPK have widely been reported in the remodelling of heart disease, (Liu et al. 2005; Lei et al. 2013; Pramod & Shivakumar, 2014), with Liu et al. (2005) reporting that inhibition of p38 MAPK reduces MCSA and ICF. It has been suggested that activation of ERK1/2 promotes cardiac inflammation, hypertrophy and fibrosis (Kehat & Molkentin, 2010). In cultured cardiac fibroblasts, FKN-induced phosphorylation of ERK1/2 participates in angiotensin II-mediated inflammation, myocardial injury and dysfunction (Song et al. 2013). In human microvascular endothelial cells and human umbilical vein endothelial cells, FKN induces significant alterations in cytoskeletal structure and specifically activates MAPKs (Volin et al. 2010), suggesting that FKN can induce pathological changes in the heart by these pathways. In support of these previous studies, the present study shows that anti-FKN treatment reduces the enhanced ERK1/2 phosphorylation and p38 MAPK phosphorylation observed after MI.
In conclusion, the present study has demonstrated that anti-FKN treatment has a cardioprotective effect in both EP4-KO and WT mice subject to MI. In contrast to our original hypothesis, EP4-KO mice did not have an exaggerated response to FKN neutralization, and FKN levels after MI were similar in both strains. Our end-points focused on functional and pathophysiological changes and histological remodelling of the heart, with a preliminary examination of the molecular and cellular mechanisms and signalling pathways that lead to the cardioprotective effect of FKN neutralization in this particular model. Whether a similar approach would be protective in other models of cardiac injury or in the clinical setting remains to be elucidated.
New Findings.
-
What is the central question of this study?
What is the cardioprotective role of fractalkine neutralization in heart failure and what are the mechanisms responsible?
-
What is the main finding and its importance?
The concentration of fractalkine is increased in the left ventricle of mice with myocardial infarction, similar to the increases in plasma from heart failure patients. The present study shows a clear beneficial effect of neutralizing fractalkine in a model of myocardial infarction, which results in increased survival. Such an approach may be worthwhile in human patients.
Acknowledgements
The authors would like to thank David Taube and Gulser Gurocak for excellent technical assistance. Thanks also to Edward Shesely for the genotyping and maintenance of the EP4-KO mouse colony.
Funding
These studies were funded by a National Institutes of Health grant [5P01HL028982] (sub-project 2) to P.H.
Footnotes
Competing interests
None declared. The results presented in this paper were presented in abstract form at the Council for High Blood Pressure Research Annual Meeting, San Francisco, September 2014.
Author contributions
Study design: P.H. Myocardial infarction, Western blot, gelatinase activity and immunohistochemistry: X.G. Echocardiography: X.G., J.X. and X.-P.Y. Data analysis: E.P. Initial draft of manuscript: P.H. and X.G. Manuscript editing and final approval: X.G., J.X., X.-P.Y., E.P. and P.H.
References
- Anzai T. Post-infarction inflammation and left ventricular remodeling: a double-edged sword. Circ J. 2013;77:580–587. doi: 10.1253/circj.cj-13-0013. [DOI] [PubMed] [Google Scholar]
- Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- Escher F, Vetter R, Küuhl U, Westermann D, Schultheiss HP, Tschöpe C. Fractalkine in human inflammatory cardiomyopathy. Heart. 2011;97:733–739. doi: 10.1136/hrt.2010.205716. [DOI] [PubMed] [Google Scholar]
- Fox KF, Cowie MR, Wood DA, Coats AJ, Gibbs JS, Underwood SR, Turner RM, Poole-Wilson PA, Davies SW, Sutton GC. Coronary artery disease as the cause of incident heart failure in the population. Eur Heart J. 2001;22:228–236. doi: 10.1053/euhj.2000.2289. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Executive summary: heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- Harding P, Yang XP, He Q, LaPointe MC. Lack of microsomal prostaglandin E synthase-1 reduces cardiac function following angiotensin II infusion. Am J Physiol Heart Circ Physiol. 2011;300:H1053–H1061. doi: 10.1152/ajpheart.00772.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding P, Yang XP, Yang J, Shesely E, He Q, LaPointe MC. Gene expression profiling of dilated cardiomyopathy in older male EP4 knockout mice. Am J Physiol Heart Circ Physiol. 2010;298:H623–H632. doi: 10.1152/ajpheart.00746.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husberg C, Nygård S, Finsen AV, Damås JK, Frigessi A, Oie E, Waehre A, Gullestad L, Aukrust P, Yndestad A, Christensen G. Cytokine expression profiling of the myocardium reveals a role for CX3CL1 (fractalkine) in heart failure. J Mol Cell Cardiol. 2008;45:261–269. doi: 10.1016/j.yjmcc.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Kehat I, Molkentin JD. Extracellular signal-regulated kinase 1/2 (ERK1/2) signaling in cardiac hypertrophy. Ann N Y Acad Sci. 2010;1188:96–102. doi: 10.1111/j.1749-6632.2009.05088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J, Xue S, Wu W, Zhou S, Zhang Y, Yuan G, Wang J. Sdc1 overexpression inhibits the p38 MAPK pathway and lessens fibrotic ventricular remodeling in MI rats. Inflammation. 2013;36:603–615. doi: 10.1007/s10753-012-9582-y. [DOI] [PubMed] [Google Scholar]
- Li J, Guo Y, Luan X, Qi T, Li D, Chen Y, Ji X, Zhang Y, Chen W. Independent roles of monocyte chemoattractant protein-1, regulated on activation, normal T-cell expressed and secreted and fractalkine in the vulnerability of coronary atherosclerotic plaques. Circ J. 2012;76:2167–2173. doi: 10.1253/circj.cj-11-1457. [DOI] [PubMed] [Google Scholar]
- Liu YH, Wang D, Rhaleb NE, Yang XP, Xu J, Sankey SS, Rudolph AE, Carretero OA. Inhibition of p38 mitogen-activated protein kinase protects the heart against cardiac remodeling in mice with heart failure resulting from myocardial infarction. J Card Fail. 2005;11:74–81. doi: 10.1016/j.cardfail.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Njerve IU, Solheim S, Lunde K, Hoffmann P, Arnesen H, Seljeflot I. Fractalkine levels are elevated early after PCI-treated ST-elevation myocardial infarction; no influence of autologous bone marrow derived stem cell injection. Cytokine. 2014;69:131–135. doi: 10.1016/j.cyto.2014.05.022. [DOI] [PubMed] [Google Scholar]
- Pan Y, Lloyd C, Zhou H, Dolich S, Deeds J, Gonzalo JA, Vath J, Gosselin M, Ma J, Dussault B, Woolf E, Alperin G, Culpepper J, Gutierrez-Ramos JC, Gearing D. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature. 1997;387:611–617. doi: 10.1038/42491. [DOI] [PubMed] [Google Scholar]
- Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Pramod S, Shivakumar K. Mechanisms in cardiac fibroblast growth: an obligate role for Skp2 and FOXO3a in ERK1/2 MAPK-dependent regulation of p27kip1. Am J Physiol Heart Circ Physiol. 2014;306:H844–H855. doi: 10.1152/ajpheart.00933.2013. [DOI] [PubMed] [Google Scholar]
- Qian JY, Harding P, Liu Y, Shesely E, Yang XP, LaPointe MC. Reduced cardiac remodeling and function in cardiac-specific EP4 receptor knockout mice with myocardial infarction. Hypertension. 2008;51:560–566. doi: 10.1161/HYPERTENSIONAHA.107.102590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LA, Nataraj C, Thomas DW, Howell DN, Griffiths R, Bautch V, Patel DD, Feng L, Coffman TM. A role for fractalkine and its receptor (CX3CR1) in cardiac allograft rejection. J Immunol. 2000;165:6067–6072. doi: 10.4049/jimmunol.165.11.6067. [DOI] [PubMed] [Google Scholar]
- Rossi DL, Hardiman G, Copeland NG, Gilbert DJ, Jenkins N, Zlotnik A, Bazan JF. Cloning and characterization of a new type of mouse chemokine. Genomics. 1998;47:163–170. doi: 10.1006/geno.1997.5058. [DOI] [PubMed] [Google Scholar]
- Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. doi: 10.1016/j.yjmcc.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Zhang ZZ, Zhong JC, Yu XY, Oudit GY, Jin HY, Lu L, Xu YL, Kassiri Z, Shen WF, Gao PJ, Zhu DL. Loss of angiotensin-converting enzyme 2 exacerbates myocardial injury via activation of the CTGF-fractalkine signaling pathway. Circ J. 2013;77:2997–3006. doi: 10.1253/circj.cj-13-0805. [DOI] [PubMed] [Google Scholar]
- Stefanon I, Valero-Muñoz M, Fernandes AA, Ribeiro RF, Jr, Rodríguez C, Miana M, Martínez-González J, Spalenza JS, Lahera V, Vassallo PF, Cachofeiro V. Left and right ventricle late remodeling following myocardial infarction in rats. PLoS ONE. 2013;8:e64986. doi: 10.1371/journal.pone.0064986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- Taube D, Xu J, Yang XP, Undrovinas A, Peterson E, Harding P. Fractalkine depresses cardiomyocyte contractility. PLoS ONE. 2013;8:e69832. doi: 10.1371/journal.pone.0069832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujita K, Kaikita K, Hayasaki T, Honda T, Kobayashi H, Sakashita N, Suzuki H, Kodama T, Ogawa H, Takeya M. Targeted deletion of class A macrophage scavenger receptor increases the risk of cardiac rupture after experimental myocardial infarction. Circulation. 2007;115:1904–1911. doi: 10.1161/CIRCULATIONAHA.106.671198. [DOI] [PubMed] [Google Scholar]
- Umehara H, Bloom ET, Okazaki T, Nagano Y, Yoshie O, Imai T. Fractalkine in vascular biology: from basic research to clinical disease. Arterioscler Thromb Vasc Biol. 2004;24:34–40. doi: 10.1161/01.ATV.0000095360.62479.1F. [DOI] [PubMed] [Google Scholar]
- Volin MV, Huynh N, Klosowska K, Reyes RD, Woods JM. Fractalkine-induced endothelial cell migration requires MAP kinase signaling. Pathobiology. 2010;77:7–16. doi: 10.1159/000272949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan W, Liao Y, Chen B, Huang Q, Xu D, Liu Y, Bin J, Kitakaze M. Detrimental effect of fractalkine on myocardial ischaemia and heart failure. Cardiovasc Res. 2011;92:385–393. doi: 10.1093/cvr/cvr221. [DOI] [PubMed] [Google Scholar]
- Yang XP, Liu YH, Rhaleb NE, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am J Physiol Heart Circ Physiol. 1999;277:H1967–H1974. doi: 10.1152/ajpheart.1999.277.5.H1967. [DOI] [PubMed] [Google Scholar]
- Zhou JJ, Wang YM, Lee VW, Phoon RK, Zhang GY, Wang Y, Tan TK, Hu M, Wang LD, Saito M, Sawyer A, Harris DC, Alexander SI, Durkan AM. DEC205-DC targeted DNA vaccines to CX3CR1 and CCL2 are potent and limit macrophage migration. Int J Clin Exp Med. 2012;5:24–33. [PMC free article] [PubMed] [Google Scholar]