

ABSTRACT
Biofilm formation is responsible for increased antibiotic tolerance in pathogenic bacteria. Cyclic di-GMP (c-di-GMP) is a widely used second-messenger signal that plays a key role in bacterial biofilm formation. c-di-GMP is synthesized by diguanylate cyclases (DGCs), a conserved class of enzymes absent in mammals and hence considered attractive molecular targets for the development of antibiofilm agents. Here, the results of a virtual screening approach aimed at identifying small-molecule inhibitors of the DGC PleD from Caulobacter crescentus are described. A three-dimensional (3D) pharmacophore model, derived from the mode of binding of GTP to the active site of PleD, was exploited to screen the ZINC database of compounds. Seven virtual hits were tested in vitro for their ability to inhibit the activity of purified PleD by using circular dichroism spectroscopy. Two drug-like molecules with a catechol moiety and a sulfonohydrazide scaffold were shown to competitively inhibit PleD at the low-micromolar range (50% inhibitory concentration [IC50] of ∼11 μM). Their predicted binding mode highlighted key structural features presumably responsible for the efficient inhibition of PleD by both hits. These molecules represent the most potent in vitro inhibitors of PleD identified so far and could therefore result in useful leads for the development of novel classes of antimicrobials able to hamper biofilm formation.
IMPORTANCE Biofilm-mediated infections are difficult to eradicate, posing a threatening health issue worldwide. The capability of bacteria to form biofilms is almost universally stimulated by the second messenger c-di-GMP. This evidence has boosted research in the last decade for the development of new antibiofilm strategies interfering with c-di-GMP metabolism. Here, two potent inhibitors of c-di-GMP synthesis have been identified in silico and characterized in vitro by using the well-characterized DGC enzyme PleD from C. crescentus as a structural template and molecular target. Given that the protein residues implied as crucial for enzyme inhibition are found to be highly conserved among DGCs, the outcome of this study could pave the way for the future development of broad-spectrum antibiofilm compounds.
INTRODUCTION
In the last decade, the nucleotide cyclic di-GMP (c-di-GMP) has emerged as the most common bacterial second messenger able to elicit different cellular responses, including virulence, motility, adhesion, and biofilm development (1, 2). c-di-GMP promotes biofilm formation by stimulating the biosynthesis of adhesins and exopolysaccharide matrix substances and by inhibiting various forms of motility (3). Intracellular levels of c-di-GMP are modulated by the opposite activities of diguanylate cyclase (DGC) enzymes (containing the conserved GGDEF domain), which synthetize this second messenger from two GTP molecules, and of phosphodiesterase (PDE) enzymes (containing either the EAL or the HD-GYP domain), which hydrolyze it to pGpG and GMP, respectively. DCGs and PDEs usually contain signaling domains that function as sensors of environmental or cellular cues to modulate their activity and, consequently, c-di-GMP intracellular levels (4).
The lack of conserved domains involved in c-di-GMP turnover in mammalian genomes suggests that small molecules targeting DGCs may represent promising hits for the development of antibiofilm drugs. Currently, different approaches to inhibit c-di-GMP signaling have been described (5), which are based mainly on whole-cell assays or in vitro screening of small-molecule libraries (6–9). Besides these strategies, structure-based rational design represents an important tool to retrieve novel molecules and gain mechanistic knowledge to target c-di-GMP signaling in bacteria. A repertoire of such approaches belongs to studies aimed at targeting the catalytic site (10) or the inhibitory site (I site) (where c-di-GMP binds as a negative allosteric regulator) of DGCs (11–13).
Regarding the I site, a series of c-di-GMP analogues have been designed, synthesized, and tested for their ability to lock the DGC enzymes in an inactive conformation (5). However, these compounds are likely ineffective against those DGCs lacking an I site (14) and are linearized in vitro by the EAL subtype of PDEs, if one of the natural phosphodiester bonds is conserved (15).
As for the active site of DGCs, virtual screening studies were previously attempted. Virtual hits for Rv1354c, a potential target of antituberculosis drugs containing both GGDEF and EAL domains, have been identified but not tested in vitro (10). In a previous study, four molecules targeting the active site of the DGC PleD from Caulobacter crescentus were identified by virtual screening (16). These molecules were able to weakly inhibit in vitro the DGC WspR from Pseudomonas aeruginosa only at high concentrations (50% inhibitory concentration [IC50] ranging from 45 μM to 100 μM) but were able to decrease biofilm levels in both P. aeruginosa and Acinetobacter baumannii (16), providing a proof of concept that targeting DGCs is a feasible strategy to interfere with biofilm formation, thus encouraging further in silico screening campaigns.
In the present work, a virtual screening approach was undertaken to identify molecules targeting DGC enzymes, using the active site of the DGC PleD from C. crescentus as a structural template. Overall, seven compounds were selected as potential inhibitors of DGCs. Two of these compounds, inhibitors 2 and 7, drastically reduced DGC activity in vitro. At present, the molecules identified in this work represent the most efficient compounds targeting the active site of the DGC enzyme, showing an IC50 of ∼11 μM.
MATERIALS AND METHODS
Virtual screening of the ZINC database.
Pharmacophore modeling calculations were performed by using LigandScout (17). Default values for the radii of atom-based chemical features (1.0 Å) and projections (1.4 Å) were used. An excluded volume, comprising residues at the active site of PleD located within a radius 15 Å from any atom of the ligands, was inserted into the pharmacophore. The obtained pharmacophore was then used to screen the purchasable subset of the ZINC database (18), using the ZincPharmer server (19). Several runs were carried out, systematically discharging one or two features of the pharmacophore each time. The compounds retrieved from the pharmacophore searches were docked into the active site of PleD, by means of Molegro Virtual Docker (MVD) software (CLCbio). To this end, the obtained three-dimensional (3D) structure of PleD (PDB accession number 2V0N) was prepared by automatically assigning bond orders and hybridization and adding explicit hydrogens, charges, and Tripos atom types. A search space with a 15-Å radius, centered on the active-site groove, was used for docking. The grid-based MolDock score with a grid resolution of 0.30 Å was used as a scoring function, and MolDock SE was used as a docking algorithm (20). For each ligand, 10 runs were defined. The retrieved compounds were ranked according to their score (obtained with the scoring function implemented in MVD). The means and standard deviations of the scores were then calculated, and those compounds scoring at >2.0 standard deviations from mean were selected and redocked with AutoDock v.4.2.5.1 (21). To this end, the Lamarckian genetic algorithm (LGA) implemented in AutoDock was used, using the following values: number of individuals in population of 150, maximum number of energy evaluations of 2,500,000, maximum number of generations of 27 × 103, and rate of gene mutation of 0.02. All other parameters were kept at their default values. Only those compounds showing a similar (root mean square deviation [RMSD] of <2.0 Å) docked pose, as assessed by MVD and AutoDock, were kept. Finally, the obtained compounds were ranked on the basis of predicted affinity and commercial availability, and the most potent compounds were chosen for in vitro testing.
DGC expression and purification.
The pET11-PleD vector was transformed into Escherichia coli BL21(DE3)/pLysS cells, and protein expression and purification were performed as described previously (12).
Purification of the DGCs from P. aeruginosa, i.e., (i) YfiNHAMP-GGDEF (cytosolic portion containing the HAMP-GGDEF domain), (ii) YfiNGGDEF (construct containing the GGDEF domain), and (iii) WspR, was performed as previously described (14, 22).
Measurement of diguanylate cyclase activity.
The inhibitory activity of synthetized compounds was measured by circular dichroism (CD) spectroscopy as described previously (23). PleD (0.5 μM) in 20 mM Tris (pH 8.0) and 100 mM NaCl was incubated for 10 min at room temperature with metals (10 mM MgCl2 and 2.5 mM MnCl2), activated by the addition of 1 mM BeCl2 and 10 mM NaF, and kept at room temperature for 30 min. The enzyme was incubated for 15 min with different concentrations of compounds. The same experiment was performed without an inhibitor as a control (with 5% dimethyl sulfoxide [DMSO]). The diguanylate cyclase reaction was started by the addition of 100 μM GTP to the mixture, and 100 mM CaCl2 was added after 30 min to stop the enzymatic reaction. Within this time frame, the enzyme is not completely inhibited, and it is still working. Samples were analyzed by using a 1-cm quartz cuvette (Hellma) on a Jasco J-710 spectropolarimeter at 25°C. The values are the means of data from at least three independent experiments, and the errors are within 15%.
Spectra were then baseline corrected by subtracting values for the same samples without the substrate from the raw data, and the signal was adjusted to zero at 340 nm, as no optical activity is expected at this wavelength. The c-di-GMP content was extrapolated by using the calibration curve of c-di-GMP obtained in the presence of compound 2 or 7 (see Fig. S1 in the supplemental material).
The inhibitory activity of 100 μM compounds 2 and 7 against PleD was also evaluated by reverse-phase high-performance liquid chromatography (HPLC), as previously described (24), to probe the effect of the sole Mn2+ or Mg2+ ion on controlling catalysis.
Compounds 2 and 7 were also tested on the YfiNHAMP-GGDEF and WspR DGCs from P. aeruginosa, two DGCs which are in the “on” state and therefore do not require preactivation. Six micromolar YfiNHAMP-GGDEF (in a buffer of 20 mM Tris [pH 7.5], 100 mM NaCl, 1% glycerol) was incubated for 10 min at room temperature with metals (10 mM MgCl2 and 2.5 mM MnCl2). Two micromolar WspR (in a buffer of 25 mM Tris [pH 7.5], 100 mM NaCl) was incubated for 10 min at room temperature with metals (2 mM MgCl2 and 2.5 mM MnCl2). If indicated, this assay was also carried out in the presence of 100 μM or 1 mM EDTA as a chelating agent.
The inhibition assays were performed under the experimental conditions described above for PleD.
Fluorescence experiments.
Titration of YfiNGGDEF with MANT [2′/3′-O-(N-methyl-anthraniloyl)]-GTP (Life Technologies) was carried out as described in the supplemental material (see Fig. S2 in the supplemental material); displacement of MANT-GTP was then carried out following the decrease in fluorescence of the MANT-GTP/YfiNGGDEF complex upon the addition of different amounts of inhibitor 7.
Briefly, protein tryptophans were excited at 280 nm, and the fluorescence emission spectra were recorded between 400 and 540 nm, as a result of Förster resonance energy transfer (FRET) to MANT-GTP. The experiment was performed by the sequential addition of different amounts of inhibitor 7 (10 to 120 μM) to 3 μM the YfiNGGDEF/MANT-GTP complex (obtained in the presence of stoichiometric amounts of the nucleotide). The corresponding fluorescence emission signal spectra were recorded on a Fluoromax single-photon counting spectrofluorometer (Horiba Jobin-Yvon) after 8 min of incubation with the inhibitor to reach equilibrium.
RESULTS
Virtual screening for identification of novel PleD inhibitors.
On the basis of the binding mode of the substrate analogue GTP-α-S (guanosine-alpha-thio-triphosphate) bound to PleD from Caulobacter crescentus CB15 (PDB accession number 2V0N) (12), the chemical features responsible for the key binding interactions were derived by using a pharmacophore-based approach. A 3D pharmacophore query resulted in a 14-point pharmacophore hypothesis (Fig. 1), including four hydrogen bond acceptor features engaged by the triphosphate moiety of GTP-α-S (F1 to F4) and pointing to the side chain of K442 (F1) or to the main chain of the binding cleft formed by residues F330 and F331 (F2 to F4); another hydrogen bond acceptor feature engaged by the carbonyl atom of the guanine moiety with the side chain of R366 (F5); two hydrogen bond donor features engaged by the N1- and 2-amino nitrogen atoms of the guanine moiety and pointing at the side chain of D344 (F6 and F7); two aromatic centroids located at the geometric center of the bicyclical aromatic moiety of the purine ring and their normal projections (F8 and F9); two metal binding features projecting from the β,γ-bisphosphate moiety and pointing to the Mg601 ion atom (F10 and F11); and three negative charges located at the geometric center of the three phosphate moieties, respectively (F12 to F14). Finally, in order to take into account the shape of the active site of PleD, excluded volumes were derived and included in the final pharmacophore, which was then used to filter the purchasable subset of the ZINC database (∼2.3 × 107 compounds) (18). The same 14-point pharmacophore hypothesis was obtained by using another GGDEF domain (PDB accession number 4H54) in complex with GTP-α-S (25).
FIG 1.

Pharmacophore hypothesis of a competitive inhibitor of GTP bound to PleD. Protein is shown as gray lines, with the binding site shown as a Van der Waals surface and colored according to polarity. GTP-α-S is also shown as sticks. Residues interacting with the pharmacophore map are represented as sticks and colored by atom type. F1 to F5, hydrogen bond acceptors (red arrows); F6 and F7, hydrogen bond donors (green arrows); F8 and F9, aromatic centroids (blue tori); F10 and F11, metal binding groups (cyan cones); F12 to F14, negative charges (red cones). Exclusion volume spheres are not shown for clarity.
A total of 9,418 compounds matching at least 6 chemical features were retrieved in this first step. These virtual hits underwent a second docking-based filtering step by means of the MVD tool. A total of 328 molecules with significant scores (≥2.0 standard deviations from the mean) were selected and resubmitted to a further docking step by means of a second docking tool, AutoDock 4.0 (21). Thirty-five compounds showed comparable poses when docked with both AutoDock 4.0 and MVD (RMSD value of ≤2 Å). The most promising virtual screening hits were selected on the basis of their predicted affinity for PleD and commercial availability. This protocol gave us seven molecules (compounds 1 to 7) (Table 1) that were further subjected to in vitro testing.
TABLE 1.
Virtual screening hits tested in vitro on PleDa
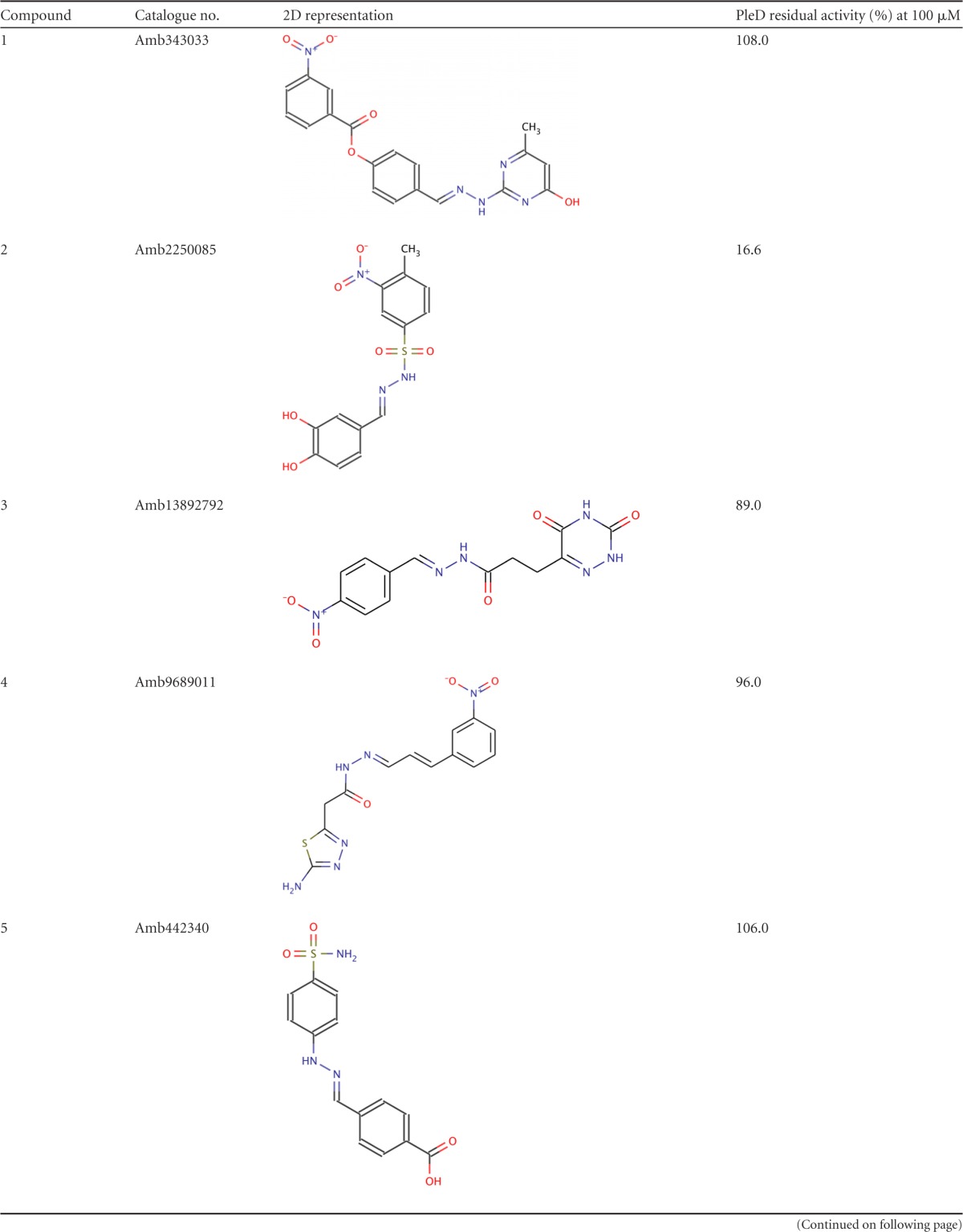
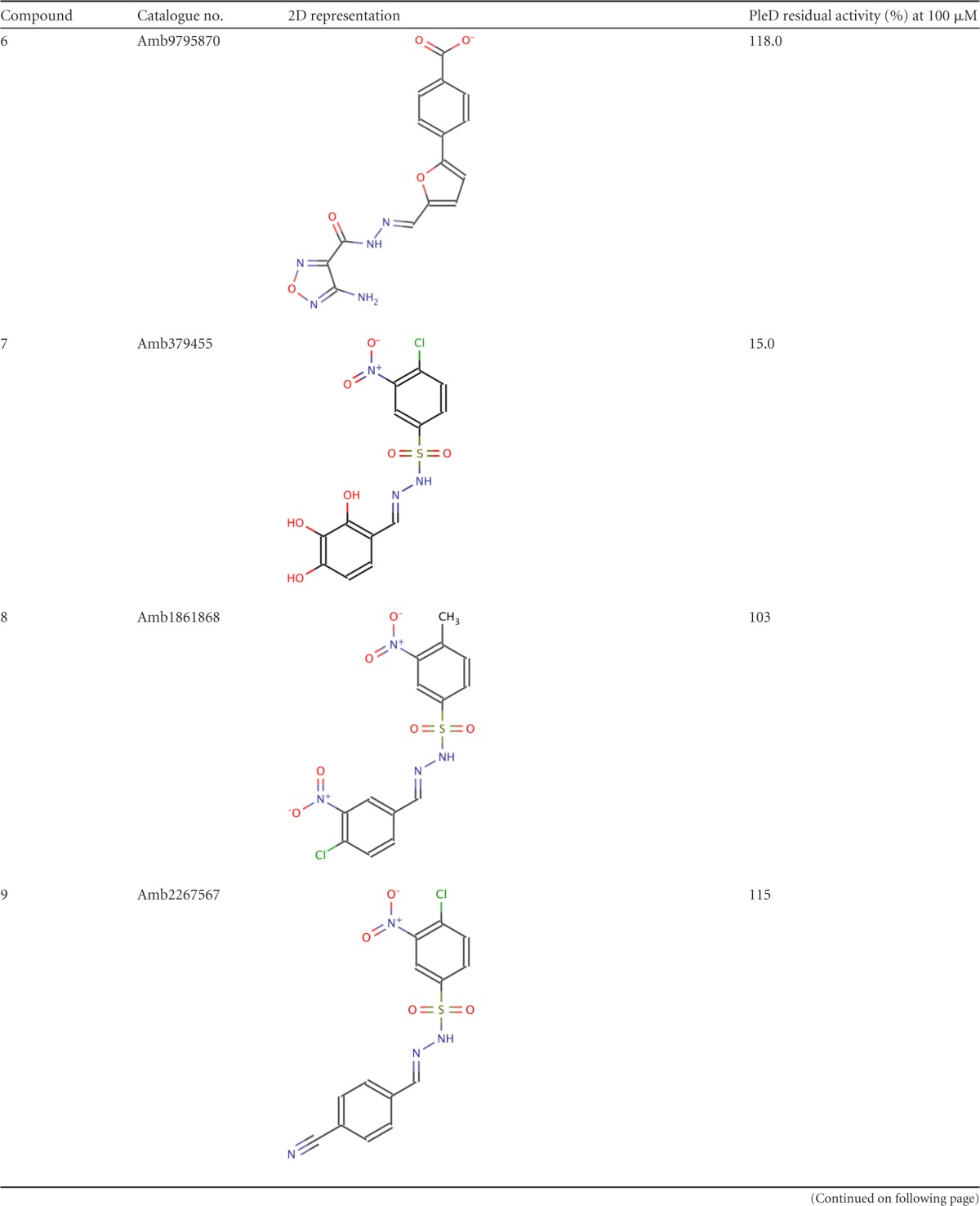

2D, two dimensional.
Effect of compounds selected by virtual screening on in vitro activity of DGC.
The seven compounds identified by in silico screening (compounds 1 to 7) (Table 1) were tested in vitro for their ability to inhibit the DGC activity of PleD from C. crescentus. Inhibitory activity was measured by using circular dichroism spectroscopy (23).
Two out of the seven compounds tested, namely, compounds 2 and 7 (Table 1), were found to significantly reduce the activity of the PleD DGC; this result was independently confirmed by assaying the residual PleD activity by HPLC (Fig. 2).
FIG 2.

PleD inhibition assay with various concentrations of inhibitors 2 (A) and 7 (B). Each point of the concentration-response curve represents the mean value and standard deviation of data from three independent experiments by CD spectroscopy. The line is the best-fit curve generated by Prism software, using the log-dose-versus-response equation. Points falling on the y axis represent the 100% residual activities obtained in the absence of an inhibitor. Data fitting allowed extrapolation of the IC50s as 11.05 ± 1.2 μM and 11.07 ± 1.1 μM for inhibitors 2 and 7, respectively. An inhibition assay with 100 μM compounds 2 and 7 was also performed by HPLC (gray points).
To quantitatively evaluate the inhibitory effects of these two compounds, the IC50 was measured by CD spectroscopy, whose real-time measurements are not affected by the heat enzyme inactivation step, contrary to HPLC analysis. Compounds 2 and 7 inhibited PleD activity, showing IC50 values of 11.1 ± 1.2 μM and 11.1 ± 1.1 μM, respectively (Fig. 2).
The high level of similarity between IC50 values displayed by compounds 2 and 7 is not surprising considering the structural similarity of these molecules (Tanimoto coefficient of >0.9), based on a sulfonohydrazide scaffold carrying a catechol moiety. Binding of compounds 2 and 7 to PleD (Fig. 3) is stabilized by polar interactions with residues surrounding the GTP binding pocket, namely, N335, D344, and R366.
FIG 3.

Predicted binding pose of the sulfonohydrazide and catechol moieties in the active site of PleD. PleD is shown as a gray ribbon. Compound 2 is shown as sticks as a reference. Conserved residues in the active site interacting with compounds 2 and 7 are indicated; the interactions are displayed in the box. GTP is shown superposed on the compounds as gray lines, for reference. The Mg601 ion is shown as a green sphere.
The metal binding catechol moiety of both compounds is predicted to chelate the Mg ion, similarly to the β-phosphate group of GTP. To probe this, we tested a series of compounds (compounds 8 to 13) with slight substitutions at the benzene ring of the catechol moiety, obtaining in every case a complete loss of inhibitory potency of the compounds.
Given the proposed role of the catechol moiety in binding the metal in the active site, we tested the effects of compounds 2 and 7 during catalysis in the presence of the sole Mn2+ or Mg2+ ion. Interestingly, compound 2 is strongly sensitive to the nature of the metal, being unable to inhibit both PleD or WspR in the presence of the sole Mg2+ ion. On the other hand, compound 7 exerts the same inhibitory role independently of the divalent ion (data not shown). Therefore, we focused our biochemical characterization on compound 7.
To exclude unspecific effects of inhibitor 7 on the cyclase activity, we performed different control experiments aimed at demonstrating the competitive binding of such a compound to the active site of DGCs. Due to the complexity of PleD (nucleotide-dependent oligomerization, extra metal binding site, and preactivation by beryllium-induced dimerization [11, 12]), some of the experiments reported below were carried out with other DGCs.
The capability of compound 7 to chelate metals may affect metal availability in the active-site pocket of DGCs, thus leading to unspecific inhibition of the cyclase activity, even though experiments have been carried out in the presence of excess metals; if so, a similar effect in the presence of excess metal-chelating agents, such as EDTA, can be expected. Nevertheless, we found that the activity of the DGC WspR (which requires magnesium for catalysis but, contrary to PleD, does not contain an extra metal binding site or beryllium metal for activation) is not affected by the presence of excess EDTA (both 100 μM and 1 mM) (data not shown), thus suggesting that, in this context, metal binding to the active site of WspR is not affected by any chelation from compound 7.
Moreover, we exclude any folding effect of compound 7 on target enzymes, by means of CD thermal melting experiments. We performed the experiment with YfiNGGDEF, which is a more suitable system for denaturation experiments than PleD or WspR, given that its oligomeric state is not nucleotide dependent (14). We found that the presence of excess compound 7 (100 μM) does not affect the secondary-structure content and the overall stability of the protein, thus indicating that the observed inhibitory property of compound 7 with DGCs is not due to unspecific folding effects (see Fig. S3 in the supplemental material); rather, the slight increase of the melting temperature (see Fig. S3, gray trace) is indicative of binding of compound 7 to the protein, with subsequent protein stabilization.
These data and the evidence that compound 7 strongly inhibits other DGCs such as WspR from P. aeruginosa (Fig. 4B) indicate that the effect of this compound on this class of protein is specific. Moreover, according to our structural model, compound 7 should target the active site of DGCs. As mentioned above, many DGCs, including PleD and WspR, are allosterically inhibited by c-di-GMP (via the I site); to exclude that compound 7 targets the I site, we tested its effect on a third DGC, i.e., the cytosolic portion of YfiN (here YfiNGGDEF-HAMP) from P. aeruginosa, which is known to be insensitive to feedback inhibition of c-di-GMP (due to the presence of a degenerated I site) (14). The residual activity of YfiNHAMP-GGDEF in the presence of 100 μM compound 7 (Fig. 4C) is close to zero, similarly to what was observed for the residual activity of PleD and WspR (Fig. 4A and B), thus confirming that the I site is not involved in such DGC inhibition.
FIG 4.

Residual DGC activity of PleD, WspR, and YfiNHAMP-GGDEF. Kinetics of PleD (A), WspR (B), and YfiNHAMP-GGDEF (C) in the presence of 100 μM compound 7 (gray traces) were determined. Control experiments without inhibitor are also reported (black traces); the kinetics shapes reported in panels A and B (black traces) are indicative of a product inhibition phenomenon occurring in PleD and WspR, due to the presence of the conserved I site, which is degenerated (and therefore c-di-GMP insensitive) in YfiNHAMP-GGDEF (C).
In light of these results, it is likely that compound 7 targets the active sites of both PleD and YfiN, whose degrees of structural similarity and conservation are very high (Fig. 5), in particular residues D344 and N335 (D304 and N295 in YfiN, respectively); therefore, we expect that this compound is a competitive inhibitor of DGCs.
FIG 5.

Structural comparison between the PleD and YfiN GGDEF domains. The PleD and YfiN GGDEF domains are shown as gray and pink ribbons, respectively. Conserved residues in the active site putatively interacting with compounds 2 and 7 are indicated; compound 2 is shown as green sticks. The Mg ion is shown as a green sphere. The location of the PleD I site is also indicated.
As previously reported (11, 26), the standard Michaelis-Menten kinetic scheme is inadequate for the kinetic profile of GGDEF enzymes, and therefore, the corresponding inhibition profile could not be informative. In order to bypass this limitation, we designed a FRET-based displacement experiment aimed at demonstrating qualitatively that inhibitor 7 competes for the GTP binding site. The experiment was done with the DGC YfiNGGDEF, for which the binding affinity for GTP is quantitatively demonstrated (14), in complex with a stoichiometric amount of the GTP analogue MANT-GTP, which saturated the protein (see Fig. S2 in the supplemental material for details), and addition of different concentrations of inhibitor 7.
Tryptophan excitation at 280 nm of the MANT-GTP/protein complex results in a peak at 440 nm (Fig. 6A, black continuous line), which is minimal in the absence of the YfiNGGDEF protein or absent in the nucleotide-free protein (see also Fig. S2 in the supplemental material). Upon the addition of increasing amounts of compound 7 (10 μM to 120 μM), we observed a fluorescence decrease (Fig. 6A, dashed lines), which reached a baseline value upon the addition of 120 μM compound 7. The hyperbolic shape of such a transition indicates that replacement equilibrium is occurring (i.e., the fluorophore MANT-GTP dissociates from and compound 7 binds to the protein) with an apparent dissociation constant value of ∼80 μM.
FIG 6.

Binding of compound 7 to the active site of YfiNGGDEF assayed by displacement of MANT-GTP. The competition experiment was carried out in the presence of a constant concentration of the ligand (MANT-GTP) and various inhibitor concentrations. The fractional saturation (ordinate axis) is determined as [PI]/([PI] + [PS]), where [PI] is the difference between Fmax and Fx and [PS] is Fx (Fmax is the signal fluorescence obtained in the absence of the inhibitor, and Fx is the signal fluorescence in the presence of the inhibitor). According to the experimental setup, we consider that the concentration of unbound protein is negligible.
DISCUSSION
It is becoming evident that c-di-GMP is a pivotal second messenger involved in bacterial virulence-related phenotypes, including virulence factor production, motility, adhesion, and biofilm development. Therefore, targeting the enzymes involved in c-di-GMP synthesis represents an appealing strategy for the development of antibiofilm drugs (5). In this study, two novel molecules with a 3-nitro-benzenesulfonamide scaffold that inhibit PleD in the low-micromolar range (IC50 of ∼11 μM) are described. To our knowledge, these are the most potent competitive inhibitors of PleD identified to date.
The active site of the DGC domain of PleD from C. crescentus (PDB accession number 2V0N) was used for in silico screening. Since the mechanism of catalysis of DGC enzymes is not yet fully understood at the atomic level, for the development of a 3D pharmacophore, we focused on the interactions between the nonhydrolyzable GTP analogue (GTP-α-S) and PleD, as found in the crystal structure. Three residues have been pinpointed for the strict specificity of the guanine base for the active site of PleD (8, 16): N335, making a hydrogen bond between the N3 of the guanine base with its amino group and a hydrogen bond between the N2 and its side-chain carbonyl group; D344, making a hydrogen bond between the N1 of the base and the oxygen of the side chain of its carboxyl group; and R366, making a hydrogen bond with the oxygen atom of the guanine ring. Notably, as assessed by the predicted binding poses, N335 and R366 also interact with the two identified inhibitors, even if the chemical entities involved are highly dissimilar compared to GTP-α-S: a hydrogen bond between the sulfohydrazine moiety of compounds 2 and 7 and N335 and another hydrogen bond between the nitro group of the inhibitors and the side chain of R366. D344 displays no interactions with the inhibitors, although it is placed at a suitable distance (∼3.6 Å) to interact with the nitro group of the compounds.
In addition, the GGDEF motif that is representative of this family of enzymes forms part of the active site, and it is involved in Mg ion binding, together with one of the oxygen atoms of the phosphate group of GTP. The catechol group of the identified inhibitors is predicted to occupy the same position of the phosphate group involved in metal chelation, and it is suitably placed to interact with the Mg ion. Indeed, catechols are well-known metal binding groups (27), and slight modifications of this moiety completely abolished the inhibitory potency of the tested compounds (compounds 8 to 13) (Table 1).
The lead compounds identified in this study show certain structural similarities with some previously identified DGC inhibitors (16). Their lengths range from 13 to 15 longest countable atomic linkages end to end. They are generally linear in shape, sharing similar steric demands.
In particular, compounds 2 and 7 share a hydrazine moiety with the previously identified inhibitors LP3134 and LP4010, which has been predicted to interact with the amino group of N335. Finally, LP3134 also contain a catechol moiety, although the latter was predicted not to be involved in any interaction with Mg.
To discard the hypothesis that these compounds target the inhibitory site of PleD, we also tested them on YfiN from P. aeruginosa, confirming that compounds 2 and 7 are also active on YfiN and that the I site is not required for their ability to inhibit DGCs.
Since bacteria usually encode multiple DGC enzymes, we also considered the possibility that the identified compounds are able to inhibit GGDEF targets other than PleD and YfiN, involved in c-di-GMP synthesis/signaling processes. Several of these targets are of great clinical interest (such as Rv1354c as a target of antituberculosis drugs [10]). Indeed, a multiple-sequence alignment of GGDEF domains from representative bacteria shows that residues at an interacting distance from compounds 2 and 7 are evolutionarily invariant (see Fig. S1 in the supplemental material). The high level of evolutionary conservation of the residues of GGDEF domains involved in protein-inhibitor contacts suggests that compounds 2 and 7 could also inhibit other proteins related to c-di-GMP synthesis/signaling and therefore represent leads for the design of broad-spectrum antibiofilm inhibitors.
Conclusions.
In summary, we have described the identification of novel catechol-containing sulfonohydrazide compounds as inhibitors of DGCs such as PleD, WspR, and YfiN and as potential lead compounds for the development of new antivirulence drugs acting on c-di-GMP signaling. In addition, we report a new and efficient pharmacophore hypothesis for targeting of the PleD active site. The identified compounds represent the most potent in vitro inhibitors of PleD known to date. The compounds described here are currently ineffective in reducing c-di-GMP intracellular levels in bacterial pathogens in which biofilm formation relies upon c-di-GMP signaling systems (see Fig. S5 and S6 in the supplemental material). Further optimization of compounds 2 and 7 is in progress and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
We thank Alessio Paone for fruitful discussion on experimental data.
Funding Statement
Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR) provided funding to Serena Rinaldo and Giordano Rampioni under grant number RBFR10LHD1. Sapienza Università di Roma (Sapienza University of Rome) provided funding to Alessandro Paiardini, Serena Rinaldo, and Francesca Cutruzzolà under grant number C26A149EC4. The Italian Cystic Fibrosis Research Foundation provided funding to Livia Leoni under grant number FFC 13/2011.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00742-15.
REFERENCES
- 1.Romling U, Gomelsky M, Galperin MY. 2005. c-di-GMP: the dawning of a novel bacterial signalling system. Mol Microbiol 57:629–639. doi: 10.1111/j.1365-2958.2005.04697.x. [DOI] [PubMed] [Google Scholar]
- 2.Hengge R. 2009. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol 7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 3.Boyd CD, O'Toole GA. 2012. Second messenger regulation of biofilm formation: breakthroughs in understanding c-di-GMP effector systems. Annu Rev Cell Dev Biol 28:439–462. doi: 10.1146/annurev-cellbio-101011-155705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caly DL, Bellini D, Walsh MA, Dow JM, Ryan RP. 2015. Targeting cyclic di-GMP signalling: a strategy to control biofilm formation? Curr Pharm Des 21:12–24. doi: 10.2174/1381612820666140905124701. [DOI] [PubMed] [Google Scholar]
- 6.Ohana P, Delmer DP, Carlson RW, Glushka J, Azadi P, Bacic T, Benziman M. 1998. Identification of a novel triterpenoid saponin from Pisum sativum as a specific inhibitor of the diguanylate cyclase of Acetobacter xylinum. Plant Cell Physiol 39:144–152. doi: 10.1093/oxfordjournals.pcp.a029351. [DOI] [PubMed] [Google Scholar]
- 7.Antoniani D, Rossi E, Rinaldo S, Bocci P, Lolicato M, Paiardini A, Raffaelli N, Cutruzzolà F, Landini P. 2013. The immunosuppressive drug azathioprine inhibits biosynthesis of the bacterial signal molecule cyclic-di-GMP by interfering with intracellular nucleotide pool availability. Appl Microbiol Biotechnol 97:7325–7336. doi: 10.1007/s00253-013-4875-0. [DOI] [PubMed] [Google Scholar]
- 8.Sambanthamoorthy K, Sloup RE, Parashar V, Smith JM, Kim EE, Semmelhack MF, Neiditch MB, Waters CM. 2012. Identification of small molecules that antagonize diguanylate cyclase enzymes to inhibit biofilm formation. Antimicrob Agents Chemother 56:5202–5211. doi: 10.1128/AAC.01396-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lieberman OJ, Orr MW, Wang Y, Lee VT. 2014. High-throughput screening using the differential radial capillary action of ligand assay identifies ebselen as an inhibitor of diguanylate cyclases. ACS Chem Biol 9:183–192. doi: 10.1021/cb400485k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui T, Zhang L, Wang X, He Z-G. 2009. Uncovering new signaling proteins and potential drug targets through the interactome analysis of Mycobacterium tuberculosis. BMC Genomics 10:118. doi: 10.1186/1471-2164-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paul R, Abel S, Wassmann P, Beck A, Heerklotz H, Jenal U. 2007. Activation of the diguanylate cyclase PleD by phosphorylation-mediated dimerization. J Biol Chem 282:29170–29177. doi: 10.1074/jbc.M704702200. [DOI] [PubMed] [Google Scholar]
- 12.Wassmann P, Chan C, Paul R, Beck A, Heerklotz H, Jenal U, Schirmer T. 2007. Structure of BeF3-modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure 15:915–927. doi: 10.1016/j.str.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 13.Malone JG, Williams R, Christen M, Jenal U, Spiers AJ, Rainey PB. 2007. The structure-function relationship of WspR, a Pseudomonas fluorescens response regulator with a GGDEF output domain. Microbiology 153:980–994. doi: 10.1099/mic.0.2006/002824-0. [DOI] [PubMed] [Google Scholar]
- 14.Giardina G, Paiardini A, Fernicola S, Franceschini S, Rinaldo S, Stelitano V, Cutruzzola F. 2013. Investigating the allosteric regulation of YfiN from Pseudomonas aeruginosa: clues from the structure of the catalytic domain. PLoS One 8:e81324. doi: 10.1371/journal.pone.0081324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Zhou J, Donaldson GP, Nakayama S, Yan L, Lam YF, Lee VT, Sintim HO. 2011. Conservative change to the phosphate moiety of cyclic diguanylic monophosphate remarkably affects its polymorphism and ability to bind DGC, PDE, and PilZ proteins. J Am Chem Soc 133:9320–9330. doi: 10.1021/ja1112029. [DOI] [PubMed] [Google Scholar]
- 16.Sambanthamoorthy K, Luo C, Pattabiraman N, Feng X, Koestler B, Waters CM, Palys TJ. 2014. Identification of small molecules inhibiting diguanylate cyclases to control bacterial biofilm development. Biofouling 30:17–28. doi: 10.1080/08927014.2013.832224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolber G, Langer T. 2005. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Infect Model 45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 18.Irwin JJ, Shoichet BK. 2005. ZINC—a free database of commercially available compounds for virtual screening. J Chem Infect Model 45:177–182. doi: 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koes DR, Camacho CJ. 2012. ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res 40:W409–W414. doi: 10.1093/nar/gks378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomsen R, Christensen MH. 2006. MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 21.Morris GM, Huey R, Olson AJ. 2008. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics Chapter 8:Unit 8.14. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 22.De N, Pirruccello M, Krasteva PV, Bae N, Raghavan RV, Sondermann H. 2008. Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol 6:e67. doi: 10.1371/journal.pbio.0060067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stelitano V, Brandt A, Fernicola S, Franceschini S, Giardina G, Pica A, Rinaldo S, Sica F, Cutruzzola F. 2013. Probing the activity of diguanylate cyclases and c-di-GMP phosphodiesterases in real-time by CD spectroscopy. Nucleic Acids Res 41:e79. doi: 10.1093/nar/gkt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stelitano V, Giardina G, Paiardini A, Castiglione N, Cutruzzola F, Rinaldo S. 2013. c-di-GMP hydrolysis by Pseudomonas aeruginosa HD-GYP phosphodiesterases: analysis of the reaction mechanism and novel roles for pGpG. PLoS One 8:e74920. doi: 10.1371/journal.pone.0074920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zahringer F, Lacanna E, Jenal U, Schirmer T, Boehm A. 2013. Structure and signaling mechanism of a zinc-sensory diguanylate cyclase. Structure 21:1149–1157. doi: 10.1016/j.str.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 26.Oliveira MC, Teixeira RD, Andrade MO, Pinheiro GM, Ramos CH, Farah CS. 2015. Cooperative substrate binding by a diguanylate cyclase. J Mol Biol 427:415–432. doi: 10.1016/j.jmb.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Hider RC, Kong X. 2010. Chemistry and biology of siderophores. Nat Prod Rep 27:637–657. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.