

ABSTRACT
An ability to sense and respond to changes in extracellular phosphate is critical for the survival of most bacteria. For Caulobacter crescentus, which typically lives in phosphate-limited environments, this process is especially crucial. Like many bacteria, Caulobacter responds to phosphate limitation through a conserved two-component signaling pathway called PhoR-PhoB, but the direct regulon of PhoB in this organism is unknown. Here we used chromatin immunoprecipitation-DNA sequencing (ChIP-Seq) to map the global binding patterns of the phosphate-responsive transcriptional regulator PhoB under phosphate-limited and -replete conditions. Combined with genome-wide expression profiling, our work demonstrates that PhoB is induced to regulate nearly 50 genes under phosphate-starved conditions. The PhoB regulon is comprised primarily of genes known or predicted to help Caulobacter scavenge for and import inorganic phosphate, including 15 different membrane transporters. We also investigated the regulatory role of PhoU, a widely conserved protein proposed to coordinate phosphate import with expression of the PhoB regulon by directly modulating the histidine kinase PhoR. However, our studies show that it likely does not play such a role in Caulobacter, as PhoU depletion has no significant effect on PhoB-dependent gene expression. Instead, cells lacking PhoU exhibit striking accumulation of large polyphosphate granules, suggesting that PhoU participates in controlling intracellular phosphate metabolism.
IMPORTANCE The transcription factor PhoB is widely conserved throughout the bacterial kingdom, where it helps organisms respond to phosphate limitation by driving the expression of a battery of genes. Most of what is known about PhoB and its target genes is derived from studies of Escherichia coli. Our work documents the PhoB regulon in Caulobacter crescentus, and comparison to the regulon in E. coli reveals significant differences, highlighting the evolutionary plasticity of transcriptional responses driven by highly conserved transcription factors. We also demonstrated that the conserved protein PhoU, which is implicated in bacterial persistence, does not regulate PhoB activity, as previously suggested. Instead, our results favor a model in which PhoU affects intracellular phosphate accumulation, possibly through the high-affinity phosphate transporter.
INTRODUCTION
Most bacteria must sense and rapidly respond to the nutrient states of their environments to survive and to proliferate. Although this capacity for adapting to extracellular changes is critical, the molecular mechanisms by which it occurs remain incompletely understood. Sensing the availability of extracellular phosphate is particularly crucial, as phosphate is required for the synthesis of many biomolecules, from ATP to phospholipids. Prior studies have demonstrated that phosphate sensing is important for maximal growth of bacteria (1), biofilm formation (2), and the virulence of some pathogens (3–6).
Most bacteria respond to phosphate limitation through a widely conserved signal transduction pathway whose connectivity and functionality remain only partly characterized (1). In this pathway, the availability of phosphate is likely sensed through changes in phosphate uptake by the high-affinity Pst transporter in conjunction with a two-component signaling pathway, PhoR-PhoB, collectively known as the Pho system. In Escherichia coli, the Pst transporter is active during growth under phosphate-replete conditions, which somehow inhibits autophosphorylation of the histidine kinase PhoR. When phosphate becomes limiting and flux through the Pst transporter is reduced, PhoR is stimulated to autophosphorylate and then transfer its phosphoryl group to PhoB. Phosphorylated PhoB undergoes a conformational change and dimerizes along its α4-β5-α5 interface (7), allowing it to then bind conserved DNA sequences called pho boxes in certain promoters, typically leading to increased transcription of target genes (8), many of which help cells cope with the decreased extracellular phosphate levels that initiated the pathway. X-ray crystallography and mutational studies indicate that PhoB binds to region 4 of σ70, stabilizing its association with the −35 region of target promoters (9, 10).
In E. coli, the expression of more than 40 genes, including the pst and pho genes themselves, changes following phosphate starvation (11). These genes were identified through a combination of reporter studies and DNA microarray analyses, but which genes are direct targets of PhoB is unclear (12, 13). More recently, a study of PhoB using chromatin immunoprecipitation with microarray technology (ChIP-chip) identified some putative direct targets but did not examine PhoB binding under high-phosphate conditions (11–13). The conservation of the Pho regulon is also unknown, as there have not yet been efforts to accurately define the entire set of genes directly regulated by PhoB in organisms other than E. coli.
Two-component systems are a predominant means by which bacteria sense and respond to external stimuli (14, 15). Although many histidine kinases bind extracellular ligands, others lack large extracellular domains and may instead respond to intracellular signals (1, 16). The conserved Gram-negative histidine kinase PhoR resides in the inner membrane but does not contain a significant periplasmic domain. PhoR has been suggested to sense the extracellular phosphate status through an interaction with the Pst transporter, which also resides in the inner membrane, but the precise mechanism by which the Pst system regulates PhoR is unclear. A protein of unknown function, PhoU, was proposed as an intermediate between the Pst and Pho systems, inhibiting PhoR when the Pst system actively transports phosphate (17, 18). phoU is widely conserved in bacteria and frequently coregulated with the pst and pho genes (17, 18). In E. coli, the expression of alkaline phosphatase and some other members of the Pho regulon are upregulated in phoU mutants (19, 20), which indicates that PhoU may function as a negative regulator of the Pho regulon. However, the effects of a phoU mutant on expression of the Pho regulon are poorly defined, and earlier studies suggested that PhoU may instead affect phosphate transport (21, 22), leaving the precise function of PhoU uncertain.
Although many bacteria use the Pst-Pho signaling pathway to respond to phosphate limitation, they likely adapt to changes in phosphate levels in different ways. For example, in the freshwater alphaproteobacterium Caulobacter crescentus, low extracellular phosphate stimulates elongation of a polar appendage called the stalk, which is a tubular extension of the cell envelope. Phosphate starvation can lead cells to extend their stalks to up to 20 times their lengths under phosphate-replete conditions (23, 24). It was initially suggested that stalk elongation may increase the nutrient-scavenging ability of phosphate-starved Caulobacter cells, but subsequent studies found that a diffusion barrier may exist between the stalk and the cell body, potentially preventing the free exchange of membrane and periplasmic proteins (25).
Here we use a combination of genome-wide expression profiling, bioinformatics, and chromatin immunoprecipitation-DNA sequencing (ChIP-Seq) to identify the direct targets of Caulobacter PhoB. The Caulobacter PhoB regulon includes nearly 50 genes, with relatively few genes in common with the E. coli PhoB regulon beyond the pst and pho genes. Active Caulobacter PhoB drives substantial changes in the repertoire of membrane transporters expressed, presumably to help cells effectively scavenge for inorganic phosphate. Our results demonstrate how a highly conserved signaling pathway can be used for vastly different programs of gene expression in different bacteria, highlighting the plasticity of bacterial regulatory networks. Further, we show that the conserved protein PhoU does not, as previously suggested from studies of E. coli, negatively regulate the PhoR-PhoB signaling pathway in Caulobacter and instead is a critical player in cellular phosphate metabolism or phosphate transport by the Pst system.
MATERIALS AND METHODS
Strains and growth conditions.
C. crescentus strains were grown in peptone-yeast extract (PYE) (rich medium), M2G (minimal medium), or M5G (low-phosphate medium), supplemented when necessary with oxytetracycline (1 μg/ml), kanamycin (25 μg/ml), or gentamicin (0.6 μg/ml). Cultures were grown at 30°C, unless otherwise noted, and diluted when necessary to maintain exponential growth. E. coli cultures used for cloning were grown at 37°C in Superbroth, supplemented when necessary with oxytetracycline (12 μg/ml), kanamycin (50 μg/ml), or gentamicin (15 μg/ml).
The phoU depletion strain was constructed by first integrating a copy of phoU at the vanillate locus using plasmid pVGFPN-4 (26), with the phoU open reading frame cloned into the NdeI and XbaI sites, which removes the green fluorescent protein (GFP) coding region from the plasmid. We subsequently deleted phoU from the pstC-pstA-pstB-phoU-phoB operon using allelic replacement, as described previously (27). The resulting strain contains phoU at the van locus and a markerless deletion of phoU in the pstC operon, in which the entire coding region of phoU is removed, except for the first and final 9 bases.
lacZ reporter plasmids were derived from pRKlac290 (28). pRKlac290 was digested with KpnI and XbaI, and a DNA fragment containing the 200 bp directly upstream of either the pstC or the pstS annotated translation start site, with flanking KpnI and XbaI cut sites, was cloned into the multiple-cloning site upstream of the lacZ open reading frame.
The pstS::Tn5 strain was obtained from Yves Brun (23) and transduced into a clean CB15N background. Lysate from this strain was also used to construct the pstS::Tn5 phoB-3×FLAG and pstS::Tn5 Pvan-phoU ΔphoU strains. The ΔphoR::tet strain was constructed previously (27). The ΔphoR Pvan-phoU ΔphoU double mutant was constructed by transduction of the ΔphoR allele into the phoU depletion strain.
Microscopy.
Cells in mid-exponential phase were fixed with 0.5% paraformaldehyde, mounted on M2G-1.5% agarose pads, and imaged as described in reference 29. To image polyphosphate granules in single cells, 12 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) was added directly to the culture medium. Culture plus DAPI was incubated at 22°C in the dark for 30 min and spotted on M2G-1% agarose pads. Fluorescence microscopy images of DAPI-stained polyphosphate granules in cells were collected at a ×630 magnification with a Leica CTR5000 microscope with a Hamamatsu ORCA-ER camera. A custom filter set (390/70-nm excitation filter, 488-nm dichroic filter, and 515-nm long-pass emission filter) was used to visualize DAPI-polyphosphate.
β-Galactosidase assays.
Strains were grown to the mid-exponential phase in medium supplemented with 1 μg/ml oxytetracycline. Assays were performed essentially as described previously (30).
Immunoblots.
Immunoblotting was performed as described previously (31). Samples were prepared in 20 μl of 1:4 sample buffer-distilled water to an optical density at 600 nm (OD600) of 0.2, resolved on 12% sodium dodecyl sulfate-polyacrylamide gels, and transferred to polyvinylidene difluoride transfer membranes (Pierce). Membranes were probed with monoclonal mouse anti-FLAG (Sigma) at a 1:1,500 dilution. Secondary horseradish peroxidase (HRP)-conjugated anti-mouse antibody (Pierce) was used at a 1:3,000 dilution.
ChIP-Seq and analysis.
Chromatin immunoprecipitation for ChIP-Seq was performed as described previously (32), with modifications. Mid-exponential-phase cultures were cross-linked in 10 mM sodium phosphate (pH 7.6) and 1% formaldehyde at room temperature for 10 min. Reactions were quenched with 0.1 M glycine at room temperature for 5 min and on ice for 15 min. Cells were washed three times in phosphate-buffered saline (PBS) and lysed with Ready-Lyse lysozyme solution (Epicentre, Madison, WI) according to the manufacturer's instructions. Lysates were diluted 1:1 in ChIP buffer (1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.1], 167 mM NaCl) with Roche protease inhibitor tablets (Roche) and incubated at 37°C for 10 min. Lysates were sonicated (Branson sonicator) on ice, with 6 bursts of 10 s each at 15% amplitude, and then cleared by centrifugation at 14,000 rpm for 5 min at 4°C. Cleared supernatants were normalized by protein content in 1 ml of ChIP buffer with 0.01% SDS and precleared with 50 μl of protein A Dynabeads (Invitrogen) (preblocked with 100 μg UltraPure bovine serum albumin [BSA] in ChIP buffer with 0.01% SDS) by rotation for 1 h at 4°C.
Ten percent of each supernatant was removed and used as the total chromatin input sample. The remaining supernatant was incubated with a 1:1,000 dilution of anti-M2 antibody overnight at 4°C. Each sample was then incubated with 50 μl of preblocked protein A Dynabeads for 6 h at 4°C, with rotation. The Dynabeads were washed consecutively at 4°C for 15 min with 1 ml of the following buffers: low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8.1], 150 mM NaCl), high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8.1], 500 mM NaCl), LiCl wash buffer (0.25 M LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl [pH 8.1]), and Tris-EDTA (TE) buffer (10 mM Tris-HCl [pH 8.1], 1 mM EDTA) (twice). Complexes were eluted twice by incubation with 250 μl freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3) at 30°C for 15 min.
Cross-links were reversed by the addition of 300 mM NaCl and 2 μl of 0.5 mg/ml RNase A and overnight incubation at 65°C. Samples were treated with 5 μl of proteinase K (20 mg/ml; NEB) in 40 mM EDTA and 40 mM Tris-HCl (pH 6.8) for 2 h at 45°C. DNA was extracted using a PCR purification kit (Qiagen) and was resuspended in 80 μl of water.
Libraries were prepared using the SPRIworks system and sequenced on an Illumina MiSeq sequencer (MIT BioMicroCenter). ChIP-Seq results were analyzed using the MACS software package (33). A total of 860,000 reads were analyzed for each sample, and peaks were called with a P value of <10−5.
To determine the amount by which a ChIP sample was enriched for individual loci, we performed quantitative PCR (qPCR) with an input DNA control for each sample; a portion of each sample was reserved before the sample was subjected to ChIP, and DNA was isolated from this input sample and analyzed by qPCR at the pstC and CC1294 loci. Fold enrichment was then calculated as the amount of the pstC or CC1294 locus found in a ChIP output sample, relative to the amount found in the input sample.
DNA microarrays.
RNA was collected from cultures grown to the mid-exponential phase in rich medium at 30°C. For the pstS mutant, RNA from the wild type was used as a reference. For the phoU depletion strain and the pstS and phoU depletion double mutant, the strains grown in the absence of vanillate were compared with the same strains grown in the presence of vanillate. Gene expression profiles were obtained as described previously (29), using custom 8×15K Agilent expression arrays, and expression values are given as the average of ratios for a given gene. Complete data sets for expression profiling and ChIP-Seq are provided in Tables S1 and S2 in the supplemental material and are available through GEO.
CFU.
Strains were grown overnight in PYE with vanillate and then washed and released into medium with or without vanillate. Cultures were subsequently grown for 30 h and diluted once every two generations to maintain mid-exponential growth. Samples were removed every 3 h and plated on PYE with vanillate. Colonies were counted after 2 days of growth for all strains except those with a pstS mutation, which have a growth defect and were counted after 3 days of growth.
Transposon mutagenesis and rescue cloning.
Electrocompetent Pvan-phoU ΔphoU cells (50 μl) were transformed with 0.5 μl of EZ-Tn5 transposon mixture (EZ-Tn5 ⟨R6Kyori/KAN-2⟩ insertion kit; Epicentre) and grown in 1 ml of PYE for 1.5 h at 30°C. Cells were then plated on PYE supplemented with kanamycin. Colonies were picked after 2, 3, and 4 days of growth at 30°C.
Colonies were restruck onto fresh plates with PYE plus kanamycin, and chromosomal DNA subsequently prepared from single colonies was cultured in PYE with kanamycin. DNA was digested with BfuCI for 2 min at room temperature and 20 min at 80°C, to yield approximately 5-kb fragments. Sheared DNA was ligated with T4 DNA ligase, and the reaction mixture was dialyzed for 1 h using 0.45-μm nitrocellulose filters (Millipore). From each dialyzed ligation reaction mixture, 1.5 μl was electroporated into 25 μl of pir-116 cells and plated on LB medium supplemented with kanamycin. DNA was extracted from the resulting colonies and sequenced using KAN-2 FP-1 and R6KAN-2 RP-1 primers (Epicentre).
RESULTS
Epitope-tagged PhoB retains wild-type function.
To map the PhoB regulon in Caulobacter, we sought to perform ChIP-Seq on PhoB. To this end, we constructed a strain in which the chromosomal copy of phoB encodes a 20-amino-acid linker and a C-terminal 3×FLAG epitope tag (phoB-3×FLAG). To test whether this version of phoB supports wild-type-like growth under both phosphate-replete and phosphate-starved conditions, we grew the phoB-3×FLAG strain in minimal medium with 10 mM phosphate (M2G) and then washed and resuspended the cells in minimal medium with either 10 mM phosphate (M2G) or 50 μM phosphate (M5G), with the latter representing phosphate-limited conditions. The growth of the phoB-3×FLAG strain was indistinguishable from that of the wild type under both conditions, in contrast to the growth of a phoB deletion strain, which grew poorly under both conditions (Fig. 1A).
FIG 1.

A strain producing PhoB with a C-terminal 3×FLAG epitope behaves like the wild type (WT) under phosphate-replete and phosphate-limited conditions. (A) Growth curves of the strains indicated, in minimal medium with 10 mM or 50 μM phosphate. (B) β-Galactosidase assays for a PpstC-lacZ (left) and PpstS-lacZ (right) reporter in minimal medium with 10 mM phosphate (top) or 50 μM phosphate (bottom). The strains indicated were grown in minimal medium with 10 mM extracellular phosphate, washed, resuspended in medium with 10 mM or 50 μM phosphate, and grown for 7 h to an OD600 between 0.3 and 0.4. For panels A and B, data points indicate the averages of results from three independent replicates, with error bars representing the standard deviations (SD). (C) Phase-contrast microscopy of the indicated strains expressing wild-type phoB. Bars, 2 μm.
We also tested whether PhoB-3×FLAG regulated PhoB-dependent genes in a manner comparable to that of the untagged PhoB. We constructed lacZ transcriptional fusions with the pstC and pstS promoters in Caulobacter and then assessed the ability of phoB-3×FLAG to induce the expression of each reporter following phosphate limitation. Cells were shifted from M2G to M5G, or mock shifted and retained in M2G, and were grown for 7 h to mid-exponential phase before β-galactosidase activity was measured. In M2G, the activity of each reporter was ∼2,000 Miller units in both the phoB-3×FLAG and wild-type strains. In M5G, β-galactosidase activity was induced to ∼10,000 Miller units for the PpstC reporter in both the wild-type and phoB-3×FLAG strains and to ∼8,000 Miller units for the PpstS reporter in both strains. These results indicate that FLAG-tagged PhoB functions as well as untagged PhoB to induce the expression of PhoB-dependent genes. As a control, we confirmed that in a phoB deletion strain, both reporters exhibited less than 2,000 Miller units of activity, consistent with these promoters being PhoB dependent (Fig. 1B).
Finally, we assessed the activity of PhoB-3×FLAG in a strain that constitutively activates the Pho system. Null mutations of pstS, which encodes the periplasmic phosphate-binding protein, block phosphate import and result in hyperactivation of the Pho regulon in Caulobacter, including stalk elongation, even during growth under phosphate-replete conditions (23). We found that in a pstS::Tn5 background, cells producing PhoB-3×FLAG also exhibited extensive stalk elongation (Fig. 1C), further supporting our conclusion that the epitope-tagged version of PhoB binds and regulates the same set of target genes as wild-type PhoB.
ChIP-Seq reveals genome-wide binding patterns of PhoB.
We performed ChIP-Seq analysis using an anti-FLAG antibody with cells expressing phoB-3×FLAG grown to mid-exponential phase in (i) peptone-yeast extract (PYE), a complex rich medium in which PhoB should be predominantly unphosphorylated and inactive, (ii) minimal defined medium containing 50 μM phosphate (M5G), in which PhoB is phosphorylated and active, as judged by the PhoB-dependent reporters for PpstC and PpstS (Fig. 1B), and (iii) PYE with cells also harboring a disruption of pstS, leading to high constitutive activation of PhoB. In each case, PhoB-bound DNA was immunoprecipitated using an anti-FLAG antibody. As controls, we performed ChIP-Seq using the same anti-FLAG antibody on strains grown under identical conditions but producing untagged wild-type PhoB.
We first used qPCR to verify that the ChIP samples for strains producing tagged PhoB were enriched for a chromosomal region (the pstC promoter) strongly predicted to be PhoB bound, based on E. coli studies and the expression analyses presented below. As a control locus, we examined the promoter of gene CC_1294 (PCC1294), whose expression is not PhoB regulated. For all three growth conditions involving cells producing PhoB-3×FLAG, the pstC locus was >20-fold enriched by ChIP relative to the input DNA (Fig. 2A), in contrast to what occurred with cells producing wild-type PhoB, for which all enrichment ratios were <2.5-fold. As expected, PCC1294 was not significantly enriched in any of the samples (Fig. 2A).
FIG 2.

ChIP-Seq reveals genome-wide binding patterns of PhoB. (A) qPCR of PpstC, a PhoB-activated promoter, and PCC1294, a negative control, using DNA samples after ChIP with an anti-FLAG antibody. Strains expressed either phoB-3×FLAG or wild-type phoB and were grown in minimal medium with 10 mM phosphate or 50 μM phosphate or in rich medium (PYE), with a pstS::Tn5 mutation introduced to constitutively activate PhoB. The y axis indicates fold enrichment of a locus in the ChIP output sample, compared to the input DNA. (B) Numbers of ChIP-Seq reads at three loci. Strains produced either PhoB-3×FLAG or wild-type PhoB and were grown in PYE or minimal medium with 50 μM phosphate; a pstS::Tn5 mutation on the chromosome is indicated when appropriate. Plots are scaled to the maximum read count for each locus. (C) Full-genome ChIP-Seq profiles for the same strains and growth conditions indicated in panel B. Samples from strains producing PhoB-3×FLAG are represented in red; samples from control strains expressing wild-type PhoB are represented in black below the x axes. The positions of genes from panel B are indicated.
We then constructed and deep sequenced libraries from the ChIP samples taken from cells producing epitope-tagged PhoB and grown in a rich medium or phosphate-limited medium or cells with an additional pstS::Tn5 mutation grown in rich medium; control ChIP samples were taken from strains treated identically but harboring the wild-type copy of phoB. Equal numbers of reads (860,000 reads) were analyzed from each sample, using the peak-calling software MACS (33), and peaks for which the P values were <10−5 were identified.
Consistent with our qPCR data, the genome-wide PhoB binding profiles (Fig. 2B and C) indicated that in most cases, PhoB binding was induced by phosphate limitation. Only 5 significant peaks were identified in the rich medium sample, while 102 significant peaks were found in the 50 μM phosphate sample and 204 were identified in the pstS::Tn5 sample, consistent with PhoB being hyperactivated in this genetic background (Fig. 3A; also see Table S1 in the supplemental material).
FIG 3.

PhoB binds to the promoters of Pho regulon genes upon phosphate limitation. (A) Overlap between the sets of genes containing significant PhoB peaks in ChIP-Seq samples from cells grown in minimal medium with 50 μM phosphate or in PYE while harboring a pstS::Tn5 mutation that mimics constitutive phosphate starvation. (B) Genes whose promoters showed >5-fold enrichment by ChIP-Seq analysis of pstS::Tn5 cells grown in rich PYE medium and a >1.7-fold expression change in pstS::Tn5 cells assayed by microarray analysis. ChIP fold enrichment is shown for the same set of genes in PYE and in minimal medium containing 50 μM phosphate. Fold enrichment relative to the input DNA is represented using colors, as indicated, and expression changes assayed by microarray analysis of the pstS::Tn5 and pstS::Tn5 ΔphoB strains are shown. The one-line annotation for each gene or operon whose promoter is bound by PhoB is listed; genes potentially in the same operon are separated by slashes. CC numbers from the original CB15 genome annotation are listed, except for genes newly annotated in the CB15N genome (with CCNA numbers). Note that small RNA genes have CCNAR numbers. (C) MEME was used to identify a motif from sequences of peaks >13-fold enriched in the pstS::Tn5 ChIP-Seq sample. The E value is 1.3 × 10−20. PhoB binding sites (pho boxes) and the putative −35 site are labeled.
Fold enrichment for individual loci in the ChIP-Seq data was determined by comparing the number of reads at a particular peak in the epitope-tagged experimental sample with the number of reads at the same locus in the non-epitope-tagged control sample. At most loci, the observed fold enrichment was greater in the pstS mutant sample than in the low-phosphate sample (Fig. 3B; also see Table S1 in the supplemental material); this may indicate that a pstS mutation leads to greater activation of PhoB than does low-phosphate growth medium. Alternatively, the difference may reflect differences in the abundances of other transcription factors, as the pstS cells were grown in rich medium while the phosphate-starved cells were grown in minimal medium. In total, 92 peaks identified in the pstS sample were also identified in the 50 μM phosphate sample (Fig. 3A). The overlap in the peaks identified for these two independent samples, in which PhoB is activated under different nutrient conditions, supports the notion that these peaks represent direct PhoB binding sites and suggests that PhoB regulates the same core set of genes at different levels of phosphate limitation. Finally, we noted that the vast majority of PhoB ChIP-Seq peaks were found in intergenic regions (see Table S1). Only 4 of the 50 highest peaks were contained within an annotated coding region. This pattern is consistent with a model in which PhoB activates transcription primarily by binding near the −35 region of promoters, outside coding regions (8).
Identification of the PhoB regulon.
To delineate high-confidence members of the PhoB regulon, we used whole-genome DNA microarrays to identify genes whose expression depends on PhoB. We harvested RNA from strains harboring either the pstS::Tn5 mutation alone or the pstS::Tn5 mutation in a ΔphoB background, with each strain being grown to mid-exponential phase in rich medium. RNAs from these strains were compared on microarrays with RNA obtained from wild-type Caulobacter grown under the same conditions. To identify PhoB-regulated genes, we selected genes that (i) had expression levels that changed at least 1.7-fold in the pstS mutant relative to the wild type but did not change in the pstS::Tn5 ΔphoB double mutant relative to the wild type and (ii) had promoters that were enriched ≥5-fold over background in the PhoB ChIP-Seq analysis of pstS::Tn5 cells. A threshold of 5-fold was chosen because promoters showing enrichment above this level in the pstS::Tn5 cells also typically showed high enrichment in the M5G ChIP sample. In total, 43 genes fit these criteria, with 32 genes in 22 putative transcriptional units being activated by PhoB and 11 genes being repressed by PhoB (Fig. 3B). There were an additional 91 genes whose expression changed at least 2-fold in the pstS::Tn5 mutant but that did not have peaks indicating ≥5-fold enrichment in the PhoB ChIP-Seq data, although some had >2-fold enrichment (see Table S2 in the supplemental material). These genes are likely indirectly regulated by PhoB. Conversely, there were 39 genes that had PhoB ChIP-Seq peaks indicating >5-fold enrichment but that did not change significantly in terms of gene expression. Some of these genes may be directly regulated by PhoB, but either a change in gene expression was not detected by DNA microarray analysis or expression of these genes may change only transiently upon phosphate starvation. Finally, we note that there were 122 genes with ChIP-Seq peaks indicating enrichment of >2-fold; some of these genes may also be bona fide PhoB targets.
We used the motif-finding program MEME (34) to identify a consensus PhoB binding site using the sequences of the 24 ChIP-Seq peaks that were >13-fold enriched in the pstS mutant. The resulting motif contains two 6-bp sites that appear to be direct repeats flanking an AT-rich region (Fig. 3C). Although similar to the PhoB consensus site predicted for E. coli (11), this Caulobacter site is 1 base pair shorter and has a higher GC content, with the latter possibly reflecting the higher GC content of the Caulobacter genome. In E. coli, the central AT-rich region was proposed to be a modified −35 binding site (11). The putative Caulobacter PhoB binding motif, or Pho box, identified here was used to predict PhoB binding sites across the Caulobacter genome using MAST (34). This analysis identified putative Pho boxes within 37 of the 50 most enriched ChIP-Seq peaks (see Table S1 in the supplemental material).
The list of 43 genes in Fig. 3B represents high-confidence members of the PhoB regulon and includes several known and expected target genes, such as the Pst transporter genes (pstCAB and pstS), phoU, and phoB itself. The PhoB regulon indicates that Caulobacter cells respond to phosphate limitation through major changes in the expression of membrane transport systems, likely to support the scavenging of inorganic phosphate from the extracellular environment while preventing unwanted efflux. In addition to upregulating the high-affinity Pst transporter, PhoB activates the expression of the phosphonate transport system PhnCDE, 6 genes annotated as TonB-dependent receptors, and the TonB accessory proteins ExbB and ExbD. The set of PhoB targets also includes genes that help to generate sources of inorganic phosphate, such as a secreted alkaline phosphatase, an exported lipoprotein, called ElpS, that may stimulate alkaline phosphatase activity (35), and another putative secreted phosphatase, PhoX. The set of genes downregulated by PhoB includes 4 TonB-dependent receptors. Whether (and how) the repression of these transporters helps cells cope with phosphate limitation is unclear, but the changes in their expression underscore the notion that Caulobacter cells respond to phosphate starvation by remodeling membrane transport capabilities.
PhoU is not a negative regulator of the Pho regulon in Caulobacter.
The activation of PhoB as a transcription factor depends on the histidine kinase PhoR, which somehow senses changes in flux through the phosphate transporter PstABC. Whether this sensing is direct is unknown. A highly conserved protein called PhoU, which often is encoded in the same operon as the pst genes, has been suggested to couple PhoR with the Pst transporter. Specifically, PhoU was suggested to repress PhoR activity under phosphate-replete conditions when the transporter is active, implying that PhoU is a negative regulator of the Pho regulon (1).
To test this hypothesis in Caulobacter, we constructed a strain in which phoU was deleted from its native locus but inserted at the van locus under the control of the Pvan promoter, permitting inducible expression of phoU by vanillate. This phoU depletion strain was cultured overnight in rich medium supplemented with vanillate, and cells were then shifted to medium without vanillate and diluted as needed to maintain exponential growth. Upon the removal of vanillate, the depletion of PhoU did not result in stalk hyperelongation, as would be expected upon loss of a negative regulator of the Pho regulon and as occurs in pstS mutants (Fig. 1C). Instead, the loss of PhoU led to enlarged and slightly filamentous cells after 8 h of depletion and modest chromosome accumulation after 16 h of depletion (Fig. 4A).
FIG 4.

Depleting PhoU does not phenocopy a pstS null mutant. (A) Phase-contrast microscopy and flow cytometry analysis of DNA contents for the strains indicated, in the presence and absence of vanillate (van). Microscopic images were taken 8 h after the removal of vanillate. Bar, 2 μm. For flow cytometry, samples were collected 16 h after the removal of vanillate. (B) Numbers of CFU of the indicated strains, grown in PYE. Numbers of CFU reported are the averages of results from two replicates and indicate the number of CFU per 1 ml normalized to an OD600 of 1.
Also in contrast to a pstS mutant, we found that the depletion of phoU was lethal. To measure viability, the phoU depletion strain was shifted to medium without vanillate and diluted approximately every two generations to maintain growth in mid-exponential phase. Samples were taken at 3-h intervals to measure CFU, normalized at each time point to the number of CFU per ml of culture at an OD600 of 1. We observed a 3-log decrease in CFU after 30 h of depletion (Fig. 4B). In contrast, when wild-type or pstS::Tn5 cells were treated in the same manner, we observed no loss in viability. Finally, we note that we were unable to delete the native copy of phoU unless another copy of phoU was present at the vanillate locus (data not shown). Collectively, these phenotypic analyses suggest that PhoU likely does not negatively regulate the Pho regulon in Caulobacter.
To directly assess whether PhoU influences the Pho regulon, we examined global patterns of gene expression in the phoU depletion strain. A culture was grown in the presence of vanillate and then shifted to medium with or without vanillate. Samples were removed at 2, 5, 7, and 16 h postshift, and RNAs from the two conditions were directly compared on DNA microarrays. Although we did not have an antibody for monitoring PhoU levels, we note that the viability of the phoU depletion strain began to decrease ∼10 h postshift, suggesting that PhoU levels had dropped significantly by that point. Moreover, even if PhoU were lost only through dilution, it would be present at <6% or <0.1% of the initial levels after 7 or 16 h of depletion, respectively, given that the phoU depletion strain accumulates (the optical density increases) at a rate comparable to that of the nondepleted control for up to 14 h.
The vast majority of genes upregulated 2-fold or more in a pstS mutant were not upregulated after phoU was depleted (Fig. 5A; also see Table S2 in the supplemental material). Expression of the pstCAB genes (but not pstS) was increased ∼2.6-fold 7 h after the shift, although this change was substantially less than that observed in the pstS mutant. After 16 h of PhoU depletion, some Pho regulon genes were upregulated (Fig. 5A); however, much greater changes in expression were observed for a multitude of other stress response genes at this time point (Fig. 5B; also see Table S2). Additionally, by the 16-h time point, an approximately 2-log decrease in viability was observed (Fig. 4B), suggesting that expression changes at this time point might have resulted nonspecifically from cell death. In support of the latter idea, we also examined gene expression changes in a phoU depletion strain harboring a suppressor mutation (see below), such that cells remain viable throughout a PhoU depletion time course. In that case, we did not observe any significant changes in the Pho regulon genes or in other genes that change following PhoU depletion, even after 16 h (Fig. 5C; also see Table S2); however, it is possible that for the Pho regulon, the genes are already maximally induced in the suppressor strain and thus cannot be further induced.
FIG 5.

Depleting PhoU does not produce the same gene expression changes seen in a pstS mutant. (A) Expression changes after depletion of PhoU for 2, 5, 7, and 16 h. Expression values are the averages of results from two replicates. Genes upregulated at least 2-fold in the pstS::Tn5 strain are shown. Green, direct PhoB targets, as determined by ChIP-Seq. The genes labeled are those that were ≥2-fold upregulated in the phoU depletion strain at the 7-h time point and found to be direct PhoB targets. (B) Venn diagrams indicating the numbers of genes whose expression changed at least 2-fold, compared to their expression in the wild type, in the pstS::Tn5 strain or in the phoU depletion strain after 7 or 16 h of depletion. (C) Expression changes in a PhoU depletion strain harboring the suppressor mutation pstS::Tn5. Data are for the same genes shown in panel A. Numbers of hours after the removal of vanillate are indicated. (D) β-Galactosidase assay of PpstC-lacZ and PpstS-lacZ reporter expression after PhoU depletion in PYE. wt, wild type. Data points indicate the averages from three independent replicates, with error bars representing the SD.
We also assayed the effects of depleting PhoU by using lacZ reporters for the pstC and pstS promoters, which are PhoB regulated (Fig. 1B and 3B). We again shifted the phoU depletion strain to noninducing conditions, and we observed a modest increase in the activity of the PpstC reporter but not the PpstS reporter after 7 h or more (Fig. 5D). In both cases, substantially higher activity was seen in the pstS mutant. Taken together, our data indicate that a loss of phoU does not induce the same expression patterns as seen in a pstS mutant in which the Pho regulon is upregulated. These data support the conclusion that PhoU does not function as a negative regulator of the Pho regulon in Caulobacter.
Mutations in both the Pst and Pho systems suppress a phoU mutant.
To further probe the function of PhoU, we isolated mutations that restore viability to a strain depleted of PhoU. We performed transposon mutagenesis of the Pvan-phoU depletion strain using kanamycin-marked Tn5. We plated transposon-mutagenized cells on rich medium with kanamycin and without vanillate, to select for mutants that are viable in the absence of phoU. We selected colonies that grew after 2 to 4 days, and we identified the sites of transposon insertion for 18 candidate suppressors (Fig. 6A). Two insertions mapped near vanR, the vanillate repressor (which regulates phoU transcription in this strain), and one mapped to cobT, a cobaltochelatase, which we did not independently verify. Seven Tn5 insertions mapped to phoR or phoB, with the remaining 8 insertions being distributed among the four components of the pst system, i.e., pstSCAB.
FIG 6.

Mutations in the pst and pho genes suppress phoU depletion lethality, and polyphosphate accumulates upon phoU depletion in Caulobacter crescentus. (A) Locations of Tn5 insertions that suppressed phoU depletion lethality are indicated by arrows. Bars denote directly adjacent but not cooperonic loci. phoU, which was deleted in this background, is shown in black. (B) Light microscopy of the noted strains in the presence and absence of vanillate. Arrows indicate dark granules that may represent polyphosphate (Fig. 7). Bars, 2 μm. (C) Numbers of CFU for the strains indicated, with (left) or without (right) vanillate, are shown, with data points representing the averages of results from two replicates and given as the number of CFU in 1 ml normalized to an OD600 of 1.
To verify that mutations in both the pst and pho systems can suppress the lethality of a phoU mutant, we independently transduced the pstS::Tn5 allele and a ΔphoR mutation into the phoU depletion strain. In both cases, the mutation introduced was epistatic to the depletion of phoU. Cells depleted of PhoU and harboring a phoR deletion had a morphology similar to that of the ΔphoR strain and no longer lost viability when shifted to medium lacking vanillate (Fig. 6B and C). Similarly, pstS::Tn5 cells depleted of PhoU exhibited the long-stalk phenotype associated with pstS::Tn5 and retained viability upon being shifted to medium without vanillate. These findings confirm that mutations disrupting either the PhoR-PhoB signaling pathway or the Pst transporter can suppress the lethality of PhoU depletion.
These genetic results corroborate our conclusion that PhoU is not a negative regulator of the Pho regulon. If it were and the lethality of phoU depletion were due to overexpression of the Pho regulon, then our screen would have been predicted to identify suppressor mutations in phoR and phoB but not in the pst genes, because such mutations, like pstS::Tn5, dramatically upregulate the Pho regulon (Fig. 3A and B). Instead, our results strongly suggest that the lethality of a PhoU mutant is suppressed by reducing the levels of the Pst transporter, which is achieved directly by disrupting pst genes or indirectly by disrupting phoR or phoB, genes that are required for expression of the pst genes (Fig. 1B and 5C). This model suggests that PhoU may participate in regulating or metabolizing cellular pools of inorganic phosphate that are taken up by the Pst system, such that, without PhoU, the inorganic phosphate imported is inappropriately metabolized or perhaps converted to a toxic form, leading to cell death. Alternatively, the activity of the Pst transporter may be increased without PhoU and cells may not tolerate the resulting excessive concentrations of inorganic phosphate.
Intriguingly, we observed the formation of large intracellular granules by phase microscopy after depletion of phoU (Fig. 7A); the granules may be comprised of polyphosphate (36). To test whether the intracellular granules observed are in fact composed of polyphosphate, we used fluorescence microscopy. When stained with DAPI, polyphosphate granules fluoresce green, while DNA fluoresces blue, and an appropriate filter set can discriminate between polyphosphate and nucleic acids (37). We found that, when grown in rich medium in the absence of vanillate, the phoU depletion strain indeed accumulated large stores of concentrated polyphosphate, visible as bright foci in most cells after 9 h (Fig. 7A, top) and as large extended structures after 22 h (Fig. 7A, bottom). In the presence of vanillate, little intracellular polyphosphate accumulation was observed with DAPI staining. Under these growth conditions, foci were rarely seen in the wild type and, when present, were substantially smaller than those seen following PhoU depletion. These results are consistent with a model in which the uptake of inorganic phosphate is unregulated in cells lacking PhoU, leading to the accumulation of large cytoplasmic pools of polyphosphate.
FIG 7.

Polyphosphate accumulates upon PhoU depletion in Caulobacter crescentus. (A) The strains indicated were grown either with or without vanillate for the times indicated. Cells were imaged by phase-contrast and epifluorescence microscopy using a filter set specific for DAPI-polyphosphate. Phase images were overlaid with the fluorescence image at 100% fluorescence excitation intensity (in green) for the 9-h time points and at a 30% excitation intensity (in magenta) for the 22-h time points. Bars, 2 μm. (B) Numbers of CFU for the strains indicated, with or without vanillate, are shown, with data points representing the averages of results from two replicates and given as the number of CFU in 1 ml normalized to an OD600 of 1.
We also asked whether the polyphosphate accumulation observed was responsible for the lethality of the phoU depletion mutant. If this were the case, then decreasing the levels of polyphosphate should restore viability. In Caulobacter, two genes, i.e., ppk1 and ppk2, are responsible for polyphosphate synthesis (38). We therefore deleted ppk1 and ppk2 in the phoU depletion strain and assayed the levels of polyphosphate using DAPI staining. These cells were largely devoid of polyphosphate foci, even after depletion of PhoU for 22 h, as expected (Fig. 7A). However, despite the absence of polyphosphate granules, cells lacking ppk1 and ppk2 and depleted of PhoU still lost viability, like the phoU depletion strain alone (Fig. 7B). These results suggest that the polyphosphate granules seen in the phoU depletion strain do not contribute significantly to cell death but reflect the fact that the cells likely have imported excessive levels of inorganic phosphate. The latter idea is consistent with the results of our suppressor screen, indicating that the lethality of a phoU mutant can be rescued by mutations that reduce the expression of the high-affinity Pst transporter and presumably prevent an accumulation of cellular inorganic phosphate. Collectively, our data suggest that PhoU is not a negative regulator of PhoR and instead regulates phosphate uptake, such that cells depleted of PhoU accumulate excessively high levels of inorganic phosphate.
DISCUSSION
Two-component signaling systems are one of the predominant forms of signal transduction in bacteria. Although these systems employ a variety of output mechanisms, most use a response regulator to enact a transcriptional response (39). However, for the vast majority of response regulators, the regulons remain uncharacterized or incompletely defined. Here we used global expression studies and ChIP-Seq to create a high-resolution map of PhoB targets in Caulobacter crescentus under both phosphate-limited and phosphate-replete conditions. Our results support a model in which phosphate limitation leads to the phosphorylation of PhoB, which enables it to bind and to regulate target genes. We found that PhoB directly activated more than 30 genes following phosphate starvation (Fig. 3B), with at least 90 other genes being indirectly activated by PhoB. We also found that PhoB negatively regulates gene expression, with microarray analysis indicating that 11 PhoB targets are likely directly repressed by PhoB (Fig. 3B).
PhoB regulates a different set of genes in Caulobacter than in E. coli.
A recent ChIP-chip-based study of the Pho regulon in E. coli (11) enables a comparison of the PhoB regulons in Caulobacter and E. coli. We find that the set of genes regulated by PhoB in Caulobacter differs substantially from that regulated in E. coli, indicating that, although the upstream signaling pathway is highly conserved, the output regulon has likely been tailored to each organism's individual needs and ecological niche. Of the 43 genes comprising the Caulobacter Pho regulon, we could identify a reciprocal best BLAST hit in E. coli for only 8 (see Table S3 in the supplemental material). Of those 8 genes, only 3 are members of the E. coli Pho regulon, including phoB itself, the pst operon, and the phosphonate (phn) transport system operon. The minimal overlap of the PhoB regulons in Caulobacter and E. coli may reflect differences in the ways in which genes were identified as PhoB targets, although this is unlikely to explain the lack of overlap entirely. In some cases, there may also be different genes in the two organisms that fulfill similar functions, particularly as both regulons encode a number of transport-related proteins. However, it generally appears that the two organisms have evolved fundamentally different Pho regulons. Some of the differences in the Caulobacter PhoB regulon relative to that of E. coli presumably account for their different physiological responses, most notably the stalk elongation that is a hallmark of phosphate-starved Caulobacter. Which genes are responsible for stalk elongation is not yet clear, but the delineation of the PhoB regulon may help guide future efforts to identify them.
PhoU does not regulate PhoR activity in Caulobacter and instead likely regulates phosphate metabolism.
Although the PhoR-PhoB signaling pathway is highly conserved throughout the bacterial kingdom and has been studied for decades, the mechanism by which the histidine kinase PhoR senses changes in extracellular phosphate levels has remained unclear. PhoU has been suggested to couple the Pst transporter to the histidine kinase PhoR, inhibiting PhoR when flux through the transporter is high (1). However, our results strongly suggest that PhoU does not function in this manner, at least in Caulobacter, because depletion of PhoU did not phenocopy a pstS mutation, which leads to hyperactivity of the PhoR-PhoB pathway. It is formally possible that PhoU has two functions, one that is essential for viability and a second that involves regulating the PhoR-PhoB pathway; if the depletion of PhoU affects the essential function first, then this could, in principle, mask effects on the PhoR-PhoB pathway. However, such a model would require that PhoU be capable of regulating PhoR-PhoB at extremely low levels, as many of the gene expression effects of PhoR hyperactivation were not seen after depletion of PhoU for 16 h. Even if PhoU were a stable protein, its levels should be <1% after 16 h due to dilution, given that the phoU depletion strain continued to grow at a rate comparable to that of the wild-type strain for ∼10 to 14 h.
Our genetic data indicate that the lethality of PhoU depletion can be rescued by mutations that block phosphate uptake, including disruptions of the genes that encode the high-affinity phosphate transporter system PstSCAB or the signaling proteins PhoR and PhoB, which stimulate expression of the transport system (Fig. 8). We can envision two general models for PhoU function, i.e., that (i) PhoU negatively regulates the phosphate import activity of the Pst transporter or (ii) PhoU regulates intracellular phosphate metabolism. We could not detect changes in radioactive phosphate uptake rates in the phoU depletion strain (data not shown). However, the phosphate uptake assay may not detect subtle differences that, over the extended period of a phoU depletion time course (Fig. 4), lead to significant differences in intracellular phosphate levels. Studies of E. coli have produced inconsistent results, as phoU mutants have been reported to have little effect on phosphate import (18), to increase phosphate import (21), and to reduce phosphate import (22). Whether PhoU affects phosphate uptake or metabolism, cell death likely results from an excessive accumulation of intracellular phosphate or other metabolites that accumulate in a phosphate-dependent manner, explaining why mutations that prevent the expression or activity of the Pst transporter, and hence slow the import of phosphate, restore viability. Additionally, we found that Caulobacter cells accumulated large polyphosphate granules following PhoU depletion (Fig. 7), as also seen in other organisms (36). The presence of these granules after PhoU depletion is consistent with a defect in the regulation of phosphate uptake. However, polyphosphate granules are not the underlying cause of cell death in the absence of PhoU, because deletion of ppk1 and ppk2 did not restore viability to the phoU mutant. Instead, only mutations that resulted in the loss of the phosphate transporter or the genes (phoR and phoB) that drive its expression rescued phoU (Fig. 6). Thus, we favor a model in which PhoU regulates the Pst transporter to prevent the toxic accumulation of inorganic phosphate or a phosphate-derived metabolite (Fig. 8). In either case, our results strongly support the conclusion that PhoU does not couple the Pst system to the histidine kinase PhoR, as is commonly asserted.
FIG 8.
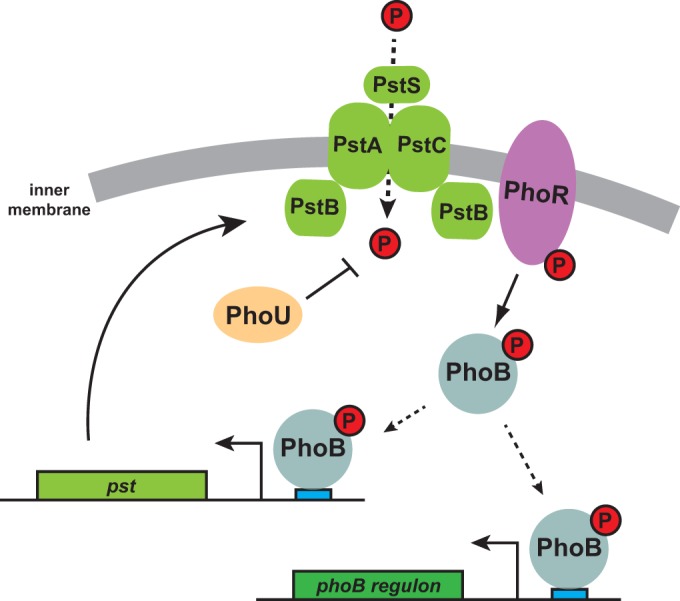
Model for PhoU function and regulation of the response to extracellular phosphate limitation in Caulobacter. The high-affinity transporter PstSCAB imports inorganic phosphate (red) into the cytoplasm. When flux through the transporter decreases, the histidine kinase PhoR is likely directly activated to autophosphorylate and then phosphotransfer to PhoB. Phosphorylated PhoB can bind to pho boxes (blue) in target genes, including the pst transporter. PhoU likely does not couple the Pst transporter to PhoR and instead is proposed to inhibit phosphate uptake by the Pst system or possibly regulate intracellular phosphate metabolism.
Concluding remarks.
We have characterized both the transcriptional output enacted in response to phosphate limitation and the signaling pathway that regulates it in Caulobacter crescentus. We have shown that PhoU does not act as a negative regulator of the Pho regulon but that it is a critical player in cellular phosphate metabolism, a role that is likely to be conserved, given the wide conservation of phoU. We have also identified the genes that PhoB regulates in response to phosphate limitation in Caulobacter crescentus; these genes show little similarity to the set of PhoB-regulated genes in E. coli, highlighting the flexibility and dynamics of transcriptional networks in bacteria. The delineation of the Caulobacter Pho regulon will enable a better understanding of how bacteria respond to phosphate limitation, including how the synthesis of a polar organelle, such as the stalk, is regulated.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Baer for help with ChIP-Seq analysis, A. Yuan for pstS::Tn5 and pstS::Tn5 ΔphoB microarray data, and B. Perchuk for assistance with viability assays.
Funding Statement
M.T.L. is an Investigator of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00658-15.
REFERENCES
- 1.Hsieh YJ, Wanner BL. 2010. Global regulation by the seven-component Pi signaling system. Curr Opin Microbiol 13:198–203. doi: 10.1016/j.mib.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'May GA, Jacobsen SM, Longwell M, Stoodley P, Mobley HL, Shirtliff ME. 2009. The high-affinity phosphate transporter Pst in Proteus mirabilis HI4320 and its importance in biofilm formation. Microbiology 155:1523–1535. doi: 10.1099/mic.0.026500-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamarche MG, Wanner BL, Crepin S, Harel J. 2008. The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol Rev 32:461–473. doi: 10.1111/j.1574-6976.2008.00101.x. [DOI] [PubMed] [Google Scholar]
- 4.Bertrand N, Houle S, LeBihan G, Poirier E, Dozois CM, Harel J. 2010. Increased Pho regulon activation correlates with decreased virulence of an avian pathogenic Escherichia coli O78 strain. Infect Immun 78:5324–5331. doi: 10.1128/IAI.00452-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobsen SM, Lane MC, Harro JM, Shirtliff ME, Mobley HLT. 2008. The high-affinity phosphate transporter Pst is a virulence factor for Proteus mirabilis during complicated urinary tract infection. FEMS Immunol Med Microbiol 52:180–193. doi: 10.1111/j.1574-695X.2007.00358.x. [DOI] [PubMed] [Google Scholar]
- 6.Pratt JT, Ismail AM, Camilli A. 2010. PhoB regulates both environmental and virulence gene expression in Vibrio cholerae. Mol Microbiol 77:1595–1605. doi: 10.1111/j.1365-2958.2010.07310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mack TR, Gao R, Stock AM. 2009. Probing the roles of the two different dimers mediated by the receiver domain of the response regulator PhoB. J Mol Biol 389:349–364. doi: 10.1016/j.jmb.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanco AG, Sola M, Gomis-Ruth FX, Coll M. 2002. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10:701–713. doi: 10.1016/S0969-2126(02)00761-X. [DOI] [PubMed] [Google Scholar]
- 9.Blanco AG, Canals A, Bernues J, Sola M, Coll M. 2011. The structure of a transcription activation subcomplex reveals how sigma-70 is recruited to PhoB promoters. EMBO J 30:3776–3785. doi: 10.1038/emboj.2011.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makino K, Amemura M, Kim SK, Nakata A, Shinagawa H. 1993. Role of the sigma 70 subunit of RNA polymerase in transcriptional activation by activator protein PhoB in Escherichia coli. Genes Dev 7:149–160. doi: 10.1101/gad.7.1.149. [DOI] [PubMed] [Google Scholar]
- 11.Yang C, Huang T-W, Wen S-Y, Chang C-Y, Tsai S-F, Wu W-F, Chang O-H. 2012. Genome-wide PhoB binding and gene expression profiles reveal the hierarchical gene regulatory network of phosphate starvation in Escherichia coli. PLoS One 7:e47314. doi: 10.1371/journal.pone.0047314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baek JH, Lee SY. 2006. Novel gene members in the Pho regulon of Escherichia coli. FEMS Microbiol Lett 264:104–109. doi: 10.1111/j.1574-6968.2006.00440.x. [DOI] [PubMed] [Google Scholar]
- 13.Metcalf WW, Steed PM, Wanner BL. 1990. Identification of phosphate starvation-inducible genes in Escherichia coli K-12 by DNA sequence analysis of psi::lacZ(Mu d1) transcriptional fusions. J Bacteriol 172:3191–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stock AM, Robinson VL, Goudreau PN. 2000. Two-component signal transduction. Annu Rev Biochem 69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 15.Capra EJ, Laub MT. 2012. Evolution of two-component signal transduction systems. Annu Rev Microbiol 66:325–347. doi: 10.1146/annurev-micro-092611-150039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krell T, Lacal J, Busch A, Siva-Jiménez H, Guazzaroni M-E, Ramos JL. 2010. Bacterial sensor kinases: diversity in the recognition of environmental signals. Annu Rev Microbiol 64:539–559. doi: 10.1146/annurev.micro.112408.134054. [DOI] [PubMed] [Google Scholar]
- 17.Baek JH, Kang YJ, Lee SY. 2007. Transcript and protein level analyses of the interactions among PhoB, PhoR, PhoU and CreC in response to phosphate starvation in Escherichia coli. FEMS Microbiol Lett 277:254–259. doi: 10.1111/j.1574-6968.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 18.Steed PM, Wanner BL. 1993. Use of the rep technique for allele replacement to construct mutants with deletions of the pstSCAB-phoU operon: evidence of a new role for the PhoU protein in the phosphate regulon. J Bacteriol 175:6797–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Zhang Y. 2007. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob Agents Chemother 51:2092–2099. doi: 10.1128/AAC.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wanner BL, Latterell P. 1980. Mutants affected in alkaline phosphatase expression: evidence for multiple positive regulators of the phosphate regulon in Escherichia coli. Genetics 96:353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rice CD, Pollard JE, Lewis ZT, McCleary WR. 2009. Employment of a promoter-swapping technique shows that PhoU modulates the activity of the PstSCAB2 ABC transporter in Escherichia coli. Appl Environ Microbiol 75:573–592. doi: 10.1128/AEM.01046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muda M, Rao NN, Torriani A. 1992. Role of PhoU in phosphate import and alkaline phosphatase regulation. J Bacteriol 174:8057–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonin M, Quardokus EM, O'Donnol D, Maddock J, Brun YV. 2000. Regulation of stalk elongation by phosphate in Caulobacter crescentus. J Bacteriol 182:337–347. doi: 10.1128/JB.182.2.337-347.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt JM, Stanier RY. 1966. The development of cellular stalks in bacteria. J Cell Biol 28:423–436. doi: 10.1083/jcb.28.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein EA, Schlimpert S, Hughes V, Brun YV, Thanbichler M, Gitai Z. 2013. Physiological role of stalk lengthening in Caulobacter crescentus. Commun Integr Biol 6:e24561. doi: 10.4161/cib.24561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thanbichler M, Iniesta AA, Shapiro L. 2007. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res 35:e137. doi: 10.1093/nar/gkm818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skerker JM, Prasol M, Perchuk B, Biondi E, Laub MT. 2005. Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a systems-level analysis. PLoS Biol 3:e334. doi: 10.1371/journal.pbio.0030334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gober JW, Shapiro L. 1992. A developmentally regulated Caulobacter flagellar promoter is activated by 3′ enhancer and IHF binding elements. Mol Biol Cell 3:913–926. doi: 10.1091/mbc.3.8.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsokos CG, Perchuk BS, Laub MT. 2011. A dynamic complex of signaling proteins uses polar localization to regulate cell-fate asymmetry in Caulobacter crescentus. Dev Cell 20:329–341. doi: 10.1016/j.devcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 31.Modell JW, Hopkins AC, Laub MT. 2011. A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes Dev 25:1328–1343. doi: 10.1101/gad.2038911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fioravanti A, Fumeaux C, Mohapatra SS, Bompard C, Brilli M, Frandi A, Castric V, Viollier PH, Biondi EG. 2013. DNA binding of the cell cycle transcriptional regulator GcrA depends on N6-adenosine methylation in Caulobacter crescentus and other Alphaproteobacteria. PLoS Genet 9:e1003541. doi: 10.1371/journal.pgen.1003541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey TJ, Bodén M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Blastier S, Hamels A, Cabeen M, Schille L, Tilquin F, Dieu M, Raes M, Matroule JY. 2010. Phosphate starvation triggers production and secretion of an extracellular lipoprotein in Caulobacter crescentus. PLoS One 5:e14198. doi: 10.1371/journal.pone.0014198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morohoshi T, Maruo T, Shirai Y, Kato J, Ikeda T, Takiguchi N, Ohtake H, Kuroda A. 2002. Accumulation of inorganic polyphosphate in phoU mutants of Escherichia coli and Synechocystis sp. strain PCC6803. Appl Environ Microbiol 68:4107–4110. doi: 10.1128/AEM.68.8.4107-4110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henry JT, Crosson S. 2013. Chromosome replication and segregation govern the biogenesis and inheritance of inorganic polyphosphate granules. Mol Biol Cell 24:3177–3186. doi: 10.1091/mbc.E13-04-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boutte CC, Henry JT, Crosson S. 2012. ppGpp and polyphosphate modulate cell cycle progression in Caulobacter crescentus. J Bacteriol 194:28–35. doi: 10.1128/JB.05932-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galperin MY. 2010. Diversity of structure and function of response regulator output domains. Curr Opin Microbiol 13:150–159. doi: 10.1016/j.mib.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.