

Abstract
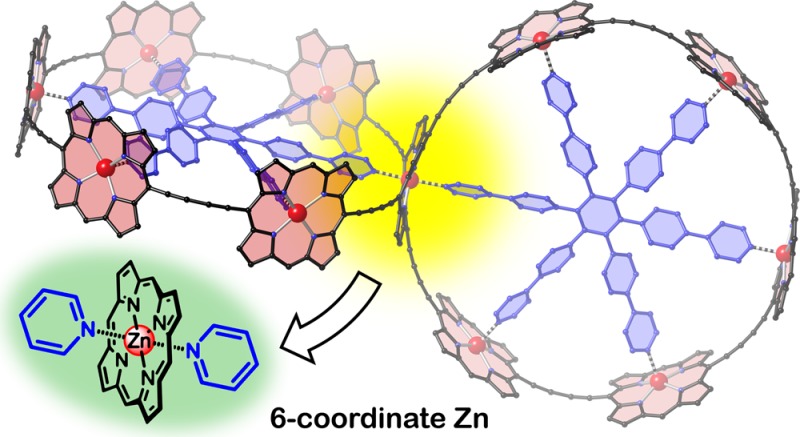
Five-coordinate geometry is the standard binding mode of zinc porphyrins with pyridine ligands. Here we show that pseudo-octahedral six-coordinate zinc porphyrin complexes can also be formed in solution, by taking advantage of the chelate effect. UV–vis–NIR titrations indicate that the strength of this second coordination is ca. 6–8 kJ mol–1. We have used the formation of six-coordinate zinc porphyrin complexes to achieve the template-directed synthesis of a 3D π-conjugated spiro-fused array of 11 porphyrin units, covalently connected in a nontrivial topology. Time-resolved fluorescence anisotropy experiments show that electronic excitation delocalizes between the two perpendicular nanorings of this spiro-system within the experimental time-resolution of 270 fs.
The coordination of pyridine ligands to zinc porphyrins has been widely used to explore the principles of cooperative molecular recognition, following pioneering studies by Tabushi and Sanders in the 1980s.1−6 The basic ground rule in this field is that a zinc porphyrin can bind one axial ligand, to become five-coordinate, but six-coordinate zinc centers, with two axial pyridine ligands, are never observed in solution (Scheme 1). Here we demonstrate that zinc porphyrins with two axial ligands can be formed in solution, when the whole system is suitably stabilized by the chelate effect. We also show that six-coordinate zinc centers can be used in the template-directed synthesis of spiro-linked porphyrin nanorings, which exhibit ultrafast energy migration between the two perpendicular fused macrocycles.
Scheme 1. Coordination of Pyridine to a Zinc Porphyrin.
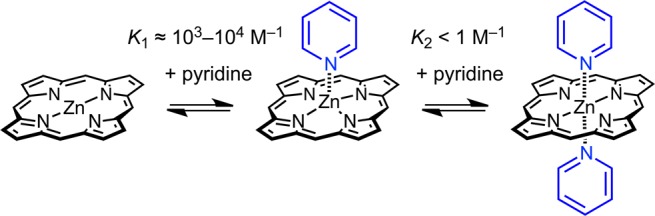
Early studies of the interaction of pyridine with zinc porphyrins in solvents such as chloroform, by UV–vis or NMR titration, found that 1:1 complexes are formed, typically with association constants in the range K1 ≈ 103–104 M–1, whereas 1:2 complexes are not detected (K2 ≤ 1 M–1).7 To the best of our knowledge, six-coordinate zinc porphyrins with two axial pyridine ligands have never previously been detected in solution, although they sometimes occur in crystal structures,8 and zinc porphyrins with two axial DABCO or pyrazine ligands have been studied in supramolecular tweezer complexes.9
We decided to probe the formation of six-coordinate zinc porphyrins, as a potential approach to 3D π-conjugated architectures, by investigating the complexation of a cross-shaped zinc porphyrin pentamer, x-P5, with a tridentate ligand T3 (Figure 1).10 Our idea was that the ability of the central zinc atom of x-P5 to bind two pyridine ligands would be reflected by the stability of the 1:2 complex, x-P5·(T3)2. As expected, the UV–vis–NIR titration of x-P5 with T3 is sharply biphasic (Figure 1c), showing formation of a strong 1:1 complex x-P5·T3, K1 = (2.8 ± 0.4) × 108 M–1,11 followed by a weaker 1:2 complex x-P5·(T3)2, K2 = (1.7 ± 0.3) × 105 M–1. Under the same conditions, the linear porphyrin trimer with a free-base porphyrin at the center, l-P3ZnH2Zn (Figure 2), forms a 1:1 complex with T3 with an association constant of KH2 = (3.3 ± 0.7) × 104 M–1. The observation that x-P5·T3 binds T3 about five times more strongly than l-P3ZnH2Zn demonstrates that there is a significant attractive interaction between the central zinc porphyrin unit of x-P5 and a second axial pyridine. After correction for statistical factors (see Supporting Information (SI)), the difference in free energies corresponding to the strength of this second axial coordination is 5.8 kJ mol–1 at 298 K, which is comparable to a typical hydrogen-bond.12 This is about 30% of the free energy of the first axial coordination to the central Zn atom in x-P5·T3, which can be estimated as 19.0 kJ mol–1 at 298 K, from the difference in ΔG of formation of x-P5·T3 and l-P3ZnH2Zn·T3.
Figure 1.
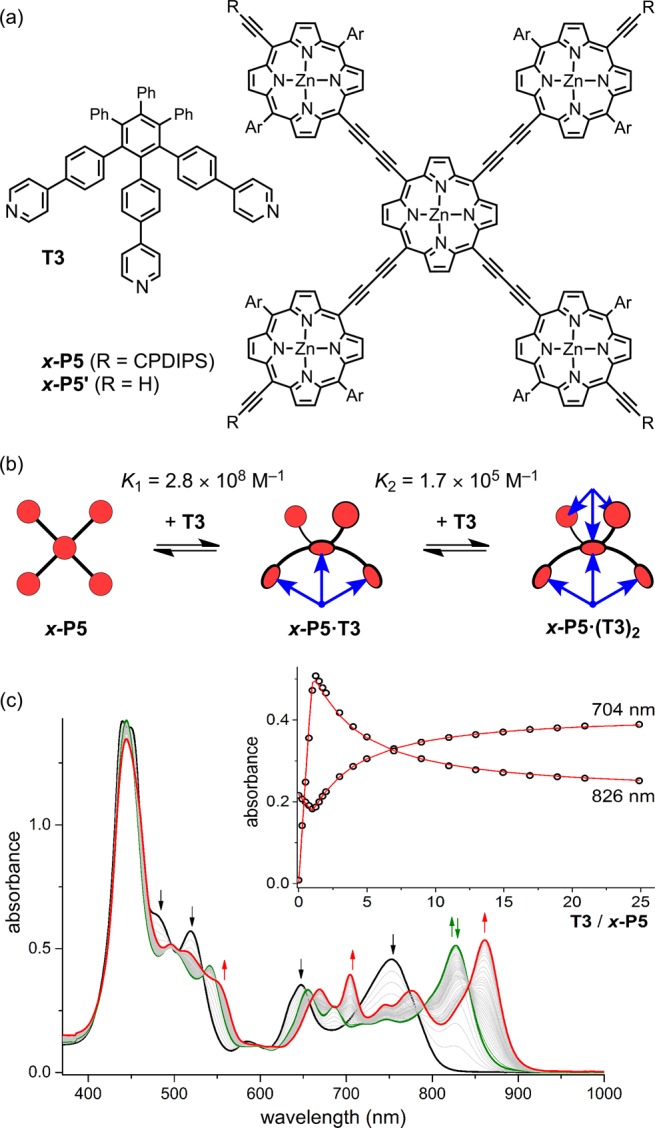
(a) Structures of T3 and x-P5 (CPDIPS = Sii-Pr2CH2CH2CH2CN; Ar = 3,5-bis(trihexylsilyl)phenyl), and (b) their two-stage complexation. (c) UV–vis–NIR titration of x-P5 with T3 in CHCl3 at 298 K. Arrows indicate increasing or decreasing absorption (black, free x-P5; green, x-P5·T3; red: x-P5·(T3)2). Inset: Binding isotherms derived from absorption at 704 and 826 nm and calculated curves for the values of K1 and K2 shown in (b).
Figure 2.
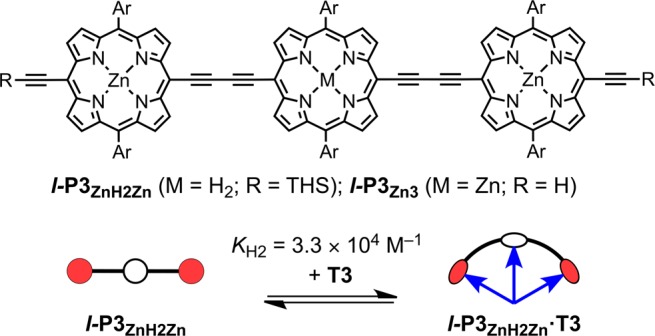
Structure of the porphyrin trimers and binding to T3 (THS = Si(C6H13)3). The formation constant of the 1:1 complex was measured in CHCl3 at 298 K.
The discovery that the porphyrin pentamer x-P5 binds two equivalents of T3 suggested that binding to the hexadentate template T6 could be used to direct the synthesis of topologically interesting 3D π-conjugated architectures, using chemistry that we developed for the synthesis of simpler nanorings.13 Palladium-catalyzed oxidative coupling of the deprotected pentamer x-P5′, bearing four terminal alkyne functionalities, with deprotected linear trimer l-P3Zn3 in the presence of T6 gave the spiro-fused porphyrin 11-mer as its template complex s-P11·(T6)2 in 15% yield (Figure 3). The six-porphyrin nanoring complex c-P6·T6 is also formed as a byproduct from coupling of two molecules of l-P3Zn3 bound to T6, however c-P6·T6 and s-P11·(T6)2 are readily separated by size-exclusion chromatography. The structure of s-P11·(T6)2 was confirmed by NMR and mass spectrometry (MALDI-ToF).
Figure 3.
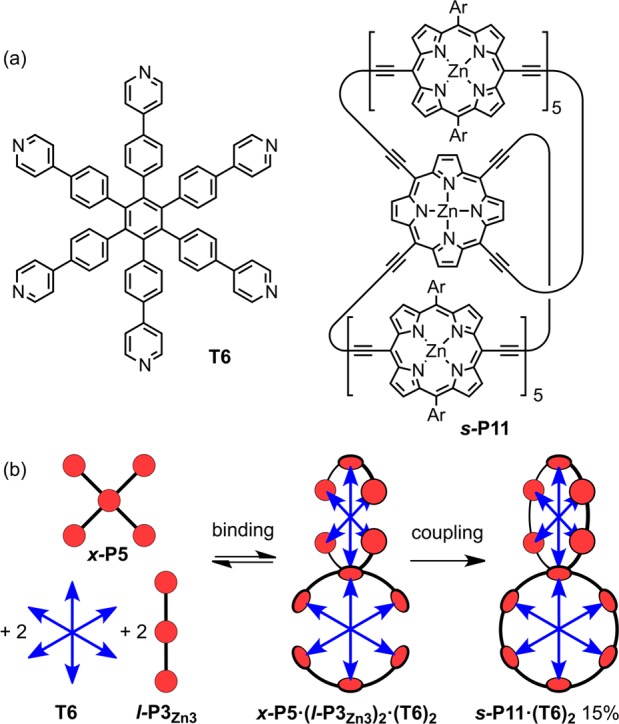
(a) Structures of T6 and s-P11. (b) Synthesis of s-P11·(T6)2 from x-P5′. Reaction conditions: Pd(PPh3)2Cl2, CuI, i-Pr2NH, CHCl3, 1,4-benzoquinone.
The 1H NMR spectrum of s-P11·(T6)2 (in CDCl3 at 298 K, assigned by COSY and NOESY spectroscopy) is consistent with the D2d symmetry of the calculated geometry (Figure 4), in which the two six-porphyrin rings are orthogonal and the central porphyrin unit has a saddle-shaped conformation. The α-pyridine protons of the template nearest to the central six-coordinate zinc atom (labeled as α1 in Figure 4) give rise to a sharp doublet at 2.68 ppm. The other three α-pyridine protons (α2–4) resonate at 2.38–2.40 ppm. The slightly weaker shielding of the central α-pyridine protons probably reflects the weaker ring current of the central porphyrin, resulting from its saddle-shaped nonplanar geometry.14 This explanation is supported by the observation that the singlet from the β-pyrrole protons of the central nonplanar porphyrin (β1) at 9.30 ppm is less deshielded than the β-pyrrole protons proximate to butadiyne substituents of the other porphyrin units (β2–6; 9.53–9.58 ppm).
Figure 4.
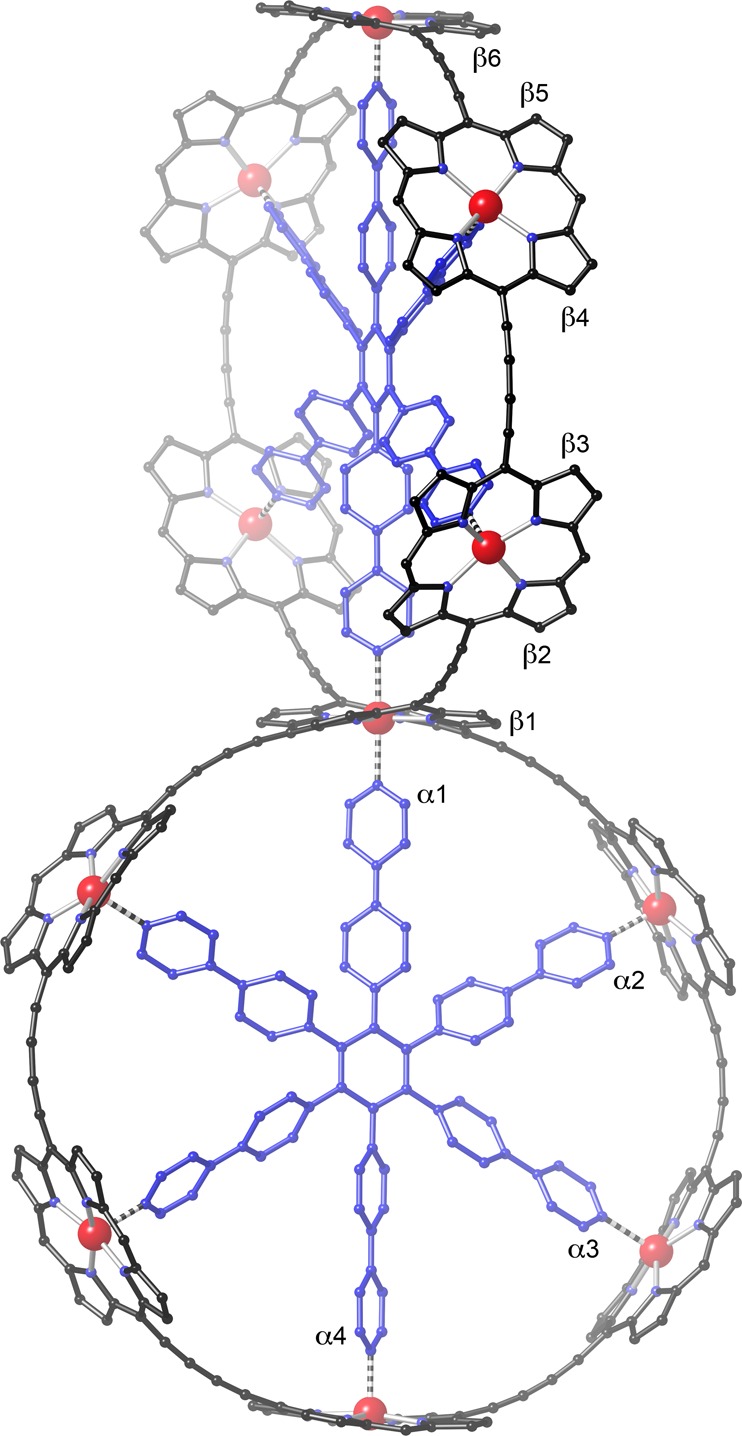
Calculated geometry of s-P11·(T6)2 (MM+ force field, HyperChem; aryl side groups were omitted to simplify the calculation and hydrogen atoms are not shown for clarity).
The UV–vis–NIR absorption and fluorescence spectra of s-P11·(T6)2 are similar to those of c-P6·T6 (Figure 5), although the absorption and emission bands of s-P11·(T6)2 are red-shifted by about 20 nm, reflecting the 3D π-conjugation. Time-resolved fluorescence measurements were performed to explore energy migration in the spiro-fused system.13b,13d,15 The results reveal ultrafast migration of excitation between the two nanorings. Figure 6 shows the fluorescence dynamics of s-P11·(T6)2, excited at 820 nm, detecting fluorescence at 915 nm polarized parallel (I||) or perpendicular (I⊥) to the excitation polarization. The time-dependent fluorescence anisotropy, γ = (I|| – I⊥)/(I|| + 2I⊥), shows no change over 0–20 ps after excitation with an average value of γ = 0.03 ± 0.02, reflecting the electronic communication between the two nanorings. We use a simple model, based on a subset of dipoles distributed across the two ring planes, which is found to predict a value of γ = 0.025 for the case of an excited state delocalized over the whole spiro-fused molecule (see SI). The excellent agreement between theoretical and experimental anisotropies demonstrates that the excited state rapidly delocalizes over all 11 porphyrin units within the 270 fs time-resolution of our experiment.
Figure 5.

UV–vis–NIR absorption (solid line) and fluorescence (dashed line) spectra of c-P6·T6 (red) and s-P11·(T6)2 (black) in toluene with 1% pyridine at 298 K. Data at 1130–1170 nm were omitted due to solvent absorption.
Figure 6.
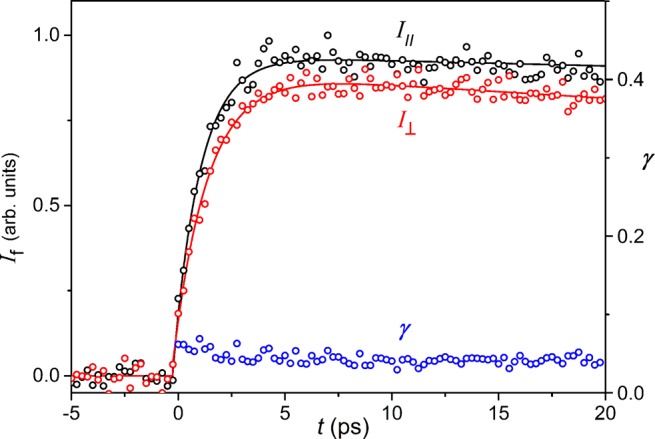
Time-resolved fluorescence decay for s-P11·(T6)2 in toluene with 1% pyridine, recorded upon excitation at 825 nm and detection at 915 nm. The sample is excited by a pulse polarized either parallel (I||, black circles) or perpendicular (I⊥ red circles) to the fluorescence detection polarization, giving the corresponding fluorescence anisotropy dynamics (γ, blue circles).
During the course of this work, we also synthesized an analogous spiro-fused nanoring, sEt-P11·(T6)2, with eight ethyl substituents on the central porphyrin (Figure 7) because the corresponding cross-pentamer xEt-P5 can be prepared in higher yield than x-P5 (see SI). The UV–vis–NIR titration of xEt-P5 with T3 shows similar behavior to titration of x-P5, with the distinct formation of a 1:1 complex xEt-P5·T3 and a 1:2 complex xEt-P5·(T3)2, K2 = (4.1 ± 1.0) × 105 M–1, which corresponds to a free energy of 7.9 kJ mol–1 for the second axial coordination at 298 K. The synthesis of spiro-fused nanoring sEt-P11·(T6)2 was achieved in 23% yield under the same conditions as s-P11·(T6)2. The central α1 pyridine proton in sEt-P11·(T6)2 exhibits a sharp signal at 4.35 ppm; this higher chemical shift, compared to s-P11·(T6)2, probably reflects the greater distortion of the octaethyl-tetraalkynyl porphyrin. The photophysical behavior of sEt-P11·(T6)2 is very similar to that of s-P11·(T6)2 (see SI); it also exhibits ultrafast fluorescence depolarization with an anisotropy of γ = 0.04 ± 0.02.
Figure 7.

Structures of the ethyl-substituted crossed porphyrin pentamer xEt-P5 and spiro-fused porphyrin 11-mer sEt-P11 (CPDIPS = Sii-Pr2CH2CH2CH2CN; Ar = 3,5-bis(trihexylsilyl)phenyl).
The template units can be removed from s-P11·(T6)2 and sEt-P11·(T6)2 by treatment with quinuclidine, as reported previously for c-P6·T6.10,13b These displacement titrations are biphasic (see details in SI), because the first T6 unit to be displaced has a weak six-coordinate zinc interaction, whereas the remaining T6 unit has only five-coordinate zinc interactions. The differences in ΔG for binding the first and second T6 units to s-P11 and sEt-P11 are 32 ± 3 and 36 ± 3 kJ mol–1, respectively, at 298 K. The first T6 unit binds each spiro-fused porphyrin oligomer about 106 times more strongly than the second, illustrating the thermodynamic penalty for formation of a six-coordinate zinc center.
In summary, we have demonstrated that zinc porphyrins can bind two axial pyridine ligands in solution to become six-coordinate, when the resulting complex is suitably stabilized by chelation. UV–vis–NIR titrations showed that the energy of this second axial Zn···N(pyridine) interaction is 6–8 kJ mol–1, which is about a third that of the first axial Zn···N(pyridine) interaction (20 kJ mol–1). This discovery led us to explore the template-directed synthesis of 3D π-conjugated systems based on a six-coordinate zinc center with hexadentate template T6, affording spiro-fused porphyrin nanorings in 15–23% yield. Time-resolved fluorescence anisotropy measurements show that electronic excitation can migrate between the two spiro-fused porphyrin nanorings within 270 fs. The ability to bind two axial ligands to a zinc porphyrin opens up new opportunities for supramolecular chemistry and for the synthesis of new 3D π-conjugated architectures.
Acknowledgments
We thank the EPSRC and the ERC (grant 320969) for support and the EPSRC UK National Mass Spectrometry Facility at Swansea University for mass spectra.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b10126.
Synthesis and characterization of new compounds; UV–vis–NIR titrations and binding data for linear and cyclic oligomer complexes; PL transients and anisotropy dynamics for spiro-nanoring compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tabushi I.; Kugimiya S.; Kinnaird M. G.; Sasaki T. J. Am. Chem. Soc. 1985, 107, 4192. 10.1021/ja00300a019. [DOI] [Google Scholar]
- Abraham R. J.; Leighton P.; Sanders J. K. M. J. Am. Chem. Soc. 1985, 107, 3472. 10.1021/ja00298a012. [DOI] [Google Scholar]
- Satake A.; Kobuke Y. Tetrahedron 2005, 61, 13. 10.1016/j.tet.2004.10.073. [DOI] [Google Scholar]
- Beletskaya I.; Tyurin V. S.; Tsivadze A. Y.; Guilard R.; Stern C. Chem. Rev. 2009, 109, 1659. 10.1021/cr800247a. [DOI] [PubMed] [Google Scholar]
- Durot S.; Taesch J.; Heitz V. Chem. Rev. 2014, 114, 8542. 10.1021/cr400673y. [DOI] [PubMed] [Google Scholar]
- a Adams H.; Chekmeneva E.; Hunter C. A.; Misuraca M. C.; Navarro C.; Turega S. M. J. Am. Chem. Soc. 2013, 135, 1853. 10.1021/ja310221t. [DOI] [PubMed] [Google Scholar]; b D’Souza F.; Amin A. N.; El-Khouly M. E.; Subbaiyan N. K.; Zandler M. E.; Fukuzumi S. J. Am. Chem. Soc. 2012, 134, 654. 10.1021/ja209718g. [DOI] [PubMed] [Google Scholar]; c Gunderson V. L.; Smeigh A. L.; Kim C. H.; Co D. T.; Wasielewski M. R. J. Am. Chem. Soc. 2012, 134, 4363. 10.1021/ja211329k. [DOI] [PubMed] [Google Scholar]
- a Miller J. R.; Dorough G. D. J. Am. Chem. Soc. 1952, 74, 3977. 10.1021/ja01136a003. [DOI] [Google Scholar]; b Kirksey C. H.; Hambright P.; Storm C. B. Inorg. Chem. 1969, 8, 2141. 10.1021/ic50080a022. [DOI] [Google Scholar]; c Cole S. J.; Curthoys G. C.; Magnusson E. A.; Phillips J. N. Inorg. Chem. 1972, 11, 1024. 10.1021/ic50111a023. [DOI] [Google Scholar]
- a Scheidt W. R.; Eigenbrot C. W.; Ogiso M.; Hatano K. Bull. Chem. Soc. Jpn. 1987, 60, 3529. 10.1246/bcsj.60.3529. [DOI] [Google Scholar]; b Krupitsky H.; Stein Z.; Goldberg I.; Strouse C. E. J. Inclusion Phenom. Mol. Recognit. Chem. 1994, 18, 177. 10.1007/BF00705820. [DOI] [Google Scholar]; c Dastidar P.; Krupitsky H.; Stein Z.; Goldberg I. J. Inclusion Phenom. Mol. Recognit. Chem. 1996, 24, 241. 10.1007/BF01133382. [DOI] [Google Scholar]; d Taylor P. N.; Wylie A. P.; Huuskonen J.; Anderson H. L. Angew. Chem., Int. Ed. 1998, 37, 986.. [DOI] [PubMed] [Google Scholar]; e Shukla A. D.; Dave P. C.; Suresh E.; Das A.; Dastidar P. J. Chem. Soc., Dalton Trans. 2000, 4459. 10.1039/b004211k. [DOI] [Google Scholar]; f Diskin-Posner Y.; Patra G. K.; Goldberg I. J. Chem. Soc., Dalton Trans. 2001, 2775. 10.1039/b104961p. [DOI] [Google Scholar]; g Ozarowski A.; Lee H. M.; Balch A. L. J. Am. Chem. Soc. 2003, 125, 12606. 10.1021/ja030221f. [DOI] [PubMed] [Google Scholar]; h Kleij A. W.; Kuil M.; Tooke D. M.; Spek A. L.; Reek J. N. H. Inorg. Chem. 2005, 44, 7696. 10.1021/ic050858v. [DOI] [PubMed] [Google Scholar]; i Ring D. J.; Aragoni M. C.; Champness N. R.; Wilson C. CrystEngComm 2005, 7, 621. 10.1039/b515083n. [DOI] [Google Scholar]; j Seidel R. W.; Goddard R.; Föcker K.; Oppel I. M. CrystEngComm 2010, 12, 387. 10.1039/B913791B. [DOI] [Google Scholar]; k Samanta S. K.; Samanta D.; Bats J. W.; Schmittel M. J. Org. Chem. 2011, 76, 7466. 10.1021/jo201252q. [DOI] [PubMed] [Google Scholar]
- a Kishore R. S. K.; Paululat T.; Schmittel M. Chem. - Eur. J. 2006, 12, 8136. 10.1002/chem.200600463. [DOI] [PubMed] [Google Scholar]; b Schmittel M.; Samanta S. K. J. Org. Chem. 2010, 75, 5911. 10.1021/jo101169f. [DOI] [PubMed] [Google Scholar]
- Hogben H. J.; Sprafke J. K.; Hoffmann M.; Pawlicki M.; Anderson H. L. J. Am. Chem. Soc. 2011, 133, 20962. 10.1021/ja209254r. [DOI] [PubMed] [Google Scholar]
- The first binding constant K1 is too strong to determine accurately from titrations of the type shown in Figure 1c. The value of 2.8 × 108 M–1 was determined by titrating x-P5·T3 with pyridine (see SI for details).
- a Anslyn E. V.; Dougherty D. A.. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006. [Google Scholar]; b Hunter C. A. Angew. Chem., Int. Ed. 2004, 43, 5310. 10.1002/anie.200301739. [DOI] [PubMed] [Google Scholar]
- a O’Sullivan M. C.; Sprafke J. K.; Kondratuk D. V.; Rinfray C.; Claridge T. D. W.; Saywell A.; Blunt M. O.; O’Shea J. N.; Beton P. H.; Malfois M.; Anderson H. L. Nature 2011, 469, 72. 10.1038/nature09683. [DOI] [PubMed] [Google Scholar]; b Sprafke J. K.; Kondratuk D. V.; Wykes M.; Thompson A. L.; Hoffmann M.; Drevinskas R.; Chen W.-H.; Yong C. K.; Kärnbratt J.; Bullock J. E.; Malfois M.; Wasielewski M. R.; Albinsson B.; Herz L. M.; Zigmantas D.; Beljonne D.; Anderson H. L. J. Am. Chem. Soc. 2011, 133, 17262. 10.1021/ja2045919. [DOI] [PubMed] [Google Scholar]; c Kondratuk D. V.; Perdigão L. M. A.; Esmail A. M. S.; O’Shea J. N.; Beton P. H.; Anderson H. L. Nat. Chem. 2015, 7, 317. 10.1038/nchem.2182. [DOI] [PubMed] [Google Scholar]; d Neuhaus P.; Cnossen A.; Gong J. Q.; Herz L. M.; Anderson H. L. Angew. Chem., Int. Ed. 2015, 54, 7344. 10.1002/anie.201502735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medforth C. J.; Muzzi C. M.; Shea K. M.; Smith K. M.; Abraham R. J.; Jia S.; Shelnutt J. A. J. Chem. Soc., Perkin Trans. 2 1997, 833. 10.1039/a603412h. [DOI] [Google Scholar]
- a Parkinson P.; Kondratuk D. V.; Menelaou C.; Gong J. Q.; Anderson H. L.; Herz L. M. J. Phys. Chem. Lett. 2014, 5, 4356. 10.1021/jz5022153. [DOI] [PubMed] [Google Scholar]; b Gong J. Q.; Parkinson P.; Kondratuk D. V.; Gil-Ramírez G.; Anderson H. L.; Herz L. M. J. Phys. Chem. C 2015, 119, 6414. 10.1021/acs.jpcc.5b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yong C.-K.; Parkinson P.; Kondratuk D. V.; Chen W.-H.; Stannard A.; Summerfield A.; Sprafke J. K.; O’Sullivan M. C.; Beton P. H.; Anderson H. L.; Herz L. M. Chem. Sci. 2015, 6, 181. 10.1039/C4SC02424A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.