

Abstract
Accumulation of oxidative damage is a common feature of neurodegeneration that together with mitochondrial dysfunction point to the fact that reactive oxygen species are major contributors to loss of neuronal homeostasis and cell death. Among several targets of oxidative stress, free radical-mediated damage to proteins is particularly important in aging and age-related neurodegenerative diseases. In the majority of cases, oxidative stress mediated post-translational modifications cause non-reversible modifications of protein structure that consistently lead to impaired function. Redox proteomics methods are powerful tools to unravel the complexity of neurodegeneration, by identifying brain proteins with oxidative post-translational modifications that are detrimental for protein function. The present review discusses the current literature showing evidence of impaired pathways linked to oxidative stress possibly involved in the neurodegenerative process leading to the development of Alzheimer-like dementia. In particular, we focus attention on dysregulated pathways that underlie neurodegeneration in both aging adults with Down syndrome (DS) and AD. Since AD pathology is age-dependent in DS and shows similarities with AD, identification of common oxidized proteins by redox proteomics in both DS and AD can improve our understanding of the overlapping mechanisms that lead from normal aging to development of AD. The most relevant proteomics findings highlight that disturbance of protein homeostasis and energy production are central mechanisms of neurodegeneration and overlap in aging DS and AD. Protein oxidation impacts crucial intracellular functions and may be considered a “leitmotif” of degenerating neurons. Therapeutic strategies aimed at preventing/reducing multiple components of processes leading to accumulation of oxidative damage will be critical in future studies.
Keywords: Down syndrome, Alzheimer disease, protein oxidation, redox proteomics, proteostasis network, energy metabolism, trisomy 21
Oxidative Stress Hypothesis of Neurodegeneration
Neurodegenerative disorders are a heterogeneous group of diseases of the nervous system that have many different etiologies. Neuropathologically, such disorders are characterized by selective abnormalities of specific regions of the brain and specific populations of neurons, but oxidative stress contributes, and eventually exacerbates, the major disease-specific pathogenic process. Oxidative stress (OS) is caused by an imbalance in the redox state of the cell, either by overproduction of reactive oxygen species and nitrogen (ROS/RNS), or by decreased antioxidant response. The high lipid content of nervous tissue, together with its high aerobic metabolic activity, leaves the brain particularly susceptible to oxidative damage [1]. In addition, those brain regions that are rich in catecholamines are selectively vulnerable to free radical generation. ROS such as superoxide anion (O2−•), hydrogen peroxide (H2O2), and hydroxyl radical (HO•), are both radical and non-radical oxygen species generated as by-products of aerobic respiration and various other catabolic and anabolic processes [2]. The major sources of free radicals is the mitochondrial oxidative phosphorylation pathway, in which electron leakage from the electron transport chain causes the formation of superoxide anion [3]. Mitochondrial Complex I and Complex III leak superoxide radical at a steady rate that approximates 1–2 % of oxygen that enters the mitochondrial electron transport chain. This radical, in turn, is converted by mitochondrial-resident MnSOD into H2O2 and O2. Detection of superoxide by EPR spin trapping and H2O2 by various fluorescence or electrochemical has been demonstrated (reviewed in [4]). Once released, H2O2 is relatively stable and can diffuse through membrane. In the cytosol, it can be efficiently removed by antioxidant systems such as catalase, glutathione peroxidase, and thioredoxin peroxidase. In addition to mitochondrial production, ROS can be released in response to different environmental stimuli such as growth factors, inflammatory cytokines, ionizing radiation, chemical oxidants, chemotherapeutics, toxins, and transition metals [5]. Other than mitochondria, a number of cytosolic enzymes are able to generate ROS [6] including the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, a plasma membrane-associated enzymes [7]. The function of NADPH oxidases is to produce superoxide from oxygen using electrons from NADPH.
At physiological conditions, intracellular ROS are kept at low but measurable levels, that result from the rate of production and the rate of scavenging by various antioxidants [8]. Indeed, ROS, at low concentration, are found to act as signaling molecules in many physiological processes, including cell growth, cell proliferation, redox homeostasis and cellular signal transduction [9], for example by activating proteins such as tyrosine kinases, mitogen-activated protein kinases, or Ras proteins. It is important to underlie that the multifaceted effects of ROS on different cellular processes suggest that oxidant species are not merely detrimental by-products, but also crucial mediators of a variety of signaling pathways [9].
The increase in energy metabolism by aerobic pathways enhances the intracellular concentration of ROS, which in turn trigger the self-sustaining lipid peroxidation cascade, inducing damage to several brain structures, mostly in the presence of compromised physiological defense systems, such as antioxidant enzymes and cell stress response. The rate of generation of ROS in different cellular types correlates with life-span, and is a major contributor in defining the rate of aging and the development of age-related diseases [6].
Over several decades, researchers demonstrated that OS is a causative or at least collateral factor in the pathogenesis of several neurodegenerative diseases [10]. Considering that aging is the most important risk factor for neurodegenenerative disorders, the free-radical theory of aging may be translated to neurodegeneration as well. Indeed, the progressive and irreversible accumulation of oxidative damage impacts the senescence process, contributing to altered physiological functions, increasing incidence of disease, along with a reduction in life span [11]. In parallel, changes in redox-responsive signaling cascades and in the expression of corresponding target genes may have a similar or even greater impact on senescence as the direct radical-specific damage of cellular components [9].
Among different targets, free radical-mediated damage to proteins is particularly crucial in aging and in age-related neurodegenerative diseases, because in the majority of cases oxidative modification is a non-reversible phenomenon that requires clearance systems for removal of oxidized/dysfunctional proteins [11]. Generally, oxidation of proteins could affect different processes including protein expression, protein turnover, cell signalling, eventually leading to cell death [12]. Another major consequence of protein oxidation is the formation of large protein aggregates, which are often toxic to cells if allowed to accumulate. Insoluble aggregates can be formed as a result of covalent crosslinks among peptide chains, as in the case of amyolid β peptide (Aβ) and hyperphosphorylated tau in AD. Deposits of aggregated, misfolded, and oxidized proteins accumulate normally over time in cells and tissues and are often present in increased amounts in a range of age-related disorders.
Once neuronal homeostasis is disturbed by the increasing burden of oxidized proteins, as a result of both physiological and pathological aging, protective mechanisms including protein degradation system sustain cellular homeostasis by repairing or removing the oxidized products [13]. However, reduced activity of these defense mechanisms may render the cell incapable of efficiently removing oxidized biomolecules, resulting in their accumulation. Two major pathways are responsible for the proteolysis of intracellular proteins, either for damage and self-renewal: the ubiquitin-proteasome system (UPS) [14] and the autophagy-lysosome pathway [15]. The UPS is located in the cytosol and the nucleus, and it is responsible for the degradation of more than 70–80% of intracellular proteins. Most of the proteins are targeted for proteasomal degradation after being tagged with a polyubiquitin chain that, in turn, is recognized by the proteasome. Experimental evidence suggests that failure of the UPS may contribute to neurodegeneration. However, additional factors, including other protein degradation pathways and mitochondrial dysfunction associated with a decline in ATP levels, may contribute to cellular viability.
In addition the UPS, the other primary pathway for protein breakdown in the cell is through autophagy. Autophagy includes three major types: macroautophagy (indicated as autophagy), microautophagy, and chaperone-mediated autophagy (CMA). All three mechanisms share a common destiny of lysosomal degradation, but are mechanistically different one from another [16–18].
Protective up-regulation of the UPS, macroautophagy, and CMA occurs in response to OS to remove oxidized proteins. However, in the presence of higher free radical levels, degradation of oxidized proteins may be inefficient [19]. Indeed, oxidative modifications induce crosslinking/misfolding that may block the entrance of protein substrates in the proteolytic cavity of the proteasome, as well as potentially inhibiting the overall activity of the proteasome [20]. In addition, oxidatively modified proteins may directly damage the autophagic degradation system and ROS can damage the lysosomal membrane [13]. Some oxidatively modified aggregated species are resistant to degradation by proteases and accumulate within lysosomes. There, the proteins resistant to proteolysis become a potential new source of free radicals, further damaging the lysosomal structure.
Intriguingly, all these processes more or less rely on ATP consumption to occur efficiently. Mitochondrial dysfunction is an early event in the pathogenesis of neurodegenerative diseases. Reduced ATP levels, increased ROS, impaired calcium buffering, and altered mitochondrial permeability are characteristic mitochondrial defects of degenerating neurons [21]. For example, in the case of the proteasome, ATP binding to the 19S proteasome component promotes the association with the 20S particle and opens a gated channel into the 20S particle that allows substrate entry and access to the peptidase sites [22]. Further, the polyubiquitination sequence is ATP dependent since ubiquitin is activated by E1 in an ATP dependent fashion. Also in the case of autophagy, the proton pumping activity of V-ATPase is responsible for acidification of the lysosome/vacuole [23].
Below we discuss evidence that supports the interplay between genetic and endogenous factors related to protein oxidation, protein aggregation and protein dysfunction, coupled with reduced energy metabolism, in development of AD. The chemistry of protein oxidation, redox proteomics approaches and results obtained on human post-mortem brain from DS and AD cases are discussed.
Protein Oxidation: Chemistry and Biology
The oxidation of proteins by ROS within a cell has long been linked with not only the normal aging process [24–26], but also with a multitude of diseases that range from cancer [27, 28] to neurodegenerative diseases such as AD [29–31]. As with other post-translational modifications (PTMs) of proteins, the oxidation or nitration of a peptide chain may alter the three-dimensional conformation of the peptide resulting in a gain or loss of function. This process inevitably affects protein structure and could lead to the alteration in the secondary and tertiary structure resulting for example in dissociation of subunits, unfolding, aggregation and backbone fragmentation [32]. Proteins can be oxidized by direct ROS attack, or by secondary oxidation products such as the reactive aldehyde, formed as final by-products of lipid peroxidation, or by glycoxidation reactions. All amino acid residues can be attacked by ROS, but methionine and cysteine residues are particularly sensitive. In the case of methionine, low levels of ROS lead to formation of methionine sulfoxide (MeSOX) that in turn can be reduced by MeSOX reductases. In addition, oxidation of sulfhydryl groups, often resulting in the formation of intra- or intermolecular disulphides, are reduced back by disulfide reductases/isomerases [33]. Since these two oxidative modifications can be enzymatically repaired in mammalian cells, it is likely they play a key regulatory role and sense the cells to changes in the redox environment. Indeed, a number of signaling pathways, such as JNK, p38 and MAPK kinases, are strongly responsive to redox regulation [9]. However, the interplay between individual redox-sensitive signaling proteins to redox-regulated processes in vivo is quite complex.
While ROS plays a much larger cellular role in both signalling and antimicrobial defense when tightly regulated by functioning ROS-scavenging mechanisms, a loss of such scavenging mechanisms or an unhealthy increase in the cellular oxidants result in damage that takes the form of protein, lipid, and nucleic acid oxidation (Figure 1) [30, 34–38].
Figure 1. ROS-induced protein modification.
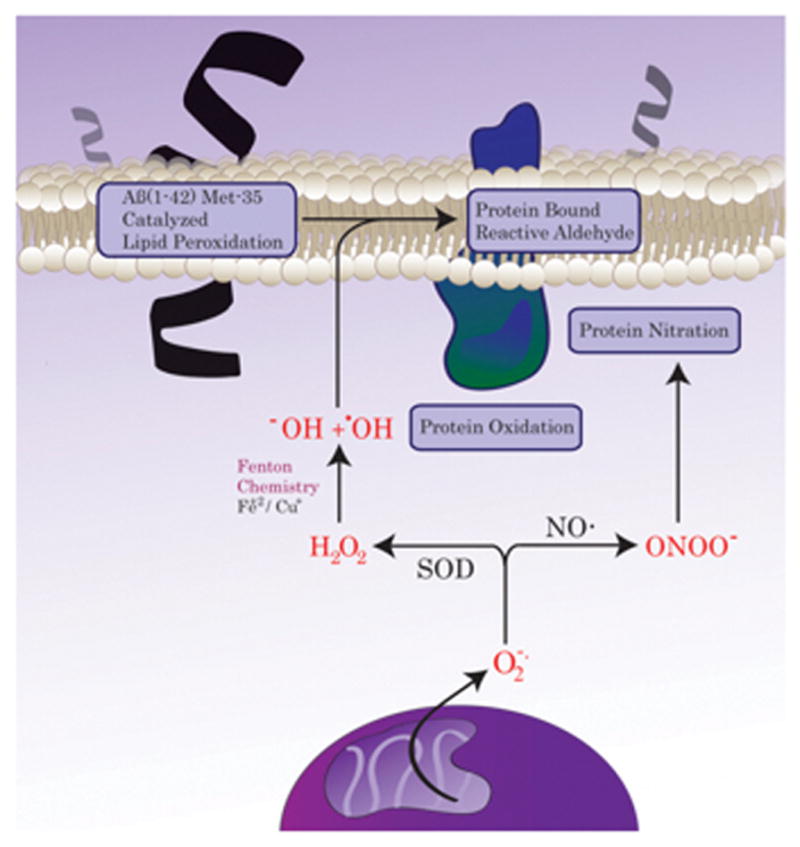
ROS/RNS are highly reactive species that can irreversibly modify proteins. Among different types of protein oxidation, protein nitration, protein carbonylation and protein buond to alkenals are the most commonly assayed. These type of modifications are crucially involved in both aging and neurodegeneration.
When ROS act directly upon a protein, protein carbonylation is a result, often forming reactive aldehydes or ketones by way of primary or a secondary mechanisms [39–41]. Primary protein carbonylation may occur via alpha-carbon hydrogen extraction, oxidation of the peptide backbone resulting in fragmentation, and oxidation of amino acid side chains. Secondary protein carbonylation on the other hand takes place with the addition of reactive aldehydes such as acrolein and 4-hydroxy-nonenal (HNE) that are produced from mechanisms of lipid peroxidation [42]. The α/β-unsaturated reactive aldehydes serve as electrophiles that react with protein side chains such as lysine, histidine, and cysteine via Michael addition. Protein carbonyls however may be more than just cell stressors, as new information points to protein carbonyls as a potential form of redox signaling through a reversible enzyme mediated system [43, 44]. In the laboratory, a common method to determine the level of protein carbonylation in a biological sample is through the use of 2,4-dinitrophenylhydrazine (2,4-DNPH) to form hydrazones which are detectable with anti-2,4-DNPH sensitive antibodies [39]. This method may be used in conjunction with 1-D or 2-D gel western blots as well as the dot/slot blot.
Unlike the aqueous environment of the cytoplasm, the lipid bilayer and its hydrophobic nature exclude many polar oxidants that would otherwise damage bilayer integrity. However, the bilayer is not impervious to oxidative stress as not only can non-polar oxidants such as H2O2 or O2 pass into the bilayer, but oxidants may also be created within the bilayer as is hypothesized in the amyloid β-peptide induced oxidative stress hypothesis in AD [45, 46]. Aβ(1-42) inserts into the bilayer and serve as a catalytic producer of ROS, initiating a process called lipid peroxidation [47, 48]. One method for the initiation of lipid peroxidation, free radical mediated lipid peroxidation, occurs when a carbon centered radical is produced on a polyunsaturated fatty acid (PUFA) by the abstraction of an allylic hydrogen by some form of radical present within the bilayer, for example the sulfuranyl free radical on Met-35 of Aβ(1-42). Oxygen, which lacks a dipole moment, diffuses into the lipid bilayer where it may react with the carbon centered radical to form a lipid peroxyl-radical [49, 50]. The lipid peroxyl-radical may then abstract an allylic hydrogen from an adjacent poly-unsaturated lipid which propagates the chain reaction and forms a lipid hydroperoxide that may then undergo cleavage forming an array of possible reactive aldehydes such as F2-isoprostane, 4-hydroxy-2-nonenal (HNE), and 2-propenal (acrolein). HNE for example is primarily produced from arachidonic acid, an omega-6 PUFA with inflammatory and signaling properties of its own [51]. In neuronal cells under Aβ toxicity it was demonstrated an increase of HNE until the concentrations of 5–10 μM within the lipid bilayer [52]. Other PUFAs that are of importance in the generation of lipid peroxidation products are linoleic acid (LA), docosohexenoic acid (DHA), and cholesterol among others [50]. While reactive aldehydes are a main by-product of lipid peroxidation, other deleterious effects such as loss of lipid asymmetry and apoptosis may follow [53]. Reactive aldehydes deplete the cell of nucleophilic compounds that serve as antioxidants (GSH, lipoic acid, thioredoxin) by covalently modifying proteins via Michael addition in a secondary protein carbonylation reaction [54]. Protein bound and free reactive aldehydes are being investigated as potential markers of oxidative stress and disease progression in conditions ranging from neurodegeneration to brain infarction [55, 56].
Nitric oxide (NO·), a molecular radical and important second messenger produced from L-arginine using one of three nitric oxide synthase (NOS) variants, is vitally important in some biological pathways such as vasodilation, inflammation, and immune response [57–59]. High concentrations of NO·, through secondary reactions with ROS, may go on to produce other reactive nitrogen species (RNS) such as NO2, N2O3, and ONOO−. These compounds react with certain metal cofactors of enzymes, the peptide backbone, and specific amino acids such as tyrosine, cysteine, and tryptophan [60]. 3-Nitrotyrosine (3-NT), a PTM of tyrosine, is a well-recognized marker of nitrosative stress. The formation of 3-NT is the product of the reaction between peroxynitrite (ONOO−) and CO2 which form both the nitrite radical (·NO2) and the carbonate radical (CO3·−) through the intermediates, nitrosoperoxycarbonate and nitrocarbonate [61]. 3-NT formation has recently been linked to systemic autoimmune disorders such as lupus through the generation of endogenous antibodies against native proteins that may be nitrated [61]. Nitration of tyrosine may also result in steric hindrance that blocks a potential phosphorylation at the 4′ para-position, affecting the potential of tyrosine to be phosphorylated resulting in a potential for decreased tyrosine signaling [41]. A decrease in tyrosine signaling may result in the progression of neurodegenerative diseases [57, 62, 63].
Cysteine is another common target of PTM by NO·, forming a reversible S-nitrosylation (R–SNO) product through a proposed interaction of a cysteine anion with NOx, ONOO−, or NO covalently bound to a metal [60]. S-nitrosylation has been found to be an important signaling modification mediated by NO·, while aberrant S-nitrosylation has been implicated in neurodegenerative diseases, among others [64, 65].
Taken together, it is clear that ROS can damage proteins, and eventually accelerate their proteolytic degradation, with two divergent purposes: i) a regulatory mechanism, for example in the degradation of the transcription factor subunit (i.e. HIF-1α and the NF-κB inhibitor IκB), the inhibition of protein tyrosine phosphatases and the regulation of vascular tone; ii) to irreversibly damage proteins leading to loss or gain of function.
Redox Proteomics Approach
In order to gather insights into the state of the proteome affected by diseases or therapies, the use of proteomics is an invaluable tool. The study of the proteomics in a given system is a complex undertaking as the proteome is fluid and dynamic and is impacted by both intrinsic and extrinsic factors that combine to shape the composition of the proteome at any given period in time. Therefore, any insights gained from applications of proteomics must have all factors taken into account that could influence the expression and/or PTMs on the proteins in the system. Such factors could take the form of stressors such as age, oxidative insult, gene activation, drug administration, or disease. In order to control for such factors, the proteome of a treated/disease state should always be compared against an age-matched control. A common method in which to conduct an expression proteomics experiment is by way of the gel-based method. This method utilizes a 2-dimensional approach, in that proteins are separated in the first dimension according to their isoelectric points before separation in the second dimension according to their migration rates through a poly-acrylamide gel. By separating the proteins in two dimensions, each spot in the subsequent gel will likely result in one protein, unlike a separation in one dimension that may result in a band of many proteins. Protein spot density programs such as PDQuest allow for the comparison of the relative concentration of protein within a specific spot which provides insight into the levels/expression of a specific protein within the samples when compared to a control.
Redox proteomics uses similar methodology to that of expression proteomics, however two identical gels are run instead of one. After 2D separation, the first gel is set aside for spot-density analysis while the second gel is transferred to a nitrocellulose membrane via Western blot techniques to undergo immunohistochemical analysis. Once the transfer is complete, the membrane is blocked using bovine serum albumin and probed with a primary antibody to the redox modification of interest, followed by a secondary antibody to recognize the primary antibody. Once the blot is developed, spots may be analyzed for relative changes in spot density which correlates with increased or decreased oxidative modification [12, 66]. The spots on the blot may then be compared to the spots on the gel.
The identification of spot of interest is achieved through spot excision followed by tryptic digestion and tandem mass-spectrometric (MS/MS) identification. Finally, a selection of proteins is designated for confirmation via Western blot to validate the MS/MS results. An example of the redox proteomics experimental workflow is shown in Figure 2.
Figure 2. Redox proteomics.

The workflow of a redox proteomics analysis is showed in four principal steps. 1. isoelectrofocusing point. 2. Second dimension electrophoresis. 3 Protein of interest, selected by 2D image analysis software, are excised from the gel and digested with trypsin. 4. Mass Spectrometry analysis leads to protein identity.
Oxidative Stress: A Common Pathological Feature of Down Syndrome and Alzheimer Neurodegeneration
In recent years, much interest has been devoted to understand the common neuropathological features of DS and AD. The relationship between DS and AD is complex. Studies have shown that, by the age of 40, almost all people with DS have evidence of brain changes characteristic of AD, with deposition of senile plaques, containing amyloid beta-peptide (Aβ), and neurofibrillary tangles (NFTs), composed of hyperphoshorylated tau, and also cholinergic and serotonergic reduction [67–70]. DS is the most frequent chromosomal abnormality that causes intellectual disability, resulting from the presence of an extra complete or segment of chromosome 21 and is termed trisomy 21 (Chr21). The high incidence of AD symptoms and pathology in DS individuals is thought to be caused by the extra copy of chromosome 21, which encodes some of the genes already known to be associated with AD brain abnormalities, including amyloid precursor protein (APP), Cu/Zn superoxide dismutase (SOD1), cystathionine beta synthase (CBS), S100beta, among others.
Accordingly, the “gene-dosage effect” of DS suggests that phenotypic alterations result from the overexpression of a subset of genes and their encoded proteins [71, 72]. Research has also demonstrated that many DS features, including the development of AD, are due to the complex effects of multiple Chr21 genes and their interactions with genes on other chromosomes. By combining these two different hypotheses, it is likely that, in this dysregulated scenario, the effects caused by some dosage sensitive genes are amplified and result in a plethora of different phenotypic traits according to the “number and dose” of genes involved. Results obtained by the analysis of DS cases and the development of a number of mouse models of the disease support both hypotheses. Interestingly, the development of AD occurs much earlier in people with DS, with symptoms beginning to be robust by the fifth decade of life, and an incidence three to five times greater than that of the general population [73].
This complex scenario is an attractive field of research, and the precise molecular pathways by which trisomy 21 leads to the early-onset of AD remain to be elucidated. Several studies supported the view that increased OS conditions may contribute to accelerated senescence and neuropathology in DS individuals [67]. Accordingly, different markers of oxidative damage are elevated in brain tissue from DS (reviewed in [74]). Indeed, DS may be regarded as a chronic OS condition, due to the fact that higher levels of free radicals are caused by a number of trisomic genes, that directly or indirectly, participate to create an imbalance between ROS production and clearance. Among ROS-inducers SOD1, APP, the transcription factor Ets-2, S100B, carbonyl reductase map on Chr21 [74]. SOD1 catalyzes the dismutation of O2•− to O2 and H2O2, the latter in turn neutralized by catalase (CAT) and by glutathione peroxidase (GPX) to water [21]. CAT and GPX are generally expressed at lower levels in brain compared with other tissues [22] and this reduced “buffer” activity may contribute to the inefficient removal of increasing levels of H2O2 in DS. In turn, accumulation of H2O2, in the presence of Fe(II) or Cu(I), leads to hydroxyl radical formation that damage membrane lipids, proteins and nucleic acids, as well, the direct interaction of Fe(II) or Cu(I) with lipid hydroperoxides can also generate OH radical.
However, as also shown by a proteomics study from Gulesserian et al. [75] increased OS in fetal DS brain is not only a consequence of SOD1 overexpression, which alone cannot explain the generalized increase of oxidative damage. To search for other major OS-inducers, another gene triplicated in DS, APP was examined. As expected by a gene-dosage effect, DS individuals with a triplication of APP gene have increased production of Aβ. Both the levels of Aβ(1-42) and Aβ(1-40) are higher in DS plasma than controls [76] and also the ratio of Aβ(1-42)/Aβ(1-40) is lower in DS than in controls. Aβ and tau lesions affect several brain regions in DS, including prefrontal cortex, hippocampus, basal ganglia, thalamus, hypothalamus and midbrain and are believed to underlie the development of cognitive decline and dementia. In DS fetal tissue are found increased APP and elevated Aβ levels that lead to mitochondrial dysfunction [77]. Interestingly, Aβ itself can be oxidized, and plaques in the aged DS brain contain a significant amount of oxidized Aβ [78]. Oxidized Aβ in DS brain is observed within microglia as small aggregates, suggesting reduced clearance from the brain.
Elevated OS markers have been measured in peripheral and CNS samples from DS patients and relevant animal models. Levels of TBARS, total protein carbonyls, and advanced glycation end products (AGEs) are increased in the cortex from DS fetal brain compared with controls. In addition, accumulation of 8-hydroxy-2-deoxyguanosine (8OHdG), oxidized proteins and nitrotyrosine, was observed in the cytoplasm of DS [79]. At the systemic level, the amount of isoprostane 8,12-iso-iPF2α (iPF2α), a specific marker of lipid peroxidation, is elevated in urine samples from adults with DS [80]. In addition, levels of AGEs, dityrosine, H2O2, and nitrite/nitrate are significantly higher in urine samples of DS compared with age-matched controls [81]. Recently, we also found in the amniotic fluid from DS pregnancies that the levels of protein carbonyl and lipid peroxidation were increased, coupled with reduction of GSH and Trx levels, and induction of the heat shock protein (HSP) response [82].
As an essential link to OS, mitochondrial dysfunction is observed as redox imbalances occur, due to the major role of mitochondria in oxygen metabolism, as mentioned above, and this is the case in DS. It is well recognized the mitochondrial dysfunction is a crucial event for neurodegeneration. Mitochondria are known to play a central role in many cell functions including ATP generation, intracellular Ca2+ homeostasis, ROS formation, as by-products of oxidative phosphorylation, and apoptosis. Neurons are particularly dependent on mitochondria because of their high energy demand, and it is likely that neurons are sensitive to mitochondrial dysfunction. The role of mitochondrial dysfunction in AD is well-established [21, 77, 83, 84]. Moreover, altered mitochondrial activity has been reported in DS fibroblasts [85] and mitochondrial DNA mutations were found in DS brain tissue [86]. In addition, both DS neurons and astrocytes display an abnormal pattern of protein processing consistent with chronic energy deficits [87]. These deficits are further complicated by the fact that a number of proteins that cause neurodegeneration, including Aβ, interact with mitochondria or affect mitochondrial function. Taken together, both DS and AD neurons are vulnerable to: increased ROS levels, decreased mitochondrial function, decreased ATP production and increased Aβ load. All these events may participate in a self-sustaining vicious cycle thus ultimately leaving neurons highly susceptible to death.
Redox proteomics studies performed on brain from DS and AD subjects identified a common framework of oxidized proteins that suggest a number of similarities in the neurodegenerative process. Below, the impact of oxidative damage to members of the proteostasis networks and energy metabolism are discussed in light of their putative role in development of AD and commonalities of oxidatively modified brain proteins between AD and DS with AD.
Oxidative Damage to the Proteostasis Network in DS and AD
When oxidized/misfolded proteins accumulate in sufficient quantity, they are prone to form aggregates [33]. Aggregates of this type include amyloid plaques and neurofibrillary tangles in both DS and AD. The formation of aggregates affects several intracellular pathways that negatively impact metabolism and protein turnover. It is not completely clear why or how toxic protein aggregates occur, but irreversible damage to proteins from OS seems to be involved in physiological aging and neurological degeneration [12]. One interesting aspect of this complex process is that ROS can be generated during early phases of protein self-aggregation [88]. Indeed, the production of hydrogen peroxide from Aβ has been demonstrated by a number of studies [88]. Further, early oligomeric forms of protein aggregates, with their associated redox-active metals, may directly interact with cell membranes, cell-surface receptors or various intracellular target molecules possibly through the generation of ROS [88]. Considering that ROS-damage induces protein aggregation, which in a vicious cycle further leads to ROS release, the ability of the intracellular protein quality control system (PQC) to efficiently remove toxic aggregates is vital to fight against neurodegeneration.
Preserving protein homeostasis, or “proteostasis,” requires several parallel strategies that involve refolding, degradation or clearance of misfolded polypeptides [89]. Any condition leading to increased protein misfolding load, may result in the disturbance of the proteostatis network, a crucial aspect of cell physiology. Thus, it is essential for the cell to mount a rapid and robust response to restore protein homeostasis by both the up-regulation of quality control components, and the removal of misfolded/aggregated proteins.
Central players of the proteostasis system are molecular chaperones that assist misfolded proteins to direct them, if refolding fails, to the protein-degradation system [90]. Environmental stress induces chaperone (heat-shock protein, stress protein) expression, a key protective factor for cell survival to repair cellular damage after stresses. Among this broad family, those initially identified as heat inducible were called heat shock proteins (HSPs). There are also dedicated molecular chaperones that remodel a specific substrate protein or complex.
AD is one of the best-known examples of misfolding-related neurodegenerative diseases and also been named a “protein misfolding disease”, along other brain disorders. Accumulation of chaperones may be one response of the affected neuron to eliminate Aβ and tau [91, 92].
Intriguingly, redox proteomics studies performed on human brain from DS and AD subjects demonstrated that a number of oxidized proteins are in common between the two pathologies, namely: GRP78, UCH-L1, HSC71 and GFAP. Among the components of the proteostasis network GRP78, UCH-L1, cathepsin D, V0-ATPase and GFAP were increasingly carbonylated in the frontal cortex of DS individuals at about 20 years of age compared with their age-matched controls [93]. These initial findings suggest the hypothesis that younger cases with DS may already show disturbance of the proteostasis network possibly linked to increased OS, many years before appearance of AD neuropathology.
We next extended our redox proteomics investigation to include older DS cases with AD pathology, and we identified 4-hydroxynonenal (HNE)-modified proteins. We analyzed post-mortem frontal cortex from four different groups: DS individuals with and without AD pathology compared with their age-matched controls [94]. The comparison entails both age- and genotype dependent variables, discriminating age-dependent effects with those intrinsic to DS. Specifically, we found that GRP78, GFAP, UCH-L1 and HSC71 are targets of HNE-modification in DS brain.
Chaperones/UPS
Members of the Grp family, mainly GRP78, participate in protective mechanisms activated by cells to adapt to stress of the endoplasmic reticulum (ER). Under conditions in which misfolded proteins accumulate within the lumen of the ER, the organelle enters into a state called “ER stress” and activates a series of complex coordinated signalling pathways, collectively called the unfolded protein response (UPR) [95, 96]. The increased carbonylation and HNE modification of GRP78 in DS with AD brain suggests that alteration of protein structure may possibly reduce the ability of GRP78 to bind to misfolded proteins with consequent accumulation of misfolded protein and risk of cognitive decline [93, 94]. Another member of HSP chaperone family, HSC71, is oxidatively modified by HNE binding [94]. HSC71 is involved in the degradation of proteins with abnormal conformation by binding to a particular peptide region and labelling it for proteasome-mediated proteolysis [97].
Upon activation of ER-stress response, UPS mediates ubiquitination and degradation of misfolded proteins, which occur in the cytoplasm [98]. We showed that UCH-L1 is a target of oxidative damage in DS, DS/AD, MCI and AD brains, and that its oxidative modification likely leads to a decreased function as measured by an activity assay [99, 100]. One of the major consequences of aberrant UCH-L1 activity is altered proteasome activity, which leads to accumulation of damaged proteins and consequently formation of protein aggregates [99, 101–104]. To confirm this hypothesis, the trypsin-like, chymotrypsin-like and caspase-like proteasome activities were measured and each activity was reduced in DS brain compared with controls [93].
Taken together, the redox proteomics results from our laboratory predict that in degenerating neurons: proteins with excess ubiquitinylation accumulate; the activity of the 26S proteasome is decreased; and consequent accumulation of aggregated/damaged proteins is favoured. All of these characteristics are observed in brains of subjects with both AD and DS with AD.
Autophagy
Emerging evidence highlights the role of autophagy in aging and neurodegeneration and several therapeutic strategies aimed at restoring autophagy have been tested in different models [105]. Among mediators of the autophagic cascade, we identified the V0-ATPase pump and cathepsin D as oxidatively modified, together with decreased autophagosome formation, in younger DS brain before the neuropathology of AD is evident. These modifications appear to be restricted to DS brain. However, GFAP is oxidatively modified in DS, DS/AD and AD brain [12, 100]. GFAP, in addition to be recognized as a marker of astrocytic activation, is a newly recognized regulator of the autophagy machinery and important regulator for CMA [106]. GFAP is proposed to interact at the lysosomal membrane either with the lysosome-associated membrane protein type 2A (LAMP-2A), an important component of the translocation complex, or with the elongation factor 1a (ef1a) [106]. Since CMA is one of the cellular mechanisms activated to resist OS required for targeting oxidized proteins to lysosomes, oxidation of GFAP might contribute to disruption of autophagic flux [107]. This result is also consistent with in vitro studies showing the carbonylation of GFAP in synaptosomes treated with Aβ (1–42) [108, 109].
Overall, the above results confirm a close connection between imbalance between increased protein oxidation and reduced ability to remove oxidized/misfolded proteins (Figure 3). A key player that seems to disrupt this fine-tuned equilibrium is OS that is not only a challenge to neuronal cells with increasing amounts of ROS and ROS-damaged by-products, but also contributes to a general failure of defense system through oxidative modifications, i.e., reduced activity, of selected members of the proteostasis network. This proposed scenario requires further elucidation in that some of the above-mentioned activities require ATP to occur efficiently.
Figure 3. Energy metabolism failure.

Increased protein oxidation of energy metabolic enzymes. Specifically, the oxidation of glycolytic enzymes, highlighted in blue, and TCA enzymes lead to reduced activity which culminates in reduced glucose metabolism and decreased synthesis of ATP.
Energy Metabolic Dysfunction in DS and AD Brain
Glucose is the principal source of energy for the brain, which utilizes 20% of glucose metabolism and consumes more than 30% of the inspired oxygen although the brain accounts for only 2% of the total body weight. Glucose metabolism is essential for healthy brain function and even a small interruption of glucose metabolism causes brain dysfunction and memory loss [12]. Emerging evidence supports the notion that AD is tightly linked to metabolic disorders in which brain glucose utilization and energy production are impaired. Both obesity and type II diabetes significantly increase the risks of cognitive decline and development of AD, consistent with the notion that impaired brain glucose metabolism plays a significant role in disease pathogenesis [110–112]. APP and Aβ cause decreased activity in mitochondrial respiratory chain complexes, decreased activity of several mitochondrial enzymes and also to induce ROS production [79, 113–116]. In addition, a number of studies on AD human specimens and/or animal and cell culture models suggested that increased levels of OS are able to impair key players of the glucose metabolic pathway [45, 117–119]. This metabolic and oxidative compromise may render neurons susceptible to excitotoxicity and apoptosis, and also induce hypothermia, causing abnormal tau phosphorylation through differential inhibition of kinases and phosphatases [120]. Reduced glucose metabolism might also affect autophagy and protein degradation pathways, as already discussed above in both AD and DS, which respond to alterations of cell energy metabolism [121]. Moreover, dysfunction of mitochondria has been reported to alter APP metabolism, increasing the intraneuronal accumulation of Aβ-peptide and enhancing the neuronal vulnerability [77, 84, 122].
Redox proteomics studies on AD brain demonstrated the oxidation of α-enolase, malate dehydrogenase (MDH), fructose bisphosphate aldolase A/C (FBA A/C), ATP synthase, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Similarly, pyruvate kinase (PK), MDH, α-enolase, FBA C, TPI were found increasingly oxidized in amnestic mild cognitive impairment (MCI) brain [102, 123–127], indicating that impaired glucose metabolism is an early event in the progression of AD. The oxidative modification of energy-related proteins correlates with reduced cerebral metabolic rate of glucose in brain of MCI and AD patients in FDG-PET studies suggesting that oxidative damage of protein involved in glycolysis, the Krebs cycle and ATP synthesis might be a crucial event in the reduction of glucose metabolism [121, 128]. Among the proteins found oxidatively modified in AD and MCI subjects, PK, α-enolase, MDH, GAPDH and FBA A/C also were identified by redox proteomics to be carbonylated or HNE-bound also in DS, and DS with AD brain, suggesting the common occurrence of reduced glucose utilization, mitochondrial deficit and increased OS in the development of AD-like dementia [94, 100].
Interestingly, the reported oxidation of MDH, GAPDH, and α-enolase is consistent with chronic dysfunction of energy metabolism that starts early in the DS population and increases in the presence of AD pathology in DS [94]. In agreement, redox proteomics performed in DS Tg mice [129] identified lipid-peroxidation-derived modification of a number of proteins belonging to energy metabolism pathways, such as ATP synthase mitochondrial F1 complex b subunit, α-enolase, and TPI, thus confirming the involvement of impaired energy consumption in DS. All the above-mentioned proteins rely, to varying extents, on ATP utilization and are closely related to energy metabolic pathways, and their oxidation eventually culminate in reduced ATP production, likely contributing to the loss of synapses and synaptic function at nerve terminals. Consistent with this notion, a recent study showed a significantly decreased level of synaptophysin in DS with AD brain [130]. Thus, we propose that these changes contribute to neurodegenerative processes and cognitive decline [12, 67, 74]. Specifically, the common proteins found oxidized in AD and DS brain are: FBA, GAPDH, α-enolase, PK, MDH.
FBA cleaves fructose 1,6-bisphosphate and produces the two glycolytic intermediates, glyceraldehyde 3-phospate (G3P) and dihydroxyacetone phosphate (DHAP). Fructose 1,6-bisphosphate is neuroprotective and preserves GSH in cortical neurons during OS conditions [131]. FBA catalyzes a critical step, as it generates two substrates that are used to eventually produce 2 molecules of ATP and more in Krebs cycle and electron transport chain (ETC).
GAPDH is known in glycolysis for the conversion of glyceraldehyde 3-phosphate to 1,3 bisphosphoglycerate and, consequently, in energy production. In addition, inhibition of GADPH can lead to accumulation of trioses, with subsequent nonenzymatic conversion to methyl glyoxal (MG), a highly reactive alpha-ketoaldehyde that readily oxidizes proteins, lipids, and other cellular components, leading to further cytotoxicity [132]. Further, GAPDH is known to facilitate APP and tau function normally [133], so oxidized GAPDH likely loses this ability and conceivably contributes to the hallmark neuropathology of AD and of DS with AD.
α-enolase catalyzes the penultimate step of glycolysis by converting 2-phosphoglycerate to phosphoenolpyruvate. Although the main function of enolase is its role in glycolysis, this enzyme also plays a role in plasminogen regulation (which leads to plasmin, a protease that cleaves Aβ) and activation of the MEK/extracellular regulated kinases (ERK) pro-survival pathways [134]. Therefore, loss of activity of enolase activity secondary to oxidative modification can contribute to Aβ-mediated neurotoxicity and decreased pro-survival mechanisms.
PK catalyzes the final step in glycolysis, the conversion of phosphoenolpyruvate to pyruvate with the concomitant transfer of the high-energy phosphate group from phosphoenolpyruate to ADP, thereby generating ATP. Under aerobic conditions, pyruvate can be transported to the mitochondria, where it enters the Krebs cycle and is further metabolized to produce considerably more ATP through oxidative phosphorylation [135].
MDH catalyzes the reversible oxidation of malate to oxaloacetate by NAD+ the Krebs cycle. MDH links glycolysis to the ETC by transferring NADH to NADH dehydrogenase (Complex I) through the malate-aspartate shuttle resulting in the production of ATP [12].
Protein modification of these glycolytic and Krebs cycle enzymes may disrupt neuronal energy metabolism and ion homeostasis, thereby impairing ion-motive ATPases, signal transduction, membrane asymmetry [53], and glucose and glutamate transporters [136], supporting the hypothesis of altered energy metabolism as a common theme in AD neurodegeneration.
Data from DS and early AD brain suggest that oxidative stress and relative oxidative damage of metabolic enzymes occurs before the onset of symptoms in AD and robust Aβ plaque formation [82, 113, 119]. Thus, is reasonable to suggest that reduced glucose utilization (Figure 4) and the consequent mitochondrial deficits are early OS-induced events strongly contributing to the neurodegenerative process that culminate in AD pathology and might represent one of the primary OS-related links between DS and AD pathology.
Figure 4. Reduced protein degradation.
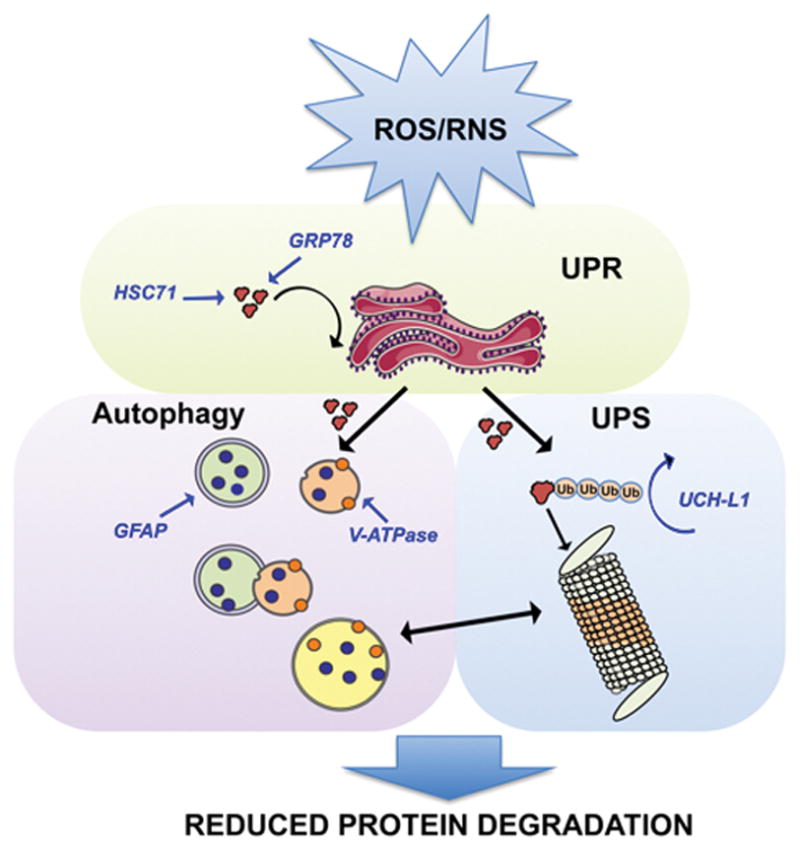
Impairment of protein degradation machinery. Redox proteomics studies identified oxidatively modified proteins in DS and AD brain that are members of the protein degradative system. Specifically, UCHL1 is involved in the proteasome pathway; GRP78 and HSC71 in unfolded protein response (UPR) and GFAP in autophagy. Dysfunction of all these three processes contributes significantly to accumulation of oxidized/misfolded proteins
Conclusions and future directions
The role of OS in neurodegeneration is well recognized, but in the case of DS and AD neuropathology, genetic similarities, due to the fact that some of the genes responsible for familial form of AD are encoded by Chr21, provide the basis to better understand specific dysregulated pathways. Results obtained by the analysis of human specimens and studies from mouse and cellular models of the disease evince a molecular link between protein oxidation/aggregation, the integrity of PQC system (proteasome, UPS and autophagy), dysfunction of energy metabolism and neurodegeneration. Many common pathological hallmarks exist between DS and AD including deposition of amyloid plaques, NFTs, increased oxidative damage, and impaired mitochondrial function, among others. Intriguingly, we propose that all these processes seem to be joined by a “leitmotif” – oxidative stress - since they are all the cause and/or the consequence of increased free radical burden. If low amount of ROS can activate the protective cellular apparatus such as the antioxidant and heat shock responses, cell cycle regulation, DNA repair, UPR and autophagy, then chronic exposure to ROS causes irreversible damage to all intracellular macromolecules. Among these, protein oxidation impairs multiple cellular functions by a largely irreversible process that results in altered, mostly reduced, protein activity. It is likely that stressed neurons have the challenge of increasing loads of oxidatively damaged proteins, which overwhelm the ability of the proteostasis network. This, in turn, promotes further accumulation of damaged proteins, increasingly prone to aggregation, ultimately resulting in neuronal death. Alteration of protein homeostasis coupled with increasing demand for protein degradation, and reduced ATP production may produce a vicious cycle that may accelerate the neurodegenerative process (Figure 5). Dementia associated with AD in DS occurs at an earlier age than that of sporadic AD, yet there are common oxidative alterations in the proteostasis network and glucose metabolism between these two conditions, implying that these processes are intimately associated with dementia and potentially targets for therapeutic intervention. In addition, other than proteostasis and glucose metabolism, redox proteomics studies allowed the identification of oxidized proteins belonging to several dysfunctional pathways among, which detoxification systems, excitotoxicity or synapse function, that highly correlates with DS and AD pathological features supporting the role of protein oxidative damage in neuron degeneration and cognitive decline. This scenario suggests that any pharmacological intervention may be more effective if it engages more than one molecular pathway, and drugs that target not only specific mechanisms but also the interplay among them might be beneficial. Current therapies are moving towards the use of formulations containing compounds able to modify several oxidative aspects of the disease. Therefore, a complete knowledge on the pathways affected by oxidative damage occurring during neurodegeneration is needed.
Figure 5. Protein oxidation overlap in DS and AD neuropathology.
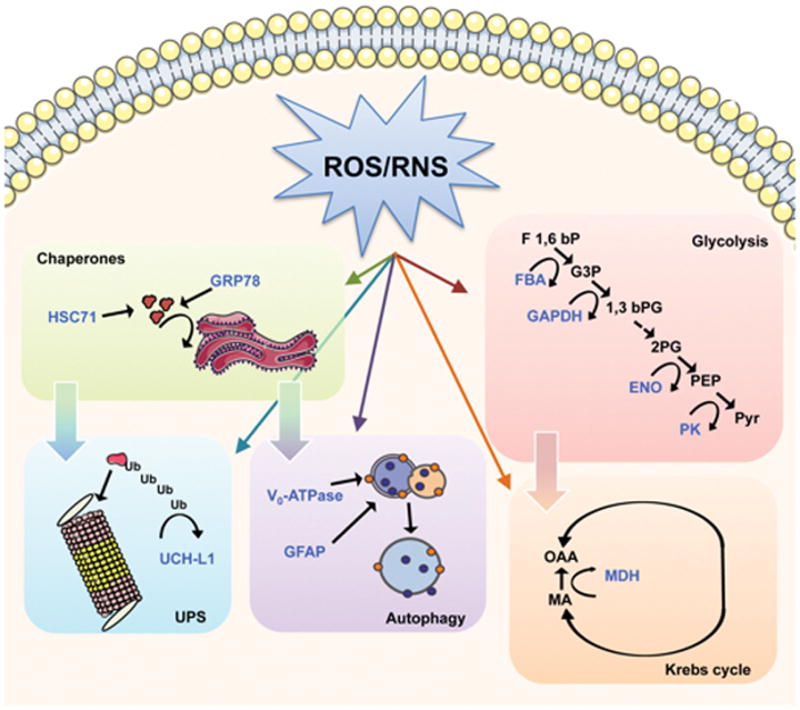
Identification of oxidized proteins in AD and DS brain suggest common dysregulated processes with oxidative stress being a “leitmotif.” These findings support a molecular link among protein oxidation/aggregation, the integrity of protein quality control (UPS and autophagy), dysfunction of energy metabolism and neurodegeneration.
Redox proteomics [137] undoubtedly played a crucial role in the detection of proteins oxidatively-modified by PCO, 3-NT and-or HNE-modified during neurodegeneration allowing to decipher the disease-specific oxidative footprint. The continuous technical advances in the field will allow in the next future to better understand the role of oxidative stress and redox signaling in biological processes related to both physiological or pathological conditions.
Acknowledgments
We thank past and present graduate students, postdoctoral scholars, visiting scientists in and collaborators with the Butterfield laboratory. This work was supported in part by a grant from NIH to DAB [AG-05119]. Research reported in this manuscript was supported by Eunice Kennedy Shriver National Institute of Child Health and Development of the National Institutes of Health under award number R01HD064993 to EH. Autopsy tissue was obtained from the UCI-ADRC (P50AG16573), from the UK ADC (P30AG28383) and from the NICHD Brain and Tissue Bank for Developmental Disorders of the University of Maryland, Baltimore, MD, contract HHSN275200900011C (N01HD90011). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Lastly, we thank Ms. Mollie Fraim for secretarial assistance.
References
- 1.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halliwell B. Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med. 1991;91:14S–22S. doi: 10.1016/0002-9343(91)90279-7. [DOI] [PubMed] [Google Scholar]
- 3.Hansford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 4.Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- 5.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 7.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 8.Sies H. Strategies of antioxidant defense. Eur J Biochem. 1993;215:213–219. doi: 10.1111/j.1432-1033.1993.tb18025.x. [DOI] [PubMed] [Google Scholar]
- 9.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 10.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 11.Perluigi M, Swomley AM, Butterfield DA. Redox proteomics and the dynamic molecular landscape of the aging brain. Ageing Res Rev. 2013 doi: 10.1016/j.arr.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Butterfield DA, Perluigi M, Reed T, Muharib T, Hughes CP, Robinson RA, Sultana R. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal. 2012;17:1610–1655. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malkus KA, Tsika E, Ischiropoulos H. Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson’s disease: how neurons are lost in the Bermuda triangle. Mol Neurodegener. 2009;4:24. doi: 10.1186/1750-1326-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grune T, Merker K, Jung T, Sitte N, Davies KJ. Protein oxidation and degradation during postmitotic senescence. Free Radic Biol Med. 2005;39:1208–1215. doi: 10.1016/j.freeradbiomed.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Metcalf DJ, Garcia-Arencibia M, Hochfeld WE, Rubinsztein DC. Autophagy and misfolded proteins in neurodegeneration. Exp Neurol. 2012;238:22–28. doi: 10.1016/j.expneurol.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Current Opinion in Cell Biology. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 18.Keller JN, Dimayuga E, Chen Q, Thorpe J, Gee J, Ding Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol. 2004;36:2376–2391. doi: 10.1016/j.biocel.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Grune T, Reinheckel T, Joshi M, Davies KJ. Proteolysis in cultured liver epithelial cells during oxidative stress. Role of the multicatalytic proteinase complex, proteasome. J Biol Chem. 1995;270:2344–2351. doi: 10.1074/jbc.270.5.2344. [DOI] [PubMed] [Google Scholar]
- 20.Friguet B, Szweda LI. Inhibition of the multicatalytic proteinase (proteasome) by 4-hydroxy-2-nonenal cross-linked protein. FEBS Lett. 1997;405:21–25. doi: 10.1016/s0014-5793(97)00148-8. [DOI] [PubMed] [Google Scholar]
- 21.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 22.Liu CW, Li X, Thompson D, Wooding K, Chang TL, Tang Z, Yu H, Thomas PJ, DeMartino GN. ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol Cell. 2006;24:39–50. doi: 10.1016/j.molcel.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mijaljica D, Prescott M, Devenish RJ. V-ATPase engagement in autophagic processes. Autophagy. 2011;7:666–668. doi: 10.4161/auto.7.6.15812. [DOI] [PubMed] [Google Scholar]
- 24.Hohn A, Konig J, Grune T. Protein oxidation in aging and the removal of oxidized proteins. J Proteomics. 2013 doi: 10.1016/j.jprot.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal S, Sohal RS. Relationship between susceptibility to protein oxidation, aging, and maximum life span potential of different species. Exp Gerontol. 1996;31:365–372. doi: 10.1016/0531-5565(95)02039-x. [DOI] [PubMed] [Google Scholar]
- 26.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 27.Yang HY, Chay KO, Kwon J, Kwon SO, Park YK, Lee TH. Comparative proteomic analysis of cysteine oxidation in colorectal cancer patients. Mol Cells. 2013;35:533–542. doi: 10.1007/s10059-013-0058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Y, Zhang L, Rong S, Qu H, Zhang Y, Chang D, Pan H, Wang W. Relation between gastric cancer and protein oxidation, DNA damage, and lipid peroxidation. Oxid Med Cell Longev. 2013;2013:543760. doi: 10.1155/2013/543760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed TT, Pierce WM, Markesbery WR, Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009;1274:66–76. doi: 10.1016/j.brainres.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer’s disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 31.Di Domenico F, Coccia R, Butterfield DA, Perluigi M. Circulating biomarkers of protein oxidation for Alzheimer disease: expectations within limits. Biochim Biophys Acta. 2011;1814:1785–1795. doi: 10.1016/j.bbapap.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Dean RT, Fu S, Stocker R, Davies MJ. Biochemistry and pathology of radical-mediated protein oxidation. Biochem J. 1997;324(Pt 1):1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunlop RA, Brunk UT, Rodgers KJ. Oxidized proteins: mechanisms of removal and consequences of accumulation. IUBMB Life. 2009;61:522–527. doi: 10.1002/iub.189. [DOI] [PubMed] [Google Scholar]
- 34.Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006;397:170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 35.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiology of Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 36.Weidner AM, Bradley MA, Beckett TL, Niedowicz DM, Dowling AL, Matveev SV, LeVine H, 3rd, Lovell MA, Murphy MP. RNA oxidation adducts 8-OHG and 8-OHA change with Abeta42 levels in late-stage Alzheimer’s disease. PLoS One. 2011;6:e24930. doi: 10.1371/journal.pone.0024930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markesbery WR, Lovell MA. DNA oxidation in Alzheimer’s disease. Antioxid Redox Signal. 2006;8:2039–2045. doi: 10.1089/ars.2006.8.2039. [DOI] [PubMed] [Google Scholar]
- 38.Perluigi M, Di Domenico F, Giorgi A, Schinina ME, Coccia R, Cini C, Bellia F, Cambria MT, Cornelius C, Butterfield DA, Calabrese V. Redox proteomics in aging rat brain: involvement of mitochondrial reduced glutathione status and mitochondrial protein oxidation in the aging process. J Neurosci Res. 2010;88:3498–3507. doi: 10.1002/jnr.22500. [DOI] [PubMed] [Google Scholar]
- 39.Cabiscol E, Tamarit J, Ros J. Protein carbonylation: proteomics, specificity and relevance to aging. Mass Spectrom Rev. 2014;33:21–48. doi: 10.1002/mas.21375. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki YJ, Carini M, Butterfield DA. Protein carbonylation. Antioxid Redox Signal. 2010;12:323–325. doi: 10.1089/ars.2009.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butterfiled DA, Stadtman ER. Protein oxidation processes in Aging Brain. Advances in Cell Aging Gerontology. 1997;2:161–191. [Google Scholar]
- 42.Stadtman ER, Levine RL. Protein oxidation. Reactive Oxygen Species: From Radiation to Molecular Biology. 2000;899:191–208. [Google Scholar]
- 43.Wong CM, Marcocci L, Liu L, Suzuki YJ. Cell signaling by protein carbonylation and decarbonylation. Antioxid Redox Signal. 2010;12:393–404. doi: 10.1089/ars.2009.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong CM, Bansal G, Marcocci L, Suzuki YJ. Proposed role of primary protein carbonylation in cell signaling. Redox Rep. 2012;17:90–94. doi: 10.1179/1351000212Y.0000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 46.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 47.Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang J, Sultana R, Bredesen DE. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med. 2010;48:136–144. doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butterfield DA, Boyd-Kimball D. The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta. 2005;1703:149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 49.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiology of Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 50.Shichiri M. The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr. 2014;54:151–160. doi: 10.3164/jcbn.14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim Biophys Acta. 2010;1801:924–929. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 53.Lange MLB, Cenini G, Piroddi M, Abdul HM, Sultana R, Galli F, Memo M, Butterfield DA. Loss of phospholipid asymmetry and elevated brain apoptotic protein levels in subjects with amnestic mild cognitive impairment and Alzheimer disease. Neurobiology of Disease. 2008;29:456–464. doi: 10.1016/j.nbd.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang X, Wu X, Choi YE, Kern JC, Kehrer JP. Effect of acrolein and glutathione depleting agents on thioredoxin. Toxicology. 2004;204:209–218. doi: 10.1016/j.tox.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 55.Igarashi K, Kashiwagi K. Protein-conjugated acrolein as a biochemical marker of brain infarction. Mol Nutr Food Res. 2011;55:1332–1341. doi: 10.1002/mnfr.201100068. [DOI] [PubMed] [Google Scholar]
- 56.Lee WC, Wong HY, Chai YY, Shi CW, Amino N, Kikuchi S, Huang SH. Lipid peroxidation dysregulation in ischemic stroke: plasma 4-HNE as a potential biomarker? Biochem Biophys Res Commun. 2012;425:842–847. doi: 10.1016/j.bbrc.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Calabrese V, Cornelius C, Rizzarelli E, Owen JB, Dinkova-Kostova AT, Butterfield DA. Nitric oxide in cell survival: a janus molecule. Antioxid Redox Signal. 2009;11:2717–2739. doi: 10.1089/ars.2009.2721. [DOI] [PubMed] [Google Scholar]
- 58.Nelson EJ, Connolly J, McArthur P. Nitric oxide and S-nitrosylation: excitotoxic and cell signaling mechanism. Biol Cell. 2003;95:3–8. doi: 10.1016/s0248-4900(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 59.Shahani N, Sawa A. Nitric oxide signaling and nitrosative stress in neurons: role for S-nitrosylation. Antioxid Redox Signal. 2011;14:1493–1504. doi: 10.1089/ars.2010.3580. [DOI] [PubMed] [Google Scholar]
- 60.Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 61.Ahsan H. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum Immunol. 2013;74:1392–1399. doi: 10.1016/j.humimm.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 62.Barone E, Di Domenico F, Cenini G, Sultana R, Cini C, Preziosi P, Perluigi M, Mancuso C, Butterfield DA. Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim Biophys Acta. 2011;1812:480–487. doi: 10.1016/j.bbadis.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alvarez B, Radi R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids. 2003;25:295–311. doi: 10.1007/s00726-003-0018-8. [DOI] [PubMed] [Google Scholar]
- 64.Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Broniowska KA, Hogg N. The chemical biology of S-nitrosothiols. Antioxid Redox Signal. 2012;17:969–980. doi: 10.1089/ars.2012.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sultana R, Butterfield DA. Oxidative Modification of Brain Proteins in Alzheimer’s Disease: Perspective on Future Studies Based on Results of Redox Proteomics Studies. Journal of Alzheimers Disease. 2013;33:S243–S251. doi: 10.3233/JAD-2012-129018. [DOI] [PubMed] [Google Scholar]
- 67.Perluigi M, Di Domenico F, Buttterfield DA. Unraveling the complexity of neurodegeneration in brains of subjects with Down syndrome: insights from proteomics. Proteomics Clin Appl. 2014;8:73–85. doi: 10.1002/prca.201300066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mann DM, Brown A, Wilks DP, Davies CA. Immunocytochemical and lectin histochemical studies of plaques and tangles in Down’s syndrome patients at different ages. Prog Clin Biol Res. 1989;317:849–856. [PubMed] [Google Scholar]
- 69.Wisniewski HM, Rabe A. Discrepancy between Alzheimer-type neuropathology and dementia in persons with Down’s syndrome. Ann N Y Acad Sci. 1986;477:247–260. doi: 10.1111/j.1749-6632.1986.tb40344.x. [DOI] [PubMed] [Google Scholar]
- 70.Lott IT. Neurological phenotypes for Down syndrome across the life span. Prog Brain Res. 2012;197:101–121. doi: 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rueda N, Florez J, Martinez-Cue C. Mouse models of Down syndrome as a tool to unravel the causes of mental disabilities. Neural Plast. 2012;2012:584071. doi: 10.1155/2012/584071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gardiner K. Gene-dosage effects in Down syndrome and trisomic mouse models. Genome Biol. 2004;5:244. doi: 10.1186/gb-2004-5-10-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schupf N, Sergievsky GH. Genetic and host factors for dementia in Down’s syndrome. Br J Psychiatry. 2002;180:405–410. doi: 10.1192/bjp.180.5.405. [DOI] [PubMed] [Google Scholar]
- 74.Perluigi M, Butterfield DA. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr Gerontol Geriatr Res. 2012;2012:724904. doi: 10.1155/2012/724904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gulesserian T, Engidawork E, Fountoulakis M, Lubec G. Antioxidant proteins in fetal brain: superoxide dismutase-1 (SOD-1) protein is not overexpressed in fetal Down syndrome. J Neural Transm Suppl. 2001:71–84. doi: 10.1007/978-3-7091-6262-0_6. [DOI] [PubMed] [Google Scholar]
- 76.Mehta PD, Capone G, Jewell A, Freedland RL. Increased amyloid beta protein levels in children and adolescents with Down syndrome. J Neurol Sci. 2007;254:22–27. doi: 10.1016/j.jns.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 77.Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron. 2002;33:677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- 78.Head E, Garzon-Rodriguez W, Johnson JK, Lott IT, Cotman CW, Glabe C. Oxidation of Abeta and plaque biogenesis in Alzheimer’s disease and Down syndrome. Neurobiol Dis. 2001;8:792–806. doi: 10.1006/nbdi.2001.0431. [DOI] [PubMed] [Google Scholar]
- 79.Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S, Smith MA. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol. 2000;59:1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- 80.Tolun AA, Scarbrough PM, Zhang H, McKillop JA, Wang F, Kishnani PS, Millington DS, Young SP, Il’yasova D. Systemic oxidative stress, as measured by urinary allantoin and F(2)-isoprostanes, is not increased in Down syndrome. Ann Epidemiol. 2012;22:892–894. doi: 10.1016/j.annepidem.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Campos C, Guzman R, Lopez-Fernandez E, Casado A. Evaluation of urinary biomarkers of oxidative/nitrosative stress in adolescents and adults with Down syndrome. Biochim Biophys Acta. 2011;1812:760–768. doi: 10.1016/j.bbadis.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 82.Perluigi M, di Domenico F, Fiorini A, Cocciolo A, Giorgi A, Foppoli C, Butterfield DA, Giorlandino M, Giorlandino C, Schinina ME, Coccia R. Oxidative stress occurs early in Down syndrome pregnancy: A redox proteomics analysis of amniotic fluid. Proteomics Clin Appl. 2011;5:167–178. doi: 10.1002/prca.201000121. [DOI] [PubMed] [Google Scholar]
- 83.Coskun PE, Wyrembak J, Derbereva O, Melkonian G, Doran E, Lott IT, Head E, Cotman CW, Wallace DC. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. J Alzheimers Dis. 2010;20(Suppl 2):S293–310. doi: 10.3233/JAD-2010-100351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lott IT, Head E, Doran E, Busciglio J. Beta-amyloid, oxidative stress and down syndrome. Curr Alzheimer Res. 2006;3:521–528. doi: 10.2174/156720506779025305. [DOI] [PubMed] [Google Scholar]
- 85.Valenti D, Manente GA, Moro L, Marra E, Vacca RA. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: involvement of the cAMP/PKA signalling pathway. Biochem J. 2011;435:679–688. doi: 10.1042/BJ20101908. [DOI] [PubMed] [Google Scholar]
- 86.Arbuzova S, Hutchin T, Cuckle H. Mitochondrial dysfunction and Down’s syndrome. Bioessays. 2002;24:681–684. doi: 10.1002/bies.10138. [DOI] [PubMed] [Google Scholar]
- 87.Helguera P, Seiglie J, Rodriguez J, Hanna M, Helguera G, Busciglio J. Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab. 2013;17:132–140. doi: 10.1016/j.cmet.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tabner BJ, El-Agnaf OM, Turnbull S, German MJ, Paleologou KE, Hayashi Y, Cooper LJ, Fullwood NJ, Allsop D. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem. 2005;280:35789–35792. doi: 10.1074/jbc.C500238200. [DOI] [PubMed] [Google Scholar]
- 89.Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3:a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morawe T, Hiebel C, Kern A, Behl C. Protein homeostasis, aging and Alzheimer’s disease. Mol Neurobiol. 2012;46:41–54. doi: 10.1007/s12035-012-8246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blair LJ, Zhang B, Dickey CA. Potential synergy between tau aggregation inhibitors and tau chaperone modulators. Alzheimers Res Ther. 2013;5:41. doi: 10.1186/alzrt207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van der Putten H, Lotz GP. Opportunities and challenges for molecular chaperone modulation to treat protein-conformational brain diseases. Neurotherapeutics. 2013;10:416–428. doi: 10.1007/s13311-013-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, Cini C, Perluigi M. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochimica Et Biophysica Acta-Molecular Basis of Disease. 2013;1832:1249–1259. doi: 10.1016/j.bbadis.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Di Domenico F, Pupo G, Tramutola A, Giorgi A, Schinina ME, Coccia R, Head E, Butterfield DA, Perluigi M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: clues for understanding the development of Alzheimer disease. Free Radic Biol Med. 2014;71:270–280. doi: 10.1016/j.freeradbiomed.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108:2777–2793. doi: 10.1002/bit.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Doyle KM, Kennedy D, Gorman AM, Gupta S, Healy SJ, Samali A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J Cell Mol Med. 2011;15:2025–2039. doi: 10.1111/j.1582-4934.2011.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kouchi Z, Sorimachi H, Suzuki K, Ishiura S. Proteasome inhibitors induce the association of Alzheimer’s amyloid precursor protein with Hsc73. Biochem Biophys Res Commun. 1999;254:804–810. doi: 10.1006/bbrc.1998.9977. [DOI] [PubMed] [Google Scholar]
- 98.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 100.Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, Cini C, Perluigi M. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain. Biochim Biophys Acta. 2013;1832:1249–1259. doi: 10.1016/j.bbadis.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J Biol Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 102.Butterfield DA, Gnjec A, Poon HF, Castegna A, Pierce WM, Klein JB, Martins RN. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer’s disease: an initial assessment. J Alzheimers Dis. 2006;10:391–397. doi: 10.3233/jad-2006-10407. [DOI] [PubMed] [Google Scholar]
- 103.Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007;1148:243–248. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hegde AN. The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010;17:314–327. doi: 10.1101/lm.1504010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hochfeld WE, Lee S, Rubinsztein DC. Therapeutic induction of autophagy to modulate neurodegenerative disease progression. Acta Pharmacol Sin. 2013;34:600–604. doi: 10.1038/aps.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell. 2010;39:535–547. doi: 10.1016/j.molcel.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boyd-Kimball D, Castegna A, Sultana R, Poon HF, Petroze R, Lynn BC, Klein JB, Butterfield DA. Proteomic identification of proteins oxidized by Abeta(1-42) in synaptosomes: implications for Alzheimer’s disease. Brain Res. 2005;1044:206–215. doi: 10.1016/j.brainres.2005.02.086. [DOI] [PubMed] [Google Scholar]
- 109.Joshi G, Sultana R, Perluigi M, Butterfield DA. In vivo protection of synaptosomes from oxidative stress mediated by Fe2+/H2O2 or 2,2-azobis-(2-amidinopropane) dihydrochloride by the glutathione mimetic tricyclodecan-9-yl-xanthogenate. Free Radical Biology and Medicine. 2005;38:1023–1031. doi: 10.1016/j.freeradbiomed.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 110.de la Monte SM. Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimer’s Disease. Current Alzheimer Research. 2012;9:35–66. doi: 10.2174/156720512799015037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.de la Monte SM. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs. 2012;72:49–66. doi: 10.2165/11597760-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease - is this type 3 diabetes? Journal of Alzheimers Disease. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 113.Swomley AM, Forster S, Keeney JT, Triplett J, Zhang Z, Sultana R, Butterfield DA. Abeta, oxidative stress in Alzheimer disease: Evidence based on proteomics studies. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbadis.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer’s disease brain: new insights from redox proteomics. Eur J Pharmacol. 2006;545:39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 115.Butterfield DA, Swomley AM, Sultana R. Amyloid beta-peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal. 2013;19:823–835. doi: 10.1089/ars.2012.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.de la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochemical Pharmacology. 2014;88:548–559. doi: 10.1016/j.bcp.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Butterfield DA. Oxidative stress in neurodegenerative disorders. Antioxid Redox Signal. 2006;8:1971–1973. doi: 10.1089/ars.2006.8.1971. [DOI] [PubMed] [Google Scholar]
- 118.Pratico D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci. 2008;29:609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 119.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 120.Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: Implications for Alzheimer’s disease. Journal of Neuroscience. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen Z, Zhong C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013;108:21–43. doi: 10.1016/j.pneurobio.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 122.Coskun PE, Busciglio J. Oxidative Stress and Mitochondrial Dysfunction in Down’s Syndrome: Relevance to Aging and Dementia. Curr Gerontol Geriatr Res. 2012;2012:383170. doi: 10.1155/2012/383170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Butterfield DA, Dalle-Donne I. Redox proteomics: from protein modifications to cellular dysfunction and disease. Mass Spectrom Rev. 2014;33:1–6. doi: 10.1002/mas.21404. [DOI] [PubMed] [Google Scholar]
- 124.Aluise CD, Robinson RA, Cai J, Pierce WM, Markesbery WR, Butterfield DA. Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease: insights into memory loss in MCI. J Alzheimers Dis. 2011;23:257–269. doi: 10.3233/JAD-2010-101083. [DOI] [PubMed] [Google Scholar]
- 125.Butterfield DA, Sultana R. Redox proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: insights into the progression of this dementing disorder. J Alzheimers Dis. 2007;12:61–72. doi: 10.3233/jad-2007-12107. [DOI] [PubMed] [Google Scholar]
- 126.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 127.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 128.Hoyer S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur J Pharmacol. 2004;490:115–125. doi: 10.1016/j.ejphar.2004.02.049. [DOI] [PubMed] [Google Scholar]
- 129.Ishihara K, Amano K, Takaki E, Ebrahim AS, Shimohata A, Shibazaki N, Inoue I, Takaki M, Ueda Y, Sago H, Epstein CJ, Yamakawa K. Increased lipid peroxidation in Down’s syndrome mouse models. J Neurochem. 2009;110:1965–1976. doi: 10.1111/j.1471-4159.2009.06294.x. [DOI] [PubMed] [Google Scholar]
- 130.Martin SB, Dowling AL, Lianekhammy J, Lott IT, Doran E, Murphy MP, Beckett TL, Schmitt FA, Head E. Synaptophysin and Synaptojanin-1 in Down Syndrome are Differentially Affected by Alzheimer’s Disease. J Alzheimers Dis. 2014 doi: 10.3233/JAD-140795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vexler ZS, Wong A, Francisco C, Manabat C, Christen S, Tauber M, Ferriero DM, Gregory G. Fructose-1,6-bisphosphate preserves intracellular glutathione and protects cortical neurons against oxidative stress. Brain Res. 2003;960:90–98. doi: 10.1016/s0006-8993(02)03777-0. [DOI] [PubMed] [Google Scholar]
- 132.Golej J, Hoeger H, Radner W, Unfried G, Lubec G. Oral administration of methylglyoxal leads to kidney collagen accumulation in the mouse. Life Sci. 1998;63:801–807. doi: 10.1016/s0024-3205(98)00336-1. [DOI] [PubMed] [Google Scholar]
- 133.Butterfield DA, Hardas SS, Lange ML. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: many pathways to neurodegeneration. J Alzheimers Dis. 2010;20:369–393. doi: 10.3233/JAD-2010-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Butterfield DA, Lange ML. Multifunctional roles of enolase in Alzheimer’s disease brain: beyond altered glucose metabolism. J Neurochem. 2009;111:915–933. doi: 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-Hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiology of Disease. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 136.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of A beta 1-42. Journal of Neurochemistry. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 137.Butterfield DA, Gu L, Domenico FD, Robinson RA. Mass spectrometry and redox proteomics: Applications in disease. Mass Spectrom Rev. 2014;33:277–301. doi: 10.1002/mas.21374. [DOI] [PubMed] [Google Scholar]