

Abstract
Progressive accumulation of extracellular potassium ions can trigger propagating waves of spreading depression (SD), which are associated with dramatic increases in extracellular potassium levels ([K+]o) and arrest in neural activity. In the central nervous system the restricted nature of the extracellular compartment creates an environment that is vulnerable to disturbances in ionic homeostasis. Here we investigate how changes in the size of the extracellular space induced by alterations in extracellular osmolarity affect locust SD. We found that hypotonic exposure increased susceptibility to experimentally induced SD evidenced by a decrease in the latency to onset and period between individual events. Hypertonic exposure was observed to delay the onset of SD or prevent the occurrence altogether. Additionally, the magnitude of extracellular K+ concentration ([K+]o) disturbance during individual SD events was significantly greater and they were observed to propagate more quickly under hypotonic conditions compared with hypertonic conditions. Our results are consistent with a conclusion that hypotonic exposure reduced the size of the extracellular compartment by causing cell swelling and thus facilitated the accumulation of K+ ions. Lastly, we found that pharmacologically reducing the accumulation of extracellular K+ using the K+ channel blocker tetraethylammonium slowed the rate of SD propagation while increasing [K+]o through inhibition of the Na-K-2Cl cotransporter increased propagation rates. Overall our findings indicate that treatments or conditions that act to reduce the accumulation of extracellular K+ help to protect against the development of SD and attenuate the spread of ionic disturbance adding to the evidence that diffusion of K+ is a leading event during locust SD.
Keywords: cell swelling, hypertonic, hypotonic, spreading depression, potassium
spreading depression (SD) is characterized by a rapid and massive depolarization of cells and an associated silencing of electrical activity that slowly propagates throughout neural tissue (Leão 1944; Pietrobon and Moskowitz 2014; Somjen 2001). SD was first discovered in the cortex of the rabbit over 70 yr ago and since has been shown to occur in both vertebrate and invertebrate nervous systems (Leão 1944; Rodgers et al. 2007; Rounds 1967). It is an ionic disturbance characterized by a failure to maintain transmembrane ionic gradients that are essential for appropriate neuronal signaling (Somjen 2001). For instance, during waves of SD there is an abrupt increase in extracellular potassium concentration ([K+]o) and a drop in extracellular sodium concentration ([Na+]o) (Rodgers et al. 2007; Somjen 2001). Under normal or healthy central nervous system (CNS) conditions, restoration of ion homeostasis and recovery of neural function are usually observed within minutes following the eruption of a SD episode (Leão 1944; Pietrobon and Moskowitz 2014; Somjen 2001). However, during periods of metabolic compromise, such as hypoxia or mitochondrial dysfunction, the disturbance worsens and can have severe consequences such as prolonged time to cell membrane repolarization or even neuron cell death (Dreier 2011; Pietrobon and Moskowitz 2014; Somjen 2001).
SD occurring in the neocortex of mammals [cortical spreading depression (CSD)] is associated with human pathologies such as migraine with aura, stroke, and traumatic brain injury and thus has been given much attention in the mammalian literature (Pietrobon and Moskowitz 2014; Somjen 2001). More recently, it was discovered that SD events occur within the CNS of Locusta migratoria and characterization of such events has demonstrated that invertebrate SD shares many similarities with CSD (Rodgers et al. 2007, 2010). Notably, the degree of ionic disruption and the propagation rate of the events are strikingly similar in both locusts and mammals. Furthermore, many conditions that initiate the development of CSD also induce SD in the locust nervous system suggesting that the cellular mechanisms underlying vertebrate and invertebrate SD are conserved (Rodgers et al. 2007, 2010). SD in the locust CNS is associated with stress-induced neural shutdown. For instance, in response to severe metabolic stress such as anoxia, hyperthermia, hypothermia, and ATP depletion, locusts enter a coma during which SD-like events can be monitored within the metathoracic ganglion (MTG) (Rodgers et al. 2007, 2010). Following acute and brief exposures to such types of stress, recovery of neural and muscular systems is observed and is dependent on the restoration of appropriate ion levels (Rodgers et al. 2007, 2010).
Despite considerable research effort, the neural underpinnings of SD are still incompletely understood. It is thought that the onset of SD is triggered by treatments or conditions that elevate [K+]o above a threshold level triggering a positive feedback system that further promotes K+ accumulation (Armstrong et al. 2009; Rodgers et al. 2009). Thus waves of SD can be experimentally induced by directly manipulating [K+]o levels. For instance, single SD events can be induced by injecting high KCl saline directly into the locust MTG (Rodgers et al. 2007; Spong et al. 2014). More interesting, repetitive waves of SD are initiated by exposing the locust CNS to ouabain, a Na+/K+-ATPase inhibitor, and thus limiting mechanisms that normally clear K+ ions from the extracellular space (Rodgers et al. 2007, 2009). Recovery relies on the restoration of normal ion levels and thus mechanisms of K+ homeostasis are thought to play a significant role during SD. Indeed, ouabain-induced SD in the locust CNS is exacerbated by disrupting mechanisms of K+ homeostasis (Spong et al. 2014; Spong and Robertson 2013). At the core, to ensure continued generation of electrical activity there must be an appropriate balance between mechanisms of K+ accumulation and mechanisms of K+ clearance within the relatively small environment of the extracellular space (Armstrong et al. 2009; Rodgers et al. 2009).
Due to the restricted nature of the extracellular compartment it is not only essential to ensure that mechanisms of ionic homeostasis are functioning optimally but the maintenance of cell volume is also important to consider. For instance, cellular swelling will cause a reduction in the size of the extracellular space enhancing the accumulation of extracellular ions due to the reduced volume for dilution (Schwartzkroin et al. 1998). During CSD the extracellular space shrinks as a consequence of neuronal swelling (Zhou et al. 2010). Furthermore, previous work using rat brain tissue demonstrated that acute cell swelling caused by exposure to hypo-osmotic solution increased hypoxia-related neuronal damage (Payne et al. 1996), highlighting the importance of controlling cell volume. Although less is known about cell swelling during locust SD, it is likely that there are important regulatory processes in place to control cell volume as changes in the size of the extracellular space could have profound effects on the severity and recovery from these events.
The main purpose of this paper was to investigate how alterations in extracellular osmolarity and the associated changes in cell volume affect locust SD. We experimentally induced SD within the locust MTG under hypotonic, isotonic, and hypertonic conditions. SD was most easily elicited and propagated more quickly under hypotonic conditions. Hypertonic exposure was found to decrease the susceptibility to SD and slow the rate of propagation. Our results are consistent with hypotonic conditions reducing the extracellular compartment by inducing cell swelling and hypertonic exposure increasing the size of the extracellular space by causing cell shrinkage. Furthermore, we show that pharmacologically manipulating [K+]o dynamics modulates the propagation of SD events in such a way as to suggest that treatments aimed at reducing the accumulation of extracellular K+ could act as a preventive measure against the spread of ionic disturbance.
MATERIALS AND METHODS
Animals.
Locusta migratoria were raised in a crowded colony located in the Animal Care Facility of the Bioscience Complex at Queen's University. The locusts were reared under a 12:12-h light-dark cycle. Room temperature was maintained at 25 ± 1°C. Animals were fed once daily and raised on a diet consisting of wheat grass, carrot slices, or tomato pieces and an ad libitum mixture of one part skim milk powder, one part torula yeast, and 13 parts bran by volume. All experiments were performed on adult male locusts aged 3–5 wk past the final molt. Before experimentation locusts were randomly chosen from the colony and held in ventilated plastic containers within the laboratory.
Semi-intact preparation.
Locusts were pinned dorsal side up onto a corkboard following a dorsal midline incision and removal of the legs, wings, and pronotum. The thoracic and abdominal cavities were continuously exposed to hypotonic, isotonic, or hypertonic saline solutions. Hypotonic saline contained the following (in mM): 147 NaCl, 10 KCl, 4 CaCl, 3 NaOH, and 10 HEPES buffer (pH 7.2; all chemicals were obtained from Sigma-Aldrich). Isotonic and hypertonic solutions were made by adding 110 or 220 mM mannitol, respectively, to the hypotonic saline and thus all solutions contained identical ion concentrations. The gut, airsacs, and fat bodies were cleared to expose the MTG, and the second spina situated between the connectives was removed. A metal plate was placed beneath the MTG and nerves 2–5 on both sides of the ganglion were severed to ensure saline and drug entry. A sliver wire was inserted into the anterior region of the thorax to ground the preparation.
Measuring extracellular K+ concentration.
K+-sensitive microelectrodes made from 1-mm diameter unfilamented glass capillary tubes (World Precision Instruments) were used to record the [K+]o from within the MTG. Capillary tubes were cleaned with methanol (99.9%), dried on a hotplate, and subsequently pulled to form tips of low resistance (5–7 MΩ). The microelectrodes were then silanized for 1 h on a hot plate (100°C) by exposure to dichlorodimethylsilane (99%; Sigma-Aldrich) vapor. Following silanization the microelectrode tips were filled with Potassium Ionophore I-Cocktail B (5% Valinomycin; Sigma-Aldrich) and back-filled with 500 mM KCl. K+-sensitive microelectrodes were stored in the dark within a glass beaker, and their tips were suspended in distilled water. Reference electrodes were prepared before each experiment from 1-mm diameter filamented glass capillary tubes (World Precision Instruments) pulled to a resistance of ∼5–7 MΩ and then back filled with 3 M KCl. The K+-sensitive and reference microelectrode pair were connected to a DUO773 two-channel intracellular/extracellular amplifier (World Precision Instruments) and calibrated using 15 mM KCl + 135 mM NaCl and 150 mM KCl solutions to obtain the voltage difference from a 10-fold change in K+ concentration. The microelectrodes were then inserted through the sheath of the MTG adjacent to one another, and K+ voltage was continuously monitored throughout the experiment. Voltage recordings were converted to [K+]o (mM) using the Nernst equation (for details, see Rodgers et al. 2007).
Recording direct current potential.
Direct current (DC) potential was monitored from two locations within the MTG using fillamented microelectrodes pulled to form low resistance tips (∼5–7 MΩ). Microelectrodes were prepared using 1-mm diameter filamented glass capillary tubes and were filled with 3 M potassium acetate (KAC) before experimentation. The two microelectrodes connected to individual amplifiers (a DUO773 two-channel intracellular/extracellular amplifier, World Precision Instruments; and a Neuroprobe amplifier, A-M Systems) were inserted through the sheath of the ganglion separated as far apart as possible (0.5–1 mm). Once inserted into the MTG the individual voltage readings were allowed to steady for ∼2 min and then were zeroed before the start of each experiment. Abrupt negative shifts in the extracellular DC potential are indicative of spreading depression events (Somjen 2001). Such drops in DC potential reflect the rapid depolarization and inward current flow that occurs during episodes of SD.
Induction of SD.
Repetitive SD was induced by bath application of either 5 × 10−4 or 10−3 M ouabain. Ouabain was dissolved in hypotonic, isotonic, or hypertonic saline and administered to semi-intact preparations using glass pipettes following a 20-min baseline recording in the corresponding saline type. Single SD events were induced by pressure injecting small volumes (∼40 nl) of 200 mM KCl directly into the MTG using a PicoSpritzer III (INTRACEL). In these experiments a within animal protocol was used where each preparation was subjected to two individual KCl-induced SD events separated by a treatment condition.
Pharmacology.
We tested the effects of tetraethylammonium (TEA; 10−3 M), a well-known blocker of voltage-dependent K+ channels, on the propagation of individual SD events. Additionally, bumetanide (10−3 M) was used to test how inhibition of the Na-K-2Cl cotransporter affected KCl-induced SD. Pharmacological agents were dissolved in hypotonic saline and bath applied to semi-intact preparations for a treatment period of 20 min. Agents were administered upon recovery from the first KCl-induced event, and the second event was induced immediately following the 20-min treatment period. Hypotonic saline was used in these experiments as SD was most easily elicited under these conditions.
Analyses of SD characteristics.
The time to the first event (min) was measured as the time from ouabain application to the downward inflection point (when monitoring DC potential) or the upward inflection point (when measuring [K+]o) of the first SD event. The period between the first and second event (min) was measured from the peak of the first [K+]o surge to the peak of the second event. The amplitude of [K+]o surges (mM) was measured by taking the difference between the [K+]o level just before the upward inflection point of the surge and the peak [K+]o (highest level of [K+]o observed). The upward and downward slopes of the first abrupt [K+]o surge were calculated at the time point of highest change to determine the rate of [K+]o increase (mM/s) and decrease (mM/s). Baseline [K+]o values (mM) represent nonsurge [K+]o levels that were taken before ouabain application and every 5 min after. The closest nonsurge value was taken if time points coincided with a [K+]o surge. The time it took for KCl-induced events to travel between recording electrodes (delay time) was obtained to examine characteristics of SD propagation. Delay time (min) was calculated using time measurements taken at half the maximum amplitude of the downward shifts in DC potential. Normalized change in delay time represents the delay time of the second KCl-induced SD event normalized relative to the first event.
Statistical analyses.
SigmaPlot 12.5 (Systat Software) was used to test data for normality and equal variance and to perform the appropriate statistical tests. To determine significant differences between two groups t-tests were used for parametric data and Mann-Whitney rank sum tests were used for nonparametric data (P < 0.05). For comparison of more than two groups a one-way ANOVA or ANOVA on ranks was used and post hoc comparisons were performed using either Holm-Sidak Multiple comparisons or the Dunn's method (P < 0.05). All data were plotted using SigmaPlot 12.5 (Systat Software). Error bars on histograms represent means ± SE. Box plots represent the 25th and 75th percentiles with a line indicating medians and whiskers extending to the 10th and 90th percentiles (outliers are shown as individual points). Parametric data are reported as means ± SE. The median (Mdn) and interquartile range (IQR) are reported for nonparametric data.
RESULTS
Susceptibility to ouabain-induced SD is affected by extracellular osmolarity.
Bath application of 5 × 10−4 M ouabain induced repetitive SD events monitored as abrupt negative shifts in DC potential (Fig. 1A). DC potential was recorded at two different locations simultaneously from within the MTG to ensure the events were propagating throughout the neuropile. SD was reliably induced in 11/11 (100%), 7/11 (∼64%), and 3/11 (∼27%) preparations treated with hypotonic, isotonic, and hypertonic saline, respectively (Fig. 1B). There was a significantly greater number of individual events observed within the 40-min treatment period under the hypotonic condition (Mdn = 4.0, IQR = 3.0, 5.2) compared with the isotonic (Mdn = 1.0, IQR = 0, 2.0) and hypertonic (Mdn = 0, IQR = 0, 1.0) saline conditions (Fig. 1C). There was a significantly shorter latency to onset of the first ouabain-induced event under hypotonic conditions (Mdn = 15.0 min, IQR = 10.9, 17.1) compared with under isotonic and hypertonic conditions (ISO: Mdn = 25.0 min, IQR = 15.6, 33.4; HYPER: Mdn = 29.4 min, IQR = 29.0, 31.1; Fig. 1D).
Fig. 1.

Ouabain-induced spreading depression (SD) in hypotonic, isotonic, and hypertonic saline conditions. A: representative traces of direct current (DC) potential recorded from 2 areas within the metathoracic ganglion during continuous bath application of 5 × 10−4 M ouabain (OUA). Arrow denotes the time at which OUA was administered. B: percentage of preparations that exhibited spreading depression following OUA application. C: number of individual SD events exhibited within the 40-min treatment period was significantly greater under hypotonic (HYPO) saline conditions compared with isotonic (ISO) and hypertonic (HYPER) saline conditions (Holm-Sidak method, P < 0.001). D: latency to onset of the 1st OUA-induced event was significantly shorter in hypotonic saline compared with isotonic and hypertonic conditions (Holm-Sidak method, P = 0.007). C and D: data are plotted as the median and upper and lower quartiles. *Significant differences. B–D: sample sizes (n) are indicated in brackets above plots.
Effects of hypotonic and hypertonic exposure on extracellular potassium dynamics during ouabain-induced SD.
The [K+]o within the MTG was recorded during treatment with either hypotonic or hypertonic saline and repetitive SD was induced by continuous bath application of 10−3M ouabain (Fig. 2, A and B). A higher dose of ouabain was used in these experiments (10−3 M compared with 5 × 10−4 M) to increase the chances of reliably inducing SD under hypertonic conditions. Under hypotonic conditions ouabain reliably induced repetitive SD in 100% of preparations. Exposure to ouabain under hypertonic conditions was found to elicit at least one SD event in 9 out of 11 preparations (∼82%). The number of individual [K+]o surges (SD events) induced by 40-min bath application of ouabain was significantly greater under hypotonic conditions (Mdn = 6.0, IQR = 4.0, 8.0) compared with hypertonic conditions (Mdn = 1.0, IQR = 1.0, 3.0; Fig. 2C). Furthermore, hypotonic exposure significantly reduced the time to the first ouabain-induced SD event (HYPO: Mdn = 7.9 min, IQR = 5.1, 13.6; HYPER: Mdn = 23.0 min, IQR = 10.9, 37.3; Fig. 2D) and the period between the first and second event (HYPO: Mdn = 5.2 min, IQR = 4.0, 7.2; HYPER: Mdn = 14.2 min, IQR = 9.5, 17.9; Fig. 2E) compared with hypertonic exposure. The amplitude of the first [K+]o event was significantly greater under hypotonic conditions (Mdn = 30.9 mM, IQR = 26.4, 39.8) compared with hypertonic conditions (Mdn = 16.0 mM, IQR = 10.4, 22.3; Fig. 2F) and was associated with faster rates of [K+]o increase (HYPO: Mdn = 1.6 mM/s, IQR = 0.9, 2.6; HYPER: Mdn = 1.0 mM/s, IQR = 0.5, 1.3; Fig. 2G). Hypotonic conditions were also associated with slightly higher rates of [K+]o clearance (HYPO: Mdn = 0.5 mM/s, IQR = 0.4, 0.8; HYPER: Mdn = 0.3 mM/s, IQR = 0.2, 0.6; Mann-Whitney rank sum test, P = 0.048; data not shown). A similar rate and degree of increase in baseline [K+]o measurements were observed over the 40-min treatment period under both hypotonic and hypertonic conditions (Fig. 2H).
Fig. 2.

OUA-induced SD and extracellular K+ concentration ([K+]o) characteristics in hypotonic and hypertonic saline conditions. A and B: representative traces of the [K+]o recorded from within the metathoracic ganglion during continuous bath application of 10−3M OUA under either hypotonic or hypertonic conditions (A and B, respectively). Arrows denote the time point at which OUA was administered. Note that the vertical lines in the recording in B are electrical artifacts of unknown origin. C: preparations treated with hypotonic saline experienced a significantly greater number of individual surges within the 40-min treatment period compared with those treated with hypertonic saline (Mann-Whitney rank sum test, P < 0.001). D: latency to onset of the 1st OUA- induced surge was significantly shorter in hypotonic saline compared with hypertonic saline (Mann-Whitney rank sum test, P = 0.012). E: period between the 1st and 2nd OUA-induced surge was significantly shorter in hypotonic saline compared with hypertonic saline (Mann-Whitney rank sum test, P = 0.009). F: amplitude of the 1st OUA-induced [K+]o surge was significantly larger in hypotonic saline compared with hypertonic saline (Mann-Whitney rank sum test, P = 0.005). G: rate of [K+]o increase during the 1st OUA-induced surge was significantly greater in the hypotonic saline condition compared with the hypertonic saline condition (Mann-Whitney rank sum test, P = 0.04). C–G: data are plotted as the median and upper and lower quartiles. *Significant differences. Sample sizes (n) are indicated in brackets above plots. H: baseline [K+]o levels (mM) during continuous bath application of OUA in hypotonic saline (white circles) or hypertonic saline (black circles). Baseline [K+]o levels were measured before OUA application (time = 0) and every 5 min after. There is a progressive increase in baseline [K+]o over the 40-min treatment period under both saline conditions. Data are plotted as means ± SE.
Effects of hypotonic and hypertonic exposure on SD propagation.
Manipulating the [K+]o within the MTG by injecting small volumes of high KCl directly into the ganglion induces single SD events (Rodgers et al. 2007), and the propagation of such events can be conveniently monitored by recording the DC potential shifts at different distances away from the injection site (Spong et al. 2014). We performed a series of within animal experiments where each preparation was subjected to two individual KCl-induced events and the delay between recording electrodes was calculated for each SD episode (Fig. 3A). The representative recordings demonstrate that the electrode closest to the injection site responds to the KCl injections first and is followed by the more distanced electrode. Preparations in the first group of experiments performed were continuously exposed to hypotonic saline and no treatment was applied. Under hypotonic conditions the delay time during the second SD event (SD2/2: Mdn = 0.2 min, IQR = 0.04, 0.2) was significantly shorter compared with the delay time during the first event (SD1/2: Mdn = 0.6 min, IQR = 0.2, 0.7; Fig. 3B). To investigate whether this was due to a preconditioning effect, a third group of preparations were tested who were only subjected to one SD event that was induced following the treatment period (SD1/1: Mdn = 0.3 min, IQR = 0.2, 0.4); however, the delay time was not significantly different from either of the two within animal measurements (Fig. 3B).
Fig. 3.

Propagation of KCl-induced SD. A: representative traces of DC potential (Elect. A and Elect. B) recorded simultaneously from different locations within the metathoracic ganglion (MTG). Preparations were subjected to 2 separate SD events triggered by ∼40 nl injections of 200 mM KCl into the neuropile of the MTG (timing of injections are indicated by the arrows on the KCl injection line). DC potential was recorded at different distances away from the injection site. The delay between Elect. A and Elect. Ai: time it takes for the KCl-induced SD event to propagate between recording electrodes as it travels from the point of origin (injection site). Aii: DC potential shifts of the 1st SD event shown in Ai to emphasize the time delay between recording electrodes. B: delay times recorded in hypotonic saline. The delay time between recording electrodes was significantly shorter during the 2nd SD event (SD2/2) compared with the 1st SD event (SD1/2) (Dunn's method, P < 0.05) whereas the mean delay time of preparations subjected to only one SD event administered at the same time point as SD2 (SD1/1) was not statistically different compared with the within animal measurements (Dunn's method, P > 0.05). C: hypertonic saline significantly increased the delay time during the 2nd SD event relative to the 1st SD event compared with hypotonic saline (Mann-Whitney rank sum test, P = 0.038). D: treatment with TEA (10−3M) significantly increased the delay time during the 2nd SD event relative to the 1st SD event compared with control conditions (Mann-Whitney rank sum test, P = 0.03). E: treatment with bumetanide significantly decreased the delay time during the 2nd SD event relative to the 1st SD event compared with controls (Mann-Whitney rank sum test, P = 0.008). *Significant differences for B–E. Sample sizes (n) are indicated in brackets above plots. F: representative recording of a bumetanide-treated preparation. Downward arrow denotes the time at which bumetanide (10−3M) was administered.
We then tested whether the reduction in delay time observed under hypotonic conditions could be manipulated by increasing the extracellular osmolarity. Preparations were either exposed to hypotonic saline for the entire experiment (HYPO/HYPO) or were subjected to the first SD event under hypotonic conditions, but then following a 10-min recovery were exposed to hypertonic saline for 20 min before inducing the second SD event (HYPO/HYPER). The normalized change in delay time was found to be significantly greater in the HYPO/HYPER condition (Mdn = 0.7, IQR = 0.3, 1.6) compared with the HYPO/HYPO condition (Mdn = 0.3, IQR = 0.1, 0.5; Fig. 3C) suggesting that treatment with hypertonic saline slowed rates of SD propagation compared with treatment with hypotonic saline.
Pharmacological manipulation of SD propagation.
Using the same protocol as described above (Fig. 3A) we tested how a 20-min treatment period with 10−3 M TEA affected the propagation of KCl-induced events. TEA treatment significantly increased the normalized change in delay time compared with control preparations (TEA: Mdn = 0.6, IQR = 0.3, 1.7; control: Mdn = 0.2, IQR = 0.1, 0.3; Fig. 3D) suggesting that TEA slowed the rate of SD propagation. Contrary to this we found that 20-min treatment with 10−3 M bumetanide, a known inhibitor of the Na-K-2Cl cotransporter, significantly reduced the normalized change in delay time compared with control preparations (bumetanide: Mdn = 0.04, IQR = 0.02, 0.1; control: Mdn = 0.3, IQR = 0.3, 0.5; Fig. 3E) indicating an increased rate of SD propagation. Additionally, bumetanide treatment consistently induced negative deflections of small amplitude in DC potential following the second SD event (Fig. 3F), which were absent in control preparations.
Bumetanide and [K+]o dynamics.
To test the effects of bumetanide on [K+]o dynamics we continuously monitored the [K+]o within the MTG while preparations were subjected to two individual KCl-induced events using the same protocol described above. Representative recordings show the individual surges in [K+]o induced by the KCl injections under control conditions (Fig. 4A) and in preparations treated with bumetanide (Fig. 4B). The second KCl injection was observed to cause more than one [K+]o surge in three of eight preparations treated with bumetanide. To determine how baseline [K+]o levels were affected we measured the overall change in [K+]o by taking the difference in [K+]o levels before the first SD event and 10 min postrecovery from the second event. The overall change in baseline [K+]o levels was found to be significantly greater for preparations treated with bumetanide (Mdn = 6.1 mM, IQR = 1.2, 7.3) compared with control preparations (Mdn = 0.2 mM, IQR = −1.0, 2.3; Fig. 4C). We also measured the change in [K+]o that occurred during the 20-min treatment period with bumetanide and found that there was also a significant increase in [K+]o during this time period compared with the change that occurred at the same time points in control preparations (butmetanide: Mdn = 2.7 mM, IQR = 1.5, 4.0; Control: Mdn = 0.6 mM, IQR = 0.5, 1.0; Fig. 4D). Bumetanide not only had an effect on baseline [K+]o levels but also affected the characteristics of the individual [K+]o surges. For instance, surge amplitude increased from 22.5 ± 3.1 mM, before bumetanide treatment, to 49.8 ± 7.2 mM following bumetanide exposure (Fig. 4E). An increase in surge amplitude was also evident in control experiments; however, the difference was not statistically significant (1st [K +]o surge: 28.5 ± 4.3 mM; 2nd [K+]o surge: 42.5 ± 6.6 mM; Fig. 4E).
Fig. 4.

The effects of bumetanide on [K+]o characteristics during KCl-induced SD. A and B: representative traces of the [K+]o recorded from within the MTG. KCl was injected at the 1 min and 31 min mark. B: downward arrow denotes the beginning of the 20 min pretreatment period with bumetanide (10−3M). C. Preparations treated with bumetanide (10−3M) exhibited a significantly greater overall change in [K+]o (mM) levels compared with controls (t-test, P = 0.009). [K+]o was measured 10 min following the 2nd KCl injection and compared with initial levels (before the 1st KCl injection). D: there was a significant increase in [K+]o during the 20-min treatment period with bumetanide (Mann-Whitney rank sum test, P = 0.005). [K+]o was measured at the beginning and end of the pretreatment period or at the same time points in the control condition. E: mean amplitude of [K+]o surges. In bumetanide-treated preparations, the mean amplitude of the 2nd surge was significantly larger than the mean amplitude of the 1st surge (Holm-Sidak method, P = 0.006). *Significant differences for C–E.
DISCUSSION
In the present paper we show that altering [K+]o dynamics within the locust MTG affects SD occurrence and propagation velocity (for an overview, refer to Fig. 5). For instance, results from both our osmolarity experiments and pharmacological manipulations demonstrate that treatments facilitating the accumulation of extracellular K+ ions predispose towards the generation of SD episodes and increase rates of SD propagation. On the other hand, reducing the rate of K+ accumulation suppresses SD onset and attenuates propagation. These results help substantiate previous models describing mechanisms underlying invertebrate SD (Armstrong et al. 2009; Rodgers et al. 2010).
Fig. 5.
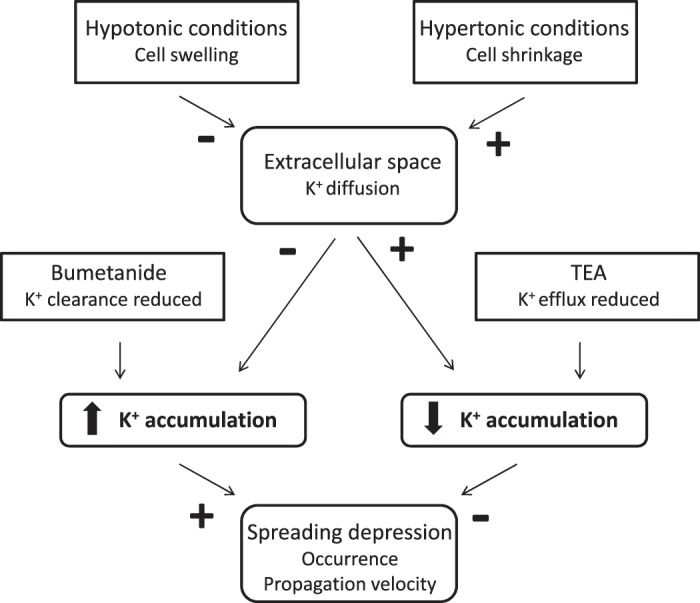
A model describing alterations in [K+]o dynamics in response to osmolarity and pharmacological manipulations and the effects they have on SD occurrence and propagation in the locust MTG. Treatments that facilitate the accumulation of extracellular K+ ions promote SD occurrence and increase propagation velocity while treatments that limit K+ accumulation suppress SD onset and slow the spread of ionic disturbance. Cell swelling under hypotonic conditions reduces the extracellular volume limiting K+ diffusion and thus promotes K+ accumulation. Hypertonic conditions increase the volume of the extracellular compartment providing more space for K+ diffusion limiting K+ accumulation. The removal of extracellular K+ ions is reduced by inhibition of the Na-K-2Cl-cotransporter with bumetanide facilitating K+ accumulation. TEA reduces K+ conductance through voltage-gated ion channels attenuating the rate of K+ accumulation.
The main focus of this paper was to investigate how a disruption in cell volume regulation and the associated changes in the volume of the CNS extracellular compartment affect SD characteristics in the locust CNS. We subjected semi-intact locust preparations to hypotonic conditions to promote cell swelling and to hypertonic conditions to promote cell shrinkage. We did not directly measure cell volume in the present experiments; however, previous imaging studies using hippocampal brain slices have demonstrated that exposure to hypo-osmotic and hyperosmotic solutions are associated with cell swelling and shrinking respectively (Andrew and MacVicar 1994). These volume changes are restricted to the astrocytes because mammalian CNS neurons lack functional aquaporins (Andrew et al. 2007). In addition, qualitative observations made during the current experiments suggest that changes in cell volume occurred in response to alterations in the osmolarity of bathing solution. For instance, under hypotonic conditions the MTG appeared swollen and the sheath was firm and easily pierced with microelectrodes whereas when preparations were bathed with hypertonic solutions the sheath of the MTG was less firm making it more difficult to pierce with microelectrodes. Moreover, inadvertent damage to the sheath under hypotonic conditions resulted in swelling of the ganglion contents out of the cut and this was not seen with iso-osmotic or hyperosmotic solutions. Thus we propose that, by manipulating the extracellular osmolarity, our treatments successfully disrupted cell volume regulation leading to changes in the volume of the extracellular compartment. During CSD in mammals the reduction in the extracellular space is largely due to the swelling of neurons (Zhou et al. 2010); however, glial cells are known to swell in response to elevated [K+]o (Walz 1987). In the current experiments we were looking at the combined effects of SD and changes in extracellular osmolarity, and therefore, under these conditions we find it likely that both neurons and glial cells contributed to alterations in the size of the extracellular space.
We first tested the effects of extracellular osmolarity on ouabain-induced SD and found that it was exacerbated under hypotonic conditions while hypertonic conditions were inhibiting. For instance, we found that ouabain-induced SD was elicited in 100% of preparations treated with hypotonic saline; however, to reliably induce SD under hypertonic conditions a higher dose of ouabain was required. Furthermore, hypo-osmotic conditions were associated with reduced latencies to onset and timing between SD episodes. Negative consequences associated with a hypotonic environment have been previously reported in mammalian nervous tissue; for example, in rat brain slices recovery of neural function following hypoxia is diminished by exposure to hypotonic conditions and prolonged exposure to hypo-osmotic stress causes damage to cortical brain cells (Andrew et al. 1997; Payne et al. 1996). Moreover, hypotonic conditions alone can lead to the eruption of spontaneous SD-like depolarizations in rat brain tissue (Chebabo et al. 1995) suggesting a hypotonic-induced disruption in the ability to maintain ionic homeostasis. In the current experiments, although we did not observe spontaneous SD before ouabain application, we did find an exacerbation of SD when ouabain was paired with hypotonic stress and thus our results also support that an impairment in the ability to maintain ionic homeostasis exists under hypotonic conditions. In contrast to lowering the osmolarity, increases in extracellular osmolarity have been reported to prevent the occurrence of SD-like depolarizations in rat brain slices suggesting that hypertonic conditions are protective (Balestrino et al. 1999; Huang et al. 1996). Our results are consistent with this as 5 × 10−4 M ouabain, a dose sufficient to induce SD in 100% and ∼64% of preparations under hypotonic and isotonic conditions, respectively, was found to induce SD in less than half of the preparations treated with hypertonic saline (∼27%). In the mammalian CNS hypo-osmotic conditions lead to hyperexcitability and promote seizure activity while increasing the extracellular osmolarity reduces brain excitability and attenuates seizure activity in both patients and brain slices (Andrew 1991). Mechanistic models of locust SD suggest that treatments or conditions that predispose towards the generation of SD act by increasing hyperexcitability (Armstrong et al. 2009). Thus our current findings that hypo-osmotic conditions promote SD while hyperosmotic conditions prevent SD are consistent with such models.
In addition to affecting the susceptibility to SD we also show that changes in the extracellular osmolarity altered K+ dynamics in such a way to suggest cell swelling under hypotonic conditions. For instance, the magnitude of ionic disturbance during individual SD events was greater under hypotonic conditions and associated with faster K+ accumulation and clearance rates. These results support cell swelling in response to hypo-osmotic solutions as a reduction in the extracellular space would magnify changes in ion concentration due to a reduced volume for dilution (Schwartzkroin et al. 1998). No differences in baseline [K+]o measurements were observed between hypotonic and hypertonic treatment groups suggesting that mechanisms of ionic homeostasis were working sufficiently to maintain appropriate ion levels at least up until the onset of individual SD events. Disrupting glial spatial buffering of K+ in the locust CNS has been shown to cause an exaggerated increase in baseline [K+]o levels compared with control preparations (Spong and Robertson 2013). Thus we propose that manipulating the extracellular osmolarity did not directly impair mechanisms of ionic homeostasis but rather changes in the size of the extracellular space can be held responsible for the differences observed. Blocking the Na+/K+-ATPase with ouabain leads to an accumulation in extracellular K+ ions that once a threshold is met triggers the positive feedback system generating a SD episode. In a reduced environment the accumulation would occur more quickly and thus shorter latencies to SD onset would be expected. On the other hand, in a larger environment not only would K+ ions accumulate more slowly but as a result the requirement for K+ clearance mechanisms would be reduced as well. Thus this would allow pumps unaffected by ouabain and other mechanisms of K+ homeostasis to maintain ionic gradients for a longer period of time delaying the onset of SD.
The trend that hypotonic conditions exacerbate SD while hypertonic conditions are protective against SD was further supported by our propagation experiments. For instance, continuous exposure to hypotonic conditions increased the rate of SD propagation, which was likely in part due to a reduction in the extracellular space. A reduced extracellular environment would be predicted to increase the propagation rate by channeling current flow as there would be less space for K+ diffusion. We show that hypertonic treatment was sufficient to slow propagation velocity and propose that by increasing the extracellular osmolarity we limited cell swelling ultimately leading to a slower spread of ionic disturbance.
A reduction in SD propagation rate was also observed following exposure to TEA. Previously, TEA has been demonstrated to slow the propagation velocity of circling spreading depression in the chicken retina (Scheller et al. 1998). Furthermore, in the locust CNS, treatment with TEA has been shown to reduce both the susceptibility to ouabain-induced SD and the amplitude of the individual [K+]o events (Rodgers et al. 2009). These findings were attributed to a reduction in K+ conductance through voltage-gated K+ channels and thus a reduced requirement for [K+]o clearance mechanisms (Rodgers et al. 2009). TEA is a well-known blocker of voltage-dependent K+ channels; however, interestingly, it has also been used as a compound to inhibit mammalian aquaporin channels (Brooks et al. 2000; Yool et al. 2010). For instance, treatment with TEA has been shown to reduce osmotically driven cell swelling in Xenopus oocytes expressing human aquaporin-1 channels (Brooks et al. 2000). Although less is known about invertebrate aquaporins, water-permeable channels have been found in insects that share similarities with vertebrate aquaporins (Campbell et al. 2008). In the current experiments TEA likely slowed the accumulation of extracellular K+ by reducing K+ conductance; however, it could also have worked to block water uptake preventing cell swelling. The exact mechanism(s) by which TEA reduced propagation rate in the current paper is not clear; however, more work designed at specifically targeting aquaporin channels merits further investigation. Nevertheless, it seems that treatments aimed at reducing the accumulation of extracellular K+ serve as a protective measure to slow the rate of SD propagation.
It has been previously demonstrated that the Na-K-2Cl cotransporter contributes to cell swelling in response to elevated [K+]o in the rat optic nerve (MacVicar et al. 2002). We predicted that if the Na-K-2Cl transporter was responsible for a component of cell swelling in the current experiments, then pharmacologically inhibiting the cotransporter with bumetanide would attenuate the rate of SD propagation; however, the opposite trend was observed. Bumetanide treatment was associated with increased propagation rates and was observed to consistently elicit low amplitude negative deflections in DC potential suggesting local disturbances in ionic homeostasis. In the vertebrate CNS, the Na-K-2Cl cotransporter has not only been implicated in volume control but has also been shown to transport Na+, K+, and Cl− ions into the cell during periods when extracellular K+ levels are high (Geck and Heinz 1986; Schwartzkroin et al. 1998; Walz and Hertz 1984). Moreover, increased rates of SD propagation have been reported following disruption to the locust blood-brain barrier, which is known to play an active role in maintaining appropriate ion levels (Kocmarek and O'Donnell 2011; Spong et al. 2014; Treherne and Schofield 1981). Thus we predicted that bumetanide treatment caused impairment in the ability to clear excess ions from the extracellular space increasing the rate of K+ accumulation and ultimately the rate of SD propagation. This was indeed supported by our K+ experiments which demonstrated that exposure to bumetanide caused a greater disturbance in the regulation of [K+]o. For example, bumetanide-treated preparations showed increased levels in baseline [K+]o over the experimental period and were associated with increased surge amplitudes. Maintenance of ionic homeostasis is critical to ensure continued generation of neural activity (Money et al. 2009; Rodgers et al. 2007; Rounds 1967; Wu and Fisher 2000) and thus elucidation of such regulatory mechanisms is of great interest. Here we provide evidence that the Na-K-2Cl cotransporter helps play a role in maintaining [K+]o in the insect nervous system.
In summary, our results demonstrate that reducing the accumulation of extracellular K+ ions can protect against both the generation of SD and the spread of ionic disturbance in locusts. Determination of what mediates the diffusion of the SD wave front is still a matter of debate although recently it has been proposed that diffusion of interstitial K+ is the leading event in mammalian CSD (Enger et al. 2015). The findings reported here suggest that the SD wave front in the invertebrate CNS is also mediated by the diffusion of extracellular K+ ions. Our results highlight the importance of controlling cell volume; however, the specific processes involved in such regulation remain unknown. Here we show that exposure to hyperosmotic conditions is protective against SD likely by increasing the volume of the CNS extracellular compartment. Interestingly, an increase in hemolymph osmolality has been shown to occur in response to mild desiccation in the goldenrod gall fly Eurosta solidaginis and this is associated with an increase in cold tolerance providing protection from chill injury (Gantz and Lee 2015). Thus we would predict that a desiccation-induced increase in hemolymph osmolality would also protect against locust SD and is an area that is worthy of future exploration.
GRANTS
We thank the Natural Sciences and Engineering Research Council of Canada for funding this research.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.E.S., K.L.M.W., and R.M.R. conception and design of research; K.E.S., B.C., and K.L.M.W. performed experiments; K.E.S., B.C., and K.L.M.W. analyzed data; K.E.S., K.L.M.W., and R.M.R. interpreted results of experiments; K.E.S. prepared figures; K.E.S. drafted manuscript; K.E.S. and R.M.R. edited and revised manuscript; K.E.S., B.C., K.L.M.W., and R.M.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. David Andrew for helpful feedback and comments on this manuscript, Dr. Chris Moyes for helpful discussions, and Kevin Cross for invaluable help during the course of this project.
REFERENCES
- Andrew RD. Seizure and acute osmotic change: clinical and neurophysiological aspects. J Neurol Sci 101: 7–18, 1991. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA. Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex 17: 787–802, 2007. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Lobinowich ME, Osehobo EP. Evidence against volume regulation by cortical brain cells during acute osmotic stress. Exp Neurol 143: 300–312, 1997. [DOI] [PubMed] [Google Scholar]
- Andrew RD, MacVicar BA. Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience 62: 371–383, 1994. [DOI] [PubMed] [Google Scholar]
- Armstrong GA, Rodgers CI, Money TG, Robertson RM. Suppression of spreading depression-like events in locusts by inhibition of the NO/cGMP/PKG pathway. J Neurosci 29: 8225–8235, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrino M, Young J, Aitken P. Block of (Na+,K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices. Brain Res 838: 37–44, 1999. [DOI] [PubMed] [Google Scholar]
- Brooks HL, Regan JW, Yool AJ. Inhibition of aquaporin-1 water permeability by tetraethylammonium: involvement of the loop E pore region. Mol Pharmacol 57: 1021–1026, 2000. [PubMed] [Google Scholar]
- Campbell EM, Ball A, Hoppler S, Bowman AS. Invertebrate aquaporins: a review. J Comp Physiol B 178: 935–955, 2008. [DOI] [PubMed] [Google Scholar]
- Chebabo SR, Hester MA, Aitken PG, Somjen GG. Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res 695: 203–216, 1995. [DOI] [PubMed] [Google Scholar]
- Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17: 439–447, 2011. [DOI] [PubMed] [Google Scholar]
- Enger R, Tang W, Vindedal GF, Jensen V, Johannes HP, Sprengel R, Looger LL, Nagelhus EA. Dynamics of ionic shifts in cortical spreading depression. Cereb Cortex 2015. April 2 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz JD, Lee RE Jr. The limits of drought-induced rapid cold-hardening: extremely brief, mild desiccation triggers enhanced freeze-tolerance in Eurosta solidaginis larvae. J Insect Physiol 73: 30–36, 2015. [DOI] [PubMed] [Google Scholar]
- Huang R, Aitken PG, Somjen GG. Hypertonic environment prevents depolarization and improves functional recovery from hypoxia in hippocampal slices. J Cereb Blood Flow Metab 16: 462–467, 1996. [DOI] [PubMed] [Google Scholar]
- Kocmarek AL, O'Donnell MJ. Potassium fluxes across the blood brain barrier of the cockroach, Periplaneta americana. J Insect Physiol 57: 127–135, 2011. [DOI] [PubMed] [Google Scholar]
- Leão AA. Spreading depression of activity in the cerebral cortex. J Neurophysiol 7: 359–390, 1944. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia 37: 114–123, 2002. [DOI] [PubMed] [Google Scholar]
- Money TG, Rodgers CI, McGregor SM, Robertson RM. Loss of potassium homeostasis underlies hyperthermic conduction failure in control and preconditioned locusts. J Neurophysiol 102: 285–293, 2009. [DOI] [PubMed] [Google Scholar]
- Payne RS, Schurr A, Rigor BM. Cell swelling exacerbates hypoxic neuronal damage in rat hippocampal slices. Brain Res 723: 210–213, 1996. [DOI] [PubMed] [Google Scholar]
- Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 15: 379–393, 2014. [DOI] [PubMed] [Google Scholar]
- Rodgers CI, Armstrong GA, Robertson RM. Coma in response to environmental stress in the locust: a model for cortical spreading depression. J Insect Physiol 56: 980–990, 2010. [DOI] [PubMed] [Google Scholar]
- Rodgers CI, Armstrong GA, Shoemaker KL, LaBrie JD, Moyes CD, Robertson RM. Stress preconditioning of spreading depression in the locust CNS. PLoS One: 2, e1366, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers CI, LaBrie JD, Robertson RM. K+ homeostasis and central pattern generation in the metathoracic ganglion of the locust. J Insect Physiol 55: 599–607, 2009. [DOI] [PubMed] [Google Scholar]
- Rounds HD. KC1-induced “spreading depression” in the cockroach. J Insect Physiol 13: 869–872, 1967. [DOI] [PubMed] [Google Scholar]
- Scheller D, Tegtmeier F, Schlue WR. Dose-dependent effects of tetraethylammonium on circling spreading depressions in chicken retina. J Neurosci Res 51: 85–89, 1998. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Baraban SC, Hochman DW. Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res 32: 275–285, 1998. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81: 1065–1096, 2001. [DOI] [PubMed] [Google Scholar]
- Spong KE, Robertson RM. Pharmacological blockade of gap junctions induces repetitive surging of extracellular potassium within the locust CNS. J Insect Physiol 59: 1031–1040, 2013. [DOI] [PubMed] [Google Scholar]
- Spong KE, Rochon-Terry G, Money TG, Robertson RM. Disruption of the blood-brain barrier exacerbates spreading depression in the locust CNS. J Insect Physiol 66: 1–9, 2014. [DOI] [PubMed] [Google Scholar]
- Treherne JE, Schofield PK. Mechanisms of ionic homeostasis in the central nervous system of an insect. J Exp Biol 95: 61–73, 1981. [DOI] [PubMed] [Google Scholar]
- Walz W. Swelling and potassium uptake in cultured astrocytes. Can J Physiol Pharmacol 65: 1051–1057, 1987. [DOI] [PubMed] [Google Scholar]
- Wu J, Fisher RS. Hyperthermic spreading depressions in the immature rat hippocampal slice. J Neurophysiol 84: 1355–1360, 2000. [DOI] [PubMed] [Google Scholar]
- Yool AJ, Brown EA, Flynn GA. Roles for novel pharmacological blockers of aquaporins in the treatment of brain oedema and cancer. Clin Exp Pharmacol Physiol 37: 403–409, 2010. [DOI] [PubMed] [Google Scholar]
- Zhou N, Gordon GR, Feighan D, MacVicar BA. Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb Cortex 20: 2614–2624, 2010. [DOI] [PubMed] [Google Scholar]