

Abstract
Introduction
Overexpression of cyclooxygenase (COX-2) is commonly observed in human cancers. In a murine model of metastatic breast cancer, we observed that COX-2 expression and enzyme activity were associated with enhanced tumorigenic and metastatic potential. In contrast to the high COX-2 expression in metastatic tumors, transplantation of poorly tumorigenic tumor cell lines to syngeneic mice results in less COX-2 expression and less COX-2 activity in vivo. Aberrant CpG island methylation, and subsequent silencing of the COX-2 promoter, has been observed in human cancer cell lines and in some human tumors of the gastrointestinal tract.
Methods
Using bisulfite modification and a methylation-specific PCR, we examined the methylation status of the COX-2 promoter in a series of four closely-related murine mammary tumors differing in COX-2 expression and metastatic potential.
Results
We showed that line 410, which does not express COX-2 in vivo, exhibited evidence of promoter methylation. Interestingly, the metastatic counterpart of this cell (line 410.4) displayed only the unmethylated COX-2 promoter, as did two additional cell lines (lines 66.1 and 67). The methylation patterns observed in vitro were maintained when these murine mammary tumor lines were transplanted to syngeneic mice. Treatment with the DNA demethylating agent 5-aza-deoxycytidine increased COX-2 mRNA, increased protein and increased enzyme activity (prostaglandin synthesis).
Conclusions
These results indicate that COX-2 promoter methylation may be one mechanism by which tumor cells regulate COX-2 expression. Upregulation of COX-2 expression in closely related metastatic lesions versus nonmetastatic lesions may represent a shift towards the unmethylated phenotype.
Keywords: cyclooxygenase, promoter methylation, breast cancer
Introduction
It is now well established that the inducible isoform of cyclooxygenase, COX-2, is commonly overexpressed in many solid tumors [1,2]. Epidemiological studies as well as clinical trials employing selective and nonselective COX-2 inhibitors indicate that COX-2 is mechanistically involved in colorectal carcinogenesis, and possibly in other sites of carcinogenesis [3-7]. In addition to early cancer development, evidence is beginning to accumulate that COX-2 may also contribute to late-stage progression (i.e. tumor metastasis) [7,8]. Early reports in breast cancer suggested a linkage between prostaglandin production and aggressive disease [9,10], but less is known regarding the specific contribution of the COX-2 isoform to behavior. Studies are beginning to emerge that suggest heightened COX-2 expression is associated with more aggressive breast cancer [11-13].
We have examined COX-2 expression in a murine model of metastatic breast cancer and have observed that COX-2 protein expression in vivo, as well as COX-2 enzyme activity (i.e. prostaglandin E2 [PGE2] synthesis), is positively correlated with more aggressive disease [14]. Furthermore, selective COX-2 or COX-1 inhibitors control metastatic disease in this model system [15].
COX-2 expression and PGE2 production were very low or absent in tumors derived from transplantation of nonmetastatic cell lines to syngeneic mice, but metastatic tumors expressed high levels of COX-2 protein, mRNA and PGE2. COX-2 expression is controlled in normal tissues at the transcriptional level. In cancer, expression of many genes is regulated by aberrant promoter methylation [16]. Several recent studies have likewise suggested that methylation of COX-2 promoter DNA at areas of CpG islands may result in aberrant COX-2 methylation in human tumors of colorectal or gastric origin [17-21]. Based on these studies, we asked whether COX-2 promoter methylation regulates gene expression in mammary epithelial tumors.
Materials and methods
Cell lines
Four murine mammary tumor cell lines (lines 410, 410.4, 66.1 and 67) were maintained in DMEM medium supplemented with 10% fetal calf serum (Gemini Bio-Products, Inc., Calabasas, CA, USA), 2 mM glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml) and 0.1 mM nonessential amino acids in a 10% CO2 humidified atmosphere.
For determination of COX expression in tumors, (1–3) × 106 viable cells of each tumor line were injected subcutaneously into syngeneic Balb/cByJ female mice (Jackson Laboratories, Bar Harbor, ME, USA). When tumors achieved an average diameter of 8 mm, the mice were sacrificed by cervical dislocation, the tumors were removed and portions were prepared for protein analysis or for DNA studies.
Bisulfite modification and methylation-specific PCR
Genomic DNA was extracted by the DNA Prep Kit (Promega, Madison, WI, USA) and was bisulfite modified with the CpGenomic DNA modification kit (Intergen, Purchase, NY, USA). Methylation-specific PCR was performed using the later described primer pairs in a volume of 50 μl containing 1 × PCR buffer, 0.2 mM dNTP, 1.5 mM MgCl2, 50 ng bisulfite-modified genomic DNA, 10 pmol each sense primer and each antisense primer, and 0.4 U Platinum Taq DNA polymerase (Gibco BRL, Gaithersburg, MD, USA).
The PCR conditions were 95°C for 5 min, then 35 cycles at 95°C for 45 s, at 60°C for 45 s and at 72°C for 45 s, and a final extension at 72°C for 4 min. Ten microliters of each PCR product were loaded on a nondenaturing 10% polyacrylamide gel, were stained with ethidium bromide and were directly visualized under UV illumination.
Mouse Cox-2 primer sequences were designed based on the published promoter sequence [22]: methylated reaction, 5'-TTTGTCGTTGCGGTTTTTGC-3' (sense) and 5'-AAAACGAACTCCACGTAACG-3' (antisense), 118 bp product; unmethylated reaction, 5'-AAAGTTTGTTGTTGTGGTTTTTGT-3' (sense) and 5'-CTATAAAACAAACTCCACATAACA-3' (antisense), 126 bp product.
For COX mRNA analysis, RNA was extracted from cultured cells using TRIAZOL reagent (Gibco BRL), was reverse transcribed and was amplified using COX-2-specific primers: forward primer, 5'-GTGGAAAAACCTCGTCCAGA-3' and reverse primer, 5'-TGATGGTGGCTGTTTTGGTA-3' (256 bp product).
5-aza-2'-deoxycytidine treatment
Cells were plated in complete medium and treated with concentrations of 5-aza-2'-deoxycytidine (5-aza) treatment from 0.1 to 1.0 μM (Sigma Chemical Co., St Louis, MO, USA) for 4 days. The medium and the drug were replaced every 24 hours. At the end of the treatment period, the medium was removed and RNA was extracted for RT-PCR or northern analysis, protein was extracted for western analysis or conditioned medium was harvested for PGE2 ELISA.
Western blotting
Cell lysates were prepared in M-Per or tumor lysates were prepared in T-Per (Pierce, Rockford, IL, USA) containing phenylmethylsulfonyl fluoride as previously described [14]. Lysates were centrifuged and 20 μg protein was denatured in Laemmli sample buffer and resolved on 10% Tris-HCl ready gels (BioRad, Hercules, CA, USA), was electrophoretically transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and was immunoblotted with Cox-2 antibody (Cayman Chemical, Ann Arbor, MI, USA) or β-actin antibody (Sigma) followed by horseradish peroxidase-conjugated second antibodies (Transduction Laboratories, Lexington, KY, USA). Specific bands were visualized by Super Signal West Pico chemiluminescent substrate (Pierce).
Prostaglandin E ELISA
Determination of PGE2 levels in cell-conditioned medium was carried out by ELISA kit following the manufacturer's instructions (Cayman Chemical). Conditioned medium was harvested and analyzed for PGE2 72 hours after the addition of 5-aza.
Results
We examined the role of COX-2 in cancer using a murine model of metastatic breast cancer. We have shown previously that metastatic potential is positively correlated with increased expression and activity of the COX-2 enzyme. Analysis of COX-2 proteins expressed in lysates of tumors revealed a strikingly different pattern for metastatic tumors versus nonmetastatic tumors (Fig. 1). COX-2 protein expression was present in vivo in metastatic populations (i.e. lines 66.1, 410.4 and 168) but was nearly absent in tumors derived from the nonmetastatic cell lines 410 and 67. Only after prolonged exposures was a faint COX-2 band visible from lysates obtained from tumor line 410 (data not shown). These data confirm our earlier report in which immmunohistochemical staining for COX-2 was evident in tumors derived from line 66.1 or from 410.4 cell lines, but not tumors derived from line 410 [14].
Figure 1.
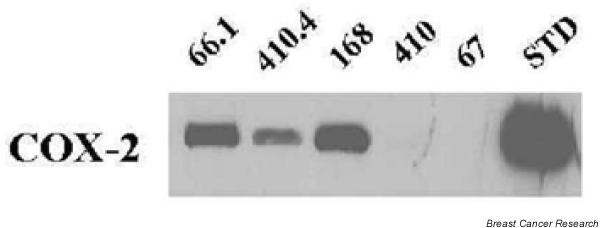
Tumors derived from the transplantation of metastatic lines 66.1, 410.4 and 168 or from nonmetastatic lines 410 and 67 were harvested. The tissue lysates were prepared and were immunoblotted with cyclooxygenase-2 antibody followed by horseradish peroxidase-conjugated secondary antibodies, and the expression was compared with 72 kDa Cox-2 protein standard (STD).
Although COX-2 expression is frequently upregulated in cancers of various histologic types, the mechanisms driving this expression are not completely elucidated. Multiple mechanisms are responsible for the regulation of gene expression. DNA methylation is an epigenetic modification that can play an important role in silencing of other genes [16]. Recent studies provide evidence of hypermethylation of CpG islands within the COX-2 promoter in both breast and colon human cancer cell lines [17-21]. To determine whether murine breast tumors employ a similar mechanism of COX-2 gene regulation, we used methylation-specific PCR to examine the methylation pattern of the COX-2 promoter in murine tumor cell lines. Figure 2a shows that three of the four cell lines (lines 66.1, 410.4 and 67) expressed only the unmethylated promoter (126 bp product), whereas both unmethylated species and methylated species were amplified from line 410 (126 bp and 118 bp, respectively).
Figure 2.
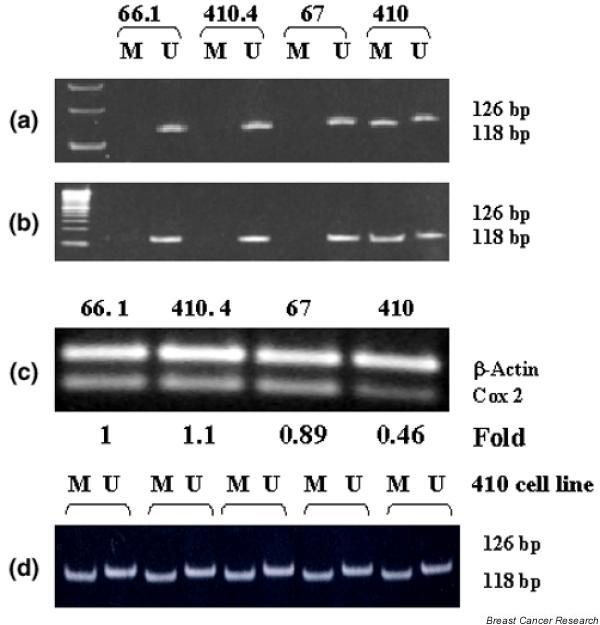
Genomic DNA was extracted from murine mammary tumor cell lines, was bisulfite modified and was PCR amplified using primer pairs specific for the murine methylated (M) (118 bp) product or the murine unmethylated (U) (126 bp) product. (a) Ten microliters of PCR product were loaded per lane of nondenaturing polyacrylamide gel and were stained with ethidium bromide. Lines 66.1, 410.4 and 67 express only the unmethylated band, whereas line 410 contains both methylated and unmethylated cyclooxygenase-2. (b) Genomic DNA was extracted from tumors derived from the transplantation of the indicated cell lines to syngeneic mice and was analyzed as in (a). The methylation pattern observed in cell lines is maintained when cells are transplanted to mice. (c) RNA was extracted from mammary tumor cell lines, was reverse transcribed and was amplified using primers specific for either murine COX-2 or β-actin. The fold increase was determined by densitometry. (d) Clonal populations were derived from line 410, and the DNA extracted, bisulfite modified and PCR amplified using primers as in (a). Each pair of methylated and unmethylated products represents an individual clone.
The methylation patterns observed in cultured tumor cells were examined in vivo. Tumor cells were transplanted to syngeneic Balb/cByJ female mice and palpable tumors were excised. DNA was extracted and analyzed by methylation-sensitive PCR as already described. This analysis shows that the methylation patterns expressed in cultured cells were maintained in solid tumors derived from these cells (Fig. 2b). Line 410 tumors thus exhibited both methylated and unmethylated species, whereas tumors derived from line 66.1, line 410.4 or line 67 expressed only the unmethylated COX-2 promoter. Figure 2c shows COX-2 and β-actin mRNA expression in the same cell lines. Consistent with the observed promoter methylation pattern, COX-2 levels were reduced in line 410 cells (approximately 50% of levels observed in line 66.1). We were concerned that the mixed methylation pattern observed in line 410 cultures might be the result of heterogeneous populations of cells. We therefore recloned line 410 and re-examined the COX promoter methylation pattern. Each clonal population retained the mixed methylation pattern (Fig. 2d).
To determine the role of methylation in silencing of COX-2, the cell line displaying evidence of promoter methylation (line 410) was treated with the DNA demethylating agent, 5-aza (0.1–1.0 μM). The effect on COX-2 mRNA and protein levels was then determined. A modest induction (1.2-fold to 2.6-fold) of COX-2 mRNA by 5-aza was observed in a dose-dependent manner (Fig. 3a). This increased COX-2 mRNA expression was associated with an increase in Cox-2 protein as well (Fig. 3b).
Figure 3.
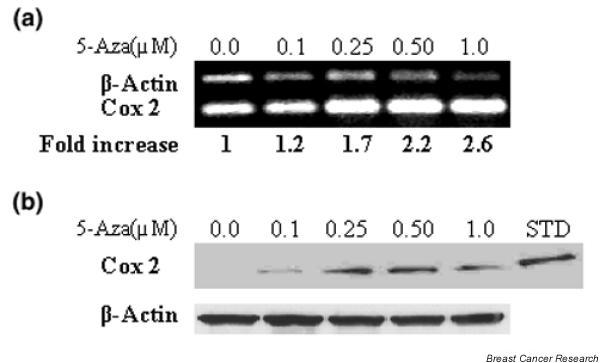
Murine line 410 was treated with 5-aza-2'-deoxycytidine (5-aza) at the concentrations indicated for 72 hours. The RNA was extracted, reverse transcribed and amplified using primers specific for either murine cyclooxygenase-2 or for β-actin. (a) The fold increase was determined by densitometry. (b) Lysates were prepared from cells treated as in (a) and were immunoblotted using COX-2-specific antibody or β-actin-specific antibody. Lane six contains a Cox-2 protein standard (STD).
PGE2 levels were assayed to determine whether the observed effects of 5-aza on the induction of COX-2 protein and mRNA are reflected in differences in enzyme activity. Line 410 cells were treated with 5-aza and, 72 hours later, the conditioned medium was harvested and analyzed for PGE2 by ELISA. As predicted by the mRNA and protein results, treatment of line 410 cells with 5-aza in concentrations of 0.1–0.4 μM resulted in a dose-dependent increase in PGE2 levels (Fig. 4) consistent with the upregulation in COX-2 protein levels.
Figure 4.
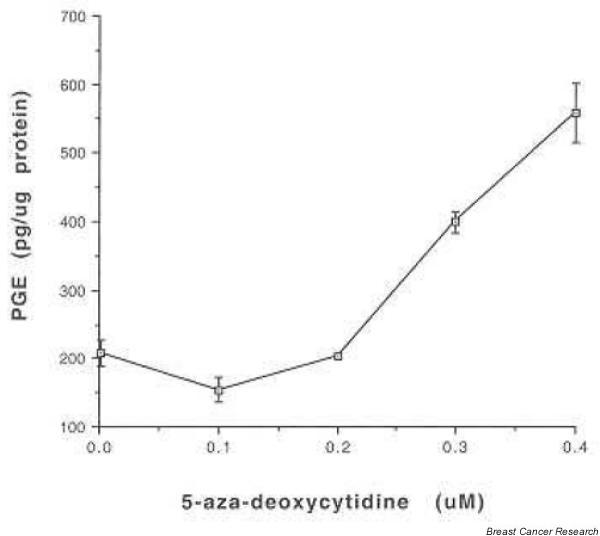
Line 410 cells were treated with various concentrations of 5-aza-2'-deoxycytidine and, 72 hours after culture initiation, conditioned medium was harvested and analyzed for prostaglandin E2 (PGE2) by ELISA.
Discussion
Overexpression of COX-2 is commonly observed in solid tumors of diverse histologic types [1,2]. COX-2 is transcriptionally regulated in normal tissues by many factors including the proinflammatory cytokines tumor necrosis factor-α and interleukin-1β as well as bacterial endotoxin. The molecular basis of COX-2 upregulation in cancer has not been established, but may in part represent the activation by stromal factors, host growth factors, and so on. Several recent reports have indicated that COX-2 promoter methylation may represent an additional regulator of expression in some cancer cell lines and tumors [17-19]. Gene silencing can occur through cytosine methylation of CpG islands. Other cancer-associated genes have been shown to be regulated by aberrant hypermethylation of 5' CpG islands in cancer [16].
Toyota and colleagues first proposed promoter methylation as a mechanism to regulate COX-2 expression in cancer [17]. They showed that a subset of colorectal cancers and adenomas reveals evidence of aberrant COX-2 methylation. Examination of a number of tumor cell lines revealed complex patterns, with some cell lines exhibiting dense methylation and other cell lines exhibiting partial methylation. Detailed methylation mapping implicated a region upstream of exon I in loss of COX-2 mRNA expression, an effect that could be reversed by treatment with 5-aza. Detailed promoter mapping in gastric carcinoma cell lines revealed that all CpG sites in the region spanning -590 to +186, with respect to the transcription initiation site, were completely methylated [18].
We have examined the role of COX-2 in a model of metastatic breast cancer. Those studies showed a strong correlation for expression and enzymatic activity of COX-2 with increased tumorigenic and metastatic potential [14,15]. When nonmetastatic cell lines (lines 410 and 67) were transplanted to syngeneic mice, no COX-2 protein could be detected by either immunohistochemistry or immunoblotting of tumor lysates. This lack of COX-2 protein expression was also associated with very weak or absent COX-2 mRNA. In contrast, metastatic tumors (lines 66.1, 410.4, 4526 and 168) express COX-2 in vivo and have much higher levels of extractable PGE2[14].
Based on studies from other laboratories demonstrating COX-2 promoter methylation as one mechanism by which COX-2 is regulated in human cancer, we sought evidence for COX-2 promoter methylation in murine mammary tumors. Using bisulfite modification and methylation-specific PCR, we compared the COX-2 methylation patterns in a series of four murine mammary tumor cell lines and tumors derived from these cell lines. We found that three of four cell lines (lines 66.1, 67 and 410.4) expressed only the unmethylated promoter, whereas both unmethylated species and methylated species were amplified in line 410. Thus, for line 410.4 and line 66.1 cells there is a correlation between expression and unmethylated status, and for line 410 there is a lack of expression and methylated status. Line 67 is the exception to this relationship and suggests that other mechanisms contribute to COX-2 regulation.
These data confirm previous studies by one of our laboratories [19] and those of Toyota and colleagues [17] showing complex COX promoter methylation patterns in human colorectal cancer cell lines, gastric epithelial cancer cell lines and breast cancer cell lines. The current findings extend these studies to indicate that COX-2 promoter methylation may also be an important mechanism in a rodent model of breast cancer. The methylation pattern observed in vitro was maintained when murine tumor cell lines were transplanted to syngeneic mice. Importantly, treatment of the cell line that exhibits methylated promoter, with 5-aza, resulted in increased COX-2 mRNA and protein consistent with a role for methylation in silencing of the COX-2 gene. This upregulation by demethylation also resulted in increased enzyme activity as indicated by increased production of PGE2.
A comparison of methylation patterns in cell lines 410 and 410.4 is of interest because of the common origin of these cells with very different phenotypes. Tumors resulting from transplantation of line 410 are thus relatively slow growing and rarely metastasize. Line 410.4 was derived from a rare metastatic lesion occurring in a mouse bearing a subcutaneous implant of line 410. Like the behavior of these tumors, the COX-2 promoter methylation patterns are also distinct. The loss of COX promoter methylation in line 410.4 versus line 410 may indicate a mechanistic role for COX-2 expression in tumor progression. The acquisition of a more aggressive and metastatic phenotype may thus be related to increased COX-2 expression in line 410.4 cells relative to line 410 cells. The current studies extend these findings to indicate that the upregulation of COX-2 we have observed during breast cancer progression may sometimes result from reduced methylation of the COX-2 promoter.
It has been suggested that the net expression of COX-2 in colorectal tumors represents a balance between the pressure to silence many genes, including tumor suppressor genes, by methylation and the pressure to express COX-2 as a mechanism to inhibit apoptosis and otherwise promote cancer growth [20]. In the case of tumors derived from line 410, little or no COX-2 mRNA or protein is detectable. Thus, in this relatively benign lesion, the balance is shifted towards silencing of this gene that, when expressed, is associated with more aggressive breast cancers. There is also evidence in human malignancies that COX-2 is sometimes hypermethylated, possibly representing a balance between methylation of tumor-promoting genes and tumor-suppressing genes [17-19,21].
A number of genes have been shown to be hypermethylated in cancer [16]. Many tumor suppressor genes as well as other cancer-related genes are inactivated by aberrant methylation of the CpG islands in the promoter region including Rb, estrogen receptor, androgen receptor, cyclin kinase inhibitors, the mismatch repair gene hMLH-1, p73, mts-1, BRCA-1, thrombospondin-1 and TIMP-3. Methylation in cancer thus often involves loss of function of an otherwise protective gene activity. Such findings are the basis for clinical studies to evaluate the therapeutic efficacy of inhibitors of DNA methylation. The current studies suggest that the COX-2 gene may be unusual in this regard, since methylation is associated with decreased tumorigenicity at least in a murine system.
It will be important to examine COX methylation in primary human breast tumors to determine whether the pattern observed in cell lines is confirmed. Furthermore, correlating levels of COX-2 expression with methylation patterns will be important to establish the role of COX promoter methylation in gene expression in human disease. Further studies are clearly indicated to clarify the role of COX-2 methylation in breast and other malignancies.
Conclusions
It is now accepted that COX-2 expression plays a role in the behavior of breast cancer and other malignancies. Less is known regarding how COX-2 is regulated in cancer, but the current study adds to the growing body of evidence that promoter methylation may be involved. Comparison of closely related metastatic and nonmetastatic counterparts suggests that the COX-2 promoter may be demethylated during tumor progression.
Competing interests
None declared.
Abbreviations
5-aza = 5-aza-2'-deoxycytidine; bp = base pairs; COX = cyclooxygenase; DMEM = Dulbecco's modified Eagle's medium; ELISA = enzyme-linked immunosorbent assay; PCR = polymerase chain reaction; PGE2 = prostaglandin E2; RT = reverse transcriptase.
Acknowledgments
Acknowledgement
This work was supported by the United States Department of Defense (to AMF).
Contributor Information
Xinrong Ma, Email: XMa@som.umaryland.edu.
Qingyuan Yang, Email: qyang001@umaryland.edu.
Keith T Wilson, Email: kwilson@umaryland.edu.
Namita Kundu, Email: nkundu@umaryland.edu.
Stephen J Meltzer, Email: Smeltzer@medicine.umaryland.edu.
Amy M Fulton, Email: afulton@umaryland.edu.
References
- Taketo MM. Cyclooxygenase-2 inhibitors in tumorigenesis (part I) J Natl Cancer Inst. 1998;90:1529–1536. doi: 10.1093/jnci/90.20.1529. [DOI] [PubMed] [Google Scholar]
- Taketo MM. Cyclooxygenase-2 inhibitors in tumorigenesis (part II) J Natl Cancer Inst. 1998;90:1609–1620. doi: 10.1093/jnci/90.21.1609. [DOI] [PubMed] [Google Scholar]
- Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW., Jr Aspirin use and risk of fatal cancer. Cancer Res. 1993;53:1322–1327. [PubMed] [Google Scholar]
- Harris RE, Namboodiri KK, Farrar WB. Epidemiological study of nonsteroidal antiinflammatory drugs and breast cancer. Oncol Rep. 1995;2:591–592. doi: 10.3892/or.2.4.591. [DOI] [PubMed] [Google Scholar]
- Harris RE, Namboodiri KK, Stellman SD, Wynder EL. Breast cancer and NSAID use: heterogeneity of effect in a case-control study. Prevent Med. 1995;24:119–120. doi: 10.1006/pmed.1995.1022. [DOI] [PubMed] [Google Scholar]
- Harris RE, Namboodiri KK, Farrar WB. Nonsteroidal antiinflammatory drugs and breast cancer. Epidemiology. 1996;7:203–205. doi: 10.1097/00001648-199603000-00017. [DOI] [PubMed] [Google Scholar]
- Murata H, Kawano S, Tsuji S, Tsuji M, Sawaoka H, Kimura Y, Shiozaki H, Hori M. Cyclooxygenase-2 overexpression enhances lymphatic invasion and metastasis in human gastric carcinoma. Am J Gastroenterol. 1999;94:451–455. doi: 10.1016/S0002-9270(98)00757-6. [DOI] [PubMed] [Google Scholar]
- Fujita T, Matsui M, Takaku K, Uetake H, Ichikawa W, Taketo MM, Sugihara K. Size- and invasion-dependent increase in cyclooxygenase 2 levels in human colorectal carcinomas. Cancer Res. 1998;58:4823–4826. [PubMed] [Google Scholar]
- Bennett A, Berstock DA, Raja B, Stamford IF. Survival time after surgery is inversely related to the amounts of prostaglandins extracted from human breast cancers. Br J Pharmacol. 1979;66:451–455. [PMC free article] [PubMed] [Google Scholar]
- Rolland PH, Martin PM, Jacquemier J, Rolland AM, Toga M. Prostaglandin in human breast cancer: evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. J Natl Cancer Inst. 1980;64:1061–1066. [PubMed] [Google Scholar]
- Liu X-H, Rose DP. Differential expression and regulation of cyclooxygenase-1 and -2 in two human breast cancer cell lines. Cancer Res. 1996;56:5125–5127. [PubMed] [Google Scholar]
- Parrett ML, Harris RL, Joarder FS, Ross MS, Clausen KP, Robertson FM. Cyclooxygenase-2 gene expression in human breast cancer. Int J Oncol. 1997;10:503–507. doi: 10.3892/ijo.10.3.503. [DOI] [PubMed] [Google Scholar]
- Ristimaki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, Joensuu H, Isola J. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002;62:632–635. [PubMed] [Google Scholar]
- Kundu N, Yang Q, Dorsey R, Fulton AM. Increased cyclooxygenase-2 expression and activity in a murine model of metastatic breast cancer. Int J Cancer. 2001;93:681–686. doi: 10.1002/ijc.1397. [DOI] [PubMed] [Google Scholar]
- Kundu N, Fulton AM. Selective cyclooxygenase-1 or cox-2 inhibitors control metastatic disease in a murine model of breast cancer. Cancer Res. 2002;62:2343–2346. [PubMed] [Google Scholar]
- Momparler R, Bovenzi V. DNA methylation and cancer. J Cell Physiol. 2000;183:145–154. doi: 10.1002/(SICI)1097-4652(200005)183:2<145::AID-JCP1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Toyota M, Shen L, Ohe-Toyata M, Hamilton SR, Sinicrope FA, Issa J-P. Aberrant methylation of the cyclooxygenase-2 CpG island in colorectal tumors. Cancer Res. 2000;60:4044–4048. [PubMed] [Google Scholar]
- Song SH, Jong H-S, Choi HH, Inoue H, Tanabe T, Kim NK, Bang Y-J. Transcriptional silencing of cyclooxygense-2 by hyper-methylation of the 5' CpG island in human gastric carcinoma cells. Cancer Res. 2001;61:4628–4635. [PubMed] [Google Scholar]
- Akhtar M, Cheng Y, Magno RM, Ashktorab H, Smoot DT, Meltzer SJ, Wilson KT. Promoter methylation regulates Helicobacter pylori-stimulated cyclooxygenase-2 expression in gastric epithelial cells. Cancer Res. 2001;61:2399–2403. [PubMed] [Google Scholar]
- Shen L, Ahuja N, Shen Y, Habib NA, Toyota M, Rashid A, Issa JP. DNA methylation and environmental exposures in human hepatocellular carcinoma. J Natl Cancer Inst. 2002;94:755–761. doi: 10.1093/jnci/94.10.755. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Itoh F, Toyota M, Suzuki H, Yamamoto H, Fujita M, Hosokawa M, Imai K. Aberrant methylation and histone deacetylation of cyclooxygenase 2 in gastric cancer. Int J Cancer. 2002;97:272–277. doi: 10.1002/ijc.1612. [DOI] [PubMed] [Google Scholar]
- Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]