

Abstract
Objective(s):
EMAP-like Protein 5 (EML5) is a new echinoderm microtubule-associated protein that is expressed in the central nervous system. The aim of this study was to investigate the expression profile of EML5 in the anterior temporal neocortex of patients presenting with intractable epilepsy (IE).
Materials and Methods:
Western blot assays were performed to determine EML5 expression in 36 surgically resected anterior temporal neocortices of patients with IE and eight control tissues. Immunohistochemistry and immunofluorescence were employed to explore protein expression in IE.
Results:
EML5 was highly expressed in both neurons and glial cells of the anterior temporal neocortex of IE patients, whereas only low levels of EML5 were detected in control brain tissues. Western blotting showed an enhanced expression of EML5 protein in the anterior temporal neocortex of IE (optical density (OD) = 1.8030 ± 0.1335/1.1852 ± 0.2253, P<0.05) compared with normal control tissues.
Conclusion:
The results demonstrate that highly expressed EML5 in the neurons and glial cells of the cortex of patients with epilepsy is associated with microtubular dysfunction after frequent and recurrent seizures.
Keywords: EML5, Microtubule, Seizure
Introduction
Epilepsy is a brain disorder that is characterized by an enduring predisposition for generating epileptic seizures (1). Despite the optimal use of many drugs, about thirty percent of patients with epilepsy never achieve remission with antiepileptic drugs (AEDs) therapy. Those patients are called as intractable epilepsy (IE) cases. Patients presenting with IE are at high risk of mental symptom, poor quality of life, and mortality (2-5). But the mechanisms for the development of IE remain unknown. Thus, understanding the genesis of epilepsy might provide novel molecular targets for drug therapy strategies that could potentially prevent the disorder.
The repetitive seizure activity induces the overexpression of cytoarchitectural-related proteins at the seizure lesion and contributes to cell loss, which can lead to drug resistance. The main components of the cytoskeleton are microtubules: they regulate the extension of neuronal neurites and thus facilitate morphological changes of the processes. This is achieved by structural binding proteins, including microtubule-stabilizing proteins such as MAP2 (i.e., microtubule associated protein 2) and microtubule disassembly protein. Mesial temporal lobe epilepsy (MTLE) is one of the main components of IE. It is characterized by cytoarchitec-tural abnormalities including neuronal cell loss and reactive gliosis (6-8). The cytoskeleton proteins, beta-tubulin, tubulin alpha-1 chain, and neuronal tropomodulin profilin II were markedly reduced, whereas GAP43 (growth associated protein 43), MAP2, and MAP1A were markedly increased in MTLE (6, 7, 9, 10). These results indicate that the abnormality of the cytoskeletal proteins leads to cytoskeletal impairment in patients with seizure recurrence.
EMAP-like proteins could adjust the formation, facilitate the depolymerization, inhibit the extension, and regulate the dynamics of microtubules in the brain (11). EML5 is expressed in the majority of the brain parts during development. The enhanced microtubule dynamics in neuronal processes is mainly during early stages of neurite extension (12). Although previous investigation of gene chip showed that EML5 mRNA was higher in IE, EML5 expression level in human epileptic brain is unclear (13). To determine whether this new type of microtubule disassembly protein is abnormally expressed in IE, we detected the levels of mRNA and protein in addition to the localization of EML5 in the surgically removed anterior temporal neocortex of patients with IE.
Materials and Methods
Experimental subjects
Thirty-six patients (mean age, 22.92 ± 8.94 years; range, 8-44 years) who were undergoing anterior temporal resections for medically diagnosed IE were randomly selected from the 200 patients in our epilepsy brain bank. The protocol of this study complied with the guidelines for the conduct of research involving human subjects by the National Institutes of Health (NIH, Bethesda, MD, USA) and the committee on human subjects’ research at the Chongqing Medical University of China. The patients or their relatives were asked to sign the informed and written consent for research studies.
The pre-surgical assessment consisted of a detailed history and neurological examination, interictal and ictal EEG, and neuropsychological and neuroradiological tests. The lesion was localized in all patients by brain magnetic resonance imaging (MRI) and 24 hr EEG or video-EEG. Sphenoidal electrode monitoring and intraoperative electrocorticography (ECOG) were performed to localize the epileptic lesion before resection in all patients. Surgical removal of the anterior epileptogenic zone was an alternative treatment option for the patients with epilepsy. The surgical procedure has been published previously (14). After the lesion was resected, the electrodes of the ECOG were placed on the remaining edge of tissue to ensure that the lesion was resected completely. The pathological findings in the resected neocortex included gliosis and neuronal loss. The clinical data for IE patients included in this study are shown in Table 1.
Table 1.
Treatment history and diagnosis of the 36 patients in the epilepsy group
| Patients | Age(yr) | M/F | Seizure at onset (y) | Antiepileptic drugs before surgery | Topography of studied tissue | Neuropathologic diagnosis | |
|---|---|---|---|---|---|---|---|
| 1 | 27 | F | 14 | CBZ>3m,TPM>5m, PHT=2y*, VPA=3y* | TL | R | NL |
| 2 | 13 | F | 4 | CBZ>4m,VPA=3y*, PB>1y*, PHT=9m | TL | L | NL |
| 3 | 12 | M | 6 | PHT>5m,VPA>5m*,CBZ>1y, PB>4y* | TL | L | Gliosis |
| 4 | 38 | F | 15 | PHT>5m,TPM>4m, PA>10m*,CBZ>1y | TL | R | Gliosis |
| 5 | 29 | F | 26 | PB>10m,VPA>5m*,CBZ>1y | TL | L | Gliosis |
| 6 | 17 | M | 7 | CBZ>3m,TPM>5m, PB=3y*, VPA=3y* | TL | R | NL |
| 7 | 13 | M | 8 | CBZ>4m,VPA=2y*, PB>1y*, PHT=9m | TL | L | NL |
| 8 | 38 | M | 19 | PHT>5m,VPA>5m*,CBZ>1y, PB>4y* | TL | R | NL |
| 9 | 25 | F | 10 | CBZ>1y*, VPA>4m*, PHT>3m, TPM>7m | TL | L | NL |
| 10 | 22 | F | 6 | PHT>3m, CBZ>5m*, TPM>1y*,PB>10m, VPA>15m | TL | L | Gliosis |
| 11 | 26 | M | 20 | LTG>5m, CBZ>14m, PB>8m*, VPA>9m* | TL | R | Gliosis |
| 12 | 24 | F | 19 | CBZ=5m,VPA>7m*, PB>6m, TPM>1y* | TL | R | NL |
| 13 | 22 | M | 9 | CBZ>11m*,VPA>4m*,PHT>9m, PB>7m, TPM>1y | TL | R | Gliosis |
| 14 | 35 | F | 12 | PB>4m*, CBZ>3m,VPA=5m,TPM>1y* | TL | L | Gliosis |
| 15 | 28 | M | 3 | CBZ >5m*,VPA>1y, PB>2y, TPM>1y* | TL | R | NL |
| 16 | 16 | F | 7 | TPM>6m*, VPA>10m, CBZ>8m*, PHT>8m | TL | L | Gliosis |
| 17 | 17 | M | 6 | PHT>8m *, CBZ>2y, VPA>9m,LTG>5m,TPM>4m | TL | R | NL |
| 18 | 15 | M | 9 | PHT>10m*,VPA>8m*,PB>1y* CBZ>8m,TPM>5m | TL | L | NL |
| 19 | 24 | M | 14 | PB>3m*, PRM>3m, CBZ>4m, VPA=5m* | TL | R | NL |
| 20 | 32 | F | 19 | CBZ >1y*,PB>2y,LTG>5m VPA>2y*, TPM>7m | TL | R | NL |
| 21 | 20 | F | 15 | CBZ=8m*, VPA>7m, TMP>5m *,PB>4m | TL | R | Gliosis |
| 22 | 8 | M | 1 | PB>5m, CBZ>7m*, PRM>6m | TL | L | NL |
| 23 | 44 | F | 22 | CBZ>5m*, VPA>7m*, LTG>5m, PB>7m | TL | R | NL |
| 24 | 12 | F | 6 | CBZ=1y, VPA>3m*, PB>5m, PHT>6m | TL | R | NL |
| 25 | 33 | M | 20 | CBZ >4m,VPA>4m, PHT>4m, TPM>5m*, LTG>6m | TL | R | Gliosis |
| 26 | 14 | M | 10 | VPA>5m*, CBZ>8m, PB>7m,TPM>6m,CLB>4m | TL | R | Gliosis |
| 27 | 15 | M | 12 | CBZ>2m, VPA>5m*, PHT>4m, PRM>3m | TL | R | NL |
| 28 | 21 | M | 11 | VPA>17m*, CLB>4m,CBZ>13m,PB>7m* | TL | R | NL |
| 29 | 26 | F | 5 | VPA>10m*, PHT>8m*, PRM>5m, CBZ>4y | TL | L | NL |
| 30 | 11 | F | 4 | TPM>4m,VPA>17m*, PB>10m, CBZ>12m* | TL | L | NL |
| 31 | 37 | M | 16 | VPA>8m*, PHT>3m, CBZ>8m,CLB>5m* | TL | L | NL |
| 32 | 18 | M | 13 | PB>5m*,PHT>5m*,VPA>6m*,CBZ>6m,TPM>5m | TL | L | NL |
| 33 | 29 | F | 20 | VPA >4m*,PHT>4m,TPM>5m*,PB>8m,LTG>5m | TL | L | NL |
| 34 | 16 | M | 7 | CBZ >5m*,VPA>4m*,PHT>10m*,TPM>5m,PB>5m | TL | L | NL |
| 35 | 21 | M | 8 | VPA>5m*,CLB>4m, CBZ=7m*, PB>3m,TPM>5m | TL | L | Gliosis |
| 36 | 27 | F | 14 | VPA>16m*,PHT>10m,CBZ>8m,TPM=7m,CLB>5m* | TL | L | NL |
* F, female; M, male; m, month; y, year; PHT, phenytoin; PRM, primaclone; CLB, clonazepam; VPA, valproic acid; CBZ, carbamazepine; PB, phenobarbital; TPM, topamax; LTG, lamotrigin; *AEDs which were taken before the operation; TL, temporal lobe; R, right; L, left; NL, neuronal loss
For comparison, eight histologically normal anterior temporal neocortex specimens were obtained following the anterior temporal lobectomy after increased intracranial pressure in individuals with head injury (mean age, 27.24±11.85 years; range, 17-51 years) and no history of epilepsy or exposure to AEDs. A conventional neuropathological examination did not reveal any evidence of central nervous system disease (cerebral ischemia, hemorrhage, or tumor).
Tissue processing
Tissue samples for immunohistochemistry and immunofluorescence were immediately immersion-fixed for 2 days at 4 °C with 4% paraformaldehyde in 0.1 M phosphate buffer at pH 7.4. The embedded tissues were successively sectioned at 5 μm for immunohistochemistry and 10 μm for immunofluorescence analysis. Samples for Western blot and FQ-PCR were freshly frozen on powdered dry ice and stored in liquid nitrogen until assayed.
Fluorescence quantitative polymerase chain reaction (FQ-PCR)
Total RNA was extracted with UNIzol reagent following the manufacturer’s protocol. The assay was performed as in our previous study. Briefly, complementary DNAs (cDNAs) were reverse transcribed and purified. The primer sequences were: EML5, forward primer 5’-TGATGATGTCGTCGCTGTGG-3’, and reverse primer 5’-CAGTGCCGCAACAACCTCTA-3’. The relative ratios were normalized to ribosomal 18s RNA. The reaction mixtures contained 2 µl of LightCycler FastStart DNA master SYBR Green mix, 0.5 µM of each primer, 4 mM MgCl2, 2 µl of DNA template, and excessive SYBR fluorescent dye. The reaction system was placed into a centrifugation FQ-PCR apparatus. Following activation of the polymerase at 94 °C for 2 min, 40 thermocycles were run with 10 sec of denaturation at 94 °C, 10 sec of annealing at 61 °C, and 10 sec of extension at 72 °C. The gene copy number was calculated by the Ct value approach (i.e., mean value of triplicate results), and the results were the ratios of the gene copy number to the copy number of the 18s RNA housekeeping genes.
Immunohistochemistry and immunofluorescence
All formalin-fixed paraffin-embedded sections were dewaxed in xylene and rehydrated in graded ethanol solutions. The steps were the same as described previously (13). The primary anti-EML5 antibody (1:100) was kindly provided by Professor French (12) and was applied to the sections at 4 °C overnight. The immunoreaction visualization and negative controls setup were performed according to standard procedures.
The sections were deparaffinized, and antigen recovery was performed as described above. After permeabilizing in 0.1% Triton X-100 in PBS for 20 min, the sections were blocked in PBS/2% bovine serum albumin for 20 min at room temperature, followed by incubation with anti-EML5 at 1:100 dilution in PBS at 4 °C for 24 hr. For the protein staining, the sections were incubated with fluorescein isothiocyanate for 40 min and then mounted in 50% glycerol and 50% PBS. The results were observed by confocal microscopy (Leica Microsystems Heidelberg GmbH, Germany).
Western blot
To examine the EML5 protein levels, Western blotting was performed as previously described (14). The membranes were incubated overnight in a solution of rabbit anti-EML5 IgG (1:200). Incubation with anti-β-actin antibody (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA) confirmed equal protein loading during Western blotting. The quantification of immunoblots was performed with BioRad Fluor-S™ MultiImager and Quantity One 4.6 software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All results were expressed as the mean±SEM (standard error of mean) and significant differences were determined by t-test (using the SPSS v11.0 software) for FQ-PCR, immunohistochemistry, immunofluorescence, and Western blot analysis. Any differences between groups were considered significant at an alpha value of P< 0.05.
Results
Comparison of the clinical characteristics
Patients with pharmacoresistant epilepsy were deemed suitable candidates for surgery by virtue of their intractable condition and the concordance of conventional presurgical evaluation procedures, including clinical, neuropsychological, EEG, and qualitative imaging tests. In our study, 66.7% of the patients had cortical neuronal loss. Most patients (i.e., 92.5%) had at least 5 years history of seizure recurrence. About 52.5% of patients had a clinical history of more than 10 years. Table 2 summarizes the demographic and clinical characteristics of the subjects who participated. There were no significant differences in terms of age, gender, and topography of studied tissues between the IE patients and control subjects.
Table 2.
Clinical characteristics of the subjects
| Epilepsy (n = 36) | Control (n =8) | Results | ||
|---|---|---|---|---|
| Age | 22.92 ± 8.94 | 27.24± 11.85 | t = 1.165, P =0.2506 | |
| Sex | ||||
| Female | 17 (47.2%) | 5 (62.5%) | χ2= 0.1528 P =0.6959 | |
| Male | 19(52.8%) | 3(37.5%) | ||
| Topography of studied tissue | ||||
| Left | 18 (50%) | 5 (62.5%) | χ2 = 0.0620 P = 0.8034 | |
| Right | 18 (50%) | 3 (37.5%) | ||
EML5 mRNA was increased in the anterior temporal neocortex of patients with IE
The concentrations of the FQ-PCR products amplified from epileptic tissues mRNA rapidly achieved a Ct value after 40 amplified reaction cycles when compared to control subjects. Figure 1A, shows the SYBR fluorescence amplification curve and standard curve. Figure 1B, shows the mean increase of EML5/18s between the epileptic tissues and the control group. The mean relative expression in the epileptic and control tissue was 0.08262±0.0095 and 0.01428±0.0022, respectively (i.e., more than a five-fold change).
Figure 1.
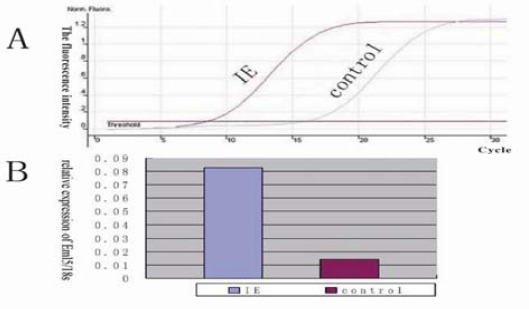
FQ-PCR analysis of EML5 mRNA in the anterior temporal lobe of IE and control patients. (A) SYBR fluorescence amplification curve; (B) The relative expression of EML5 mRNA in IE patients was higher than that in the control (greater than a five-fold change)
EML5 staining was prominent in the anterior temporal neocortex of patients with IE
Immunostaining of brain sections from IE patients with anti-EML5 antibodies revealed EML5-positive neurons and glial cells predominantly in the anterior temporal neocortex of patients with IE (Figure 2 A, C, and D). Such neurons and glial cells were detected much less frequently in the normal control anterior temporal neocortex tissue (Figure 2B and E). No immunoreactivity was observed in the temporal cortex tissue of negative controls in which the primary antibody had been omitted. The optical density (OD) values for the epileptic and control tissues were determined to be 0.4251±0.0855 and 0.1919±0.0769, respectively. The results showed markedly higher EML5 in epileptic tissue compared with the controls (P<0.05). Positive cells were immunologically cross-reactive for both glial cells and neurons (Figure 2).
Figure 2.
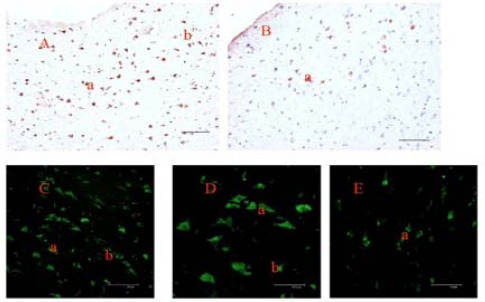
Immunohistochemical staining of the anterior temporal neocortex with antibody targeted to EML5. EML5 was abnormally expressed in the temporal neocortex of IE patients (A, C, and D), whereas minimal immunoreactive protein was found in the normal temporal neocortex (B, and E); a, neuron; b, glial cell; DAB staining (A, and B); Immunofluorescence staining (C, D, and E) (Bar: A, B, C, and E=75 μm; D=37.5 μm)
Higher EML5 protein levels occurred in the anterior temporal neocortex of patients with IE
To further confirm the increase in immune-histochemical staining of the epilepsy brain slices, we tested EML5 levels by Western blot (Figure 3). Immunoblotting was performed with an EML5 antibody. The protein was expressed as a band at 180 kDa while faintly stained bands were detected in control tissue samples, whereas relatively strong bands of EML5 were observed in the epileptic group. Another band of β-actin as control at 42 kDa was also observed in each channel, but no differences were observed among the groups. Figure 3A, shows the results of X-ray film development of EML5 staining for both groups. Figure 3B, shows the ratio of the OD of EML5 and β-actin. The OD values in the epileptic tissue and in the control tissue were 1.8030±0.1335 and 1.1852±0.2253, respectively. The results revealed a significant difference between the tissues from the epileptic and control groups (P<0.05).
Figure 3.
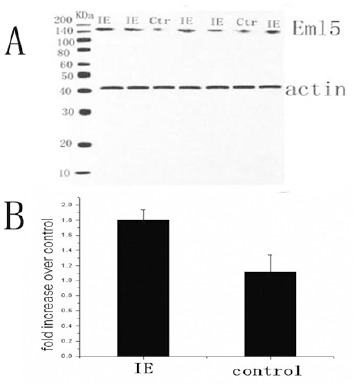
A: Western blot analysis of the EML5 expression in the anterior temporal lobe of patients with IE. A representative EML5 immunoblot that was loaded with homogenates from the control and IE cases is shown. Molecular weight markers (MW, kDa) are indicated on the left. IE, intractable epilepsy; Ctr, control. B: Semi-quantitative analysis of EML5 in brain homogenates of IE (P<0.05)
Discussion
Here, for the first time, we described the expression pattern of EML5 mRNA and protein in the anterior temporal neocortex of patients with IE. EML5 was primarily expressed in neuronal and glial cells. EML5 expression in the anterior temporal neocortex of patients with IE was markedly higher than that in the control group. Because EML5 regulates the function of microtubules, it may lead to the seizure recurrence in IE patients.
The absence of control tissue sections from normal living human hippocampus and from the temporal lobe made it difficult to investigate the complex nature of epilepsy. Because of the ethical issues and difficulty in obtaining normal human hippocampus specimens from patients undergoing resection surgeries, structurally normal brain sections obtained from head traumatic patients who had undergone anterior temporal decompression procedures were used as control samples, similar to previous study (15).
The most important findings in the pathophysiology of IE were neuronal loss and gliosis which are the consequences of seizure activity that can lead to the impairment of cells (16, 17). The neuronal loss was observed in the patients with epilepsy, particularly in patients with recurrent and state epilepsy (18-21). In our study, the high rates (66.7%) of neuronal loss might be a consequence of the long course of the disease. Most of the patients (92.5%) presented with recurrent seizures for at least 5 years, whereas 52.5% of cases had experienced recurrences more than 10 years.
IE may be self-perpetuating in some individuals and may potentially cause irreversible neuroplastic changes such as dendritic sprouting, glial proliferation, and cell death (22, 23). All reported changes have been associated with the abnormal expression of microtubule-associated protein (MAP). In addition, MAP1A, MAP2, MAP1B, and LAMB1 (laminin-beta 1) were previously reported to be differentially regulated in epileptogenesis/chronic epilepsy (8, 24). Furthermore, MAP1A and MAP1B displayed a differential affinity for tubulin. The higher MAPs levels could increase the interaction of MAPs and microtubules. For example, MAP2 can compete with MAP1B for binding to tubulin, but not with MAP1A. The longitudinal binding of MAP2/tau proteins to protofilaments leads to microtubule stabilization and does so by bridging tubulin interfaces. By contrast, MAPs can regulate their binding affinity for microtubules by phosphorylation.
EMLs are a type of microtubule disassembly protein and are expressed in the central nervous system (25, 26). Furthermore, EML4 negative cells show a completely modified microtubule network which indicate that EML4 is necessary for correct microtubule formation (27). EML5 is predominantly expressed in the gyrus dentatus, the CA1 and CA3 regions of the hippocampus, olfactory bulb, and cerebral cortex, both during neuronal development and in the adult brain (12, 27, 28). Many of these regions are sensitive to epileptic attack. Furthermore, EML5-positive neurons show enhanced microtubule dynamics compared with EML5-negative neurons (12).
Evidence to support this assumption has primarily been derived from “epileptic” brain tissues that were removed during surgical procedures from epileptic patients with drug-resistant epilepsy (3). In our study, EML5 in the anterior temporal neocortex of patients presenting with IE was markedly higher than that found in normal control tissues. The exact role of EML5 in IE remains unclear. However, from the perspective of the pathological role of EML5, we hypothesize that the overexpression of EML5 promotes the extension of neuronal dendrites and axons and then stimulates the budding of neurons. Collectively, these effects likely promote pathological neural network plasticity and excitatory synapse reconstruction.
In terms of clinical applications, this study updates our understanding of the pathogenesis of epilepsy and offers hope of finding an objective biological index for the diagnosis of drug-resistant epilepsy. There is an urgent need to identify therapeutic targets for the development of novel antiepileptic drugs and to test new ideas for the treatment of drug-resistant epilepsy.
Conclusion
Altogether, our study demonstrates that EML5 mRNA and protein are more highly expressed in the anterior temporal neocortex of patients with IE. However, the role of EML5 in seizure recurrence remains unclear. In view of the mechanism of EML5, the overexpression of EML5 may affect the assembly of microtubules, contribute to the process of neuron axonal outgrowth and sprouting, and lead to the recurrence of seizure.
Acknowledgment
The authors thank Professor French PJ (Department of Neurology, Erasmus Medical Centre, Netherlands) for providing the EML5-specific antibody. We also acknowledge the support of the patients and their families for participating in this study. We would also like to thank the support of the local ethics committee and the National Board of the Medical Affairs.
References
- 1.Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, et al. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:470–472. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- 2.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- 3.Kwan P, Brodie MJ. Refractory epilepsy: a progressive, intractable but preventable condition? Seizure. 2002;11:77–84. doi: 10.1053/seiz.2002.0593. [DOI] [PubMed] [Google Scholar]
- 4.Brodie MJ. Diagnosing and predicting refractory epilepsy. Acta Neurol Scand Suppl. 2005;181:36–39. doi: 10.1111/j.1600-0404.2005.00507.x. [DOI] [PubMed] [Google Scholar]
- 5.Kwan P, Brodie MJ. Potential role of drug transporters in the pathogenesis of medically intractable epilepsy. Epilepsia. 2005;46:224–235. doi: 10.1111/j.0013-9580.2005.31904.x. [DOI] [PubMed] [Google Scholar]
- 6.Thom M, Sisodiya SM, Beckett A, Martinian L, Lin WR, Harkness W, et al. Cytoarchitectural abnormalities in hippocampal sclerosis. J Neuropathol Exp Neurol. 2002;61:510–519. doi: 10.1093/jnen/61.6.510. [DOI] [PubMed] [Google Scholar]
- 7.Longo B, Vezzani A, Mello LE. Growth-associated protein 43 expression in hippocampal molecular layer of chronic epileptic rats treated with cycloheximide. Epilepsia. 2005;46:125–128. doi: 10.1111/j.1528-1167.2005.01019.x. [DOI] [PubMed] [Google Scholar]
- 8.Tang L, Lu Y, Zheng W, Li Y. Overexpression of MAP-2 via formation of microtubules plays an important role in the sprouting of mossy fibers in epileptic rats. J Mol Neurosci. 2014;53:103–108. doi: 10.1007/s12031-013-0204-4. [DOI] [PubMed] [Google Scholar]
- 9.An SJ, See MO, Kim HS, Park SK, Hwang IK, Won MH, et al. Accumulation of microtubule-associated proteins in the hippocampal neurons of seizure-sensitive gerbils. Mol Cells. 2003;15:200–207. [PubMed] [Google Scholar]
- 10.Yang JW, Czech T, Felizardo M, Baumgartner C, Lubec G. Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids. 2006;30:477–493. doi: 10.1007/s00726-005-0281-y. [DOI] [PubMed] [Google Scholar]
- 11.Eichenmuller B, Everley P, Palange J, Lepley D, Suprenant KA. The human EMAP-like protein-70 (ELP70) is a microtubule destabilizer that localizes to the mitotic apparatus. J Biol Chem. 2002;277:1301–1309. doi: 10.1074/jbc.M106628200. [DOI] [PubMed] [Google Scholar]
- 12.O’Connor V, Houtman SH, De Zeeuw CI, Bliss TV, French PJ. Eml5, a novel WD40 domain protein expressed in rat brain. Gene. 2004;336:127–137. doi: 10.1016/j.gene.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Xi ZQ, Xiao F, Yuan J, Wang XF, Wang L, Quan FY, et al. Gene expression analysis on anterior temporal neocortex of patients with intractable epilepsy. Synapse. 2009;63:1017–1028. doi: 10.1002/syn.20681. [DOI] [PubMed] [Google Scholar]
- 14.Mizrahi EM, Kellaway P, Grossman RG, Rutecki PA, Armstrong D, Rettig G, et al. Anterior temporal lobectomy and medically refractory temporal lobe epilepsy of childhood. Epilepsia. 1990;31:302–312. doi: 10.1111/j.1528-1157.1990.tb05380.x. [DOI] [PubMed] [Google Scholar]
- 15.Sisodiya SM, Martinian L, Scheffer GL, van der Valk P, Cross JH, Scheper RJ, et al. Major vault protein, a marker of drug resistance, is upregulated in refractory epilepsy. Epilepsia. 2003;44:1388–1396. doi: 10.1046/j.1528-1157.2003.21803.x. [DOI] [PubMed] [Google Scholar]
- 16.Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, et al. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- 17.Thom M, Zhou J, Martinian L, Sisodiya S. Quantitative post-mortem study of the hippocampus in chronic epilepsy: seizures do not inevitably cause neuronal loss. Brain. 2005;128:1344–1357. doi: 10.1093/brain/awh475. [DOI] [PubMed] [Google Scholar]
- 18.Arroyo S, Brodie MJ, Avanzini G, Baumgartner C, Chiron C, Dulac O, et al. Is refractory epilepsy preventable? Epilepsia. 2002;43:437–444. doi: 10.1046/j.1528-1157.2002.38501.x. [DOI] [PubMed] [Google Scholar]
- 19.Dawodu S, Thom M. Quantitative neuropathology of the entorhinal cortex region in patients with hippocampal sclerosis and temporal lobe epilepsy. Epilepsia. 2005;46:23–30. doi: 10.1111/j.0013-9580.2005.21804.x. [DOI] [PubMed] [Google Scholar]
- 20.Gomes FG, Gomes Da Silva S, Cavalheiro EA, Arida RM. Beneficial influence of physical exercise following status epilepticus in the immature brain of rats. Neuroscience. 2014;274C:69–81. doi: 10.1016/j.neuroscience.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 21.Nomura S, Shimakawa S, Miyamoto R, Fukui M, Tamai H. Methyl-1-phenyl-2-pyrazolin-5-one or N-acetylcysteine prevents hippocampal mossy fiber sprouting and rectifies subsequent convulsive susceptibility in a rat model of kainic acid-induced seizure ceased by pentobarbital. Brain Res. 2014;1590:65–74. doi: 10.1016/j.brainres.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 22.Pitsch J, Opitz T, Borm V, Woitecki A, Staniek M, Beck H, et al. The presynaptic active zone protein RIM1alpha controls epileptogenesis following status epilepticus. J Neurosci. 2012;32:12384–12395. doi: 10.1523/JNEUROSCI.0223-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan XX, Cai Y, Zhang XM, Luo XG, Cai H, Rose GM, et al. BACE1 elevation is associated with aberrant limbic axonal sprouting in epileptic CD1 mice. Exp Neurol. 2012;235:228–237. doi: 10.1016/j.expneurol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Y, Wang XF, Mo XA, Li JM, Yuan J, Zheng JO, et al. Expression of laminin beta1 and integrin alpha2 in the anterior temporal neocortex tissue of patients with intractable epilepsy. Int J Neurosci. 2011;121:323–328. doi: 10.3109/00207454.2011.558224. [DOI] [PubMed] [Google Scholar]
- 25.Cassimeris L. The oncoprotein 18/stathmin family of microtubule destabilizers. Curr Opin Cell Biol. 2002;14:18–24. doi: 10.1016/s0955-0674(01)00289-7. [DOI] [PubMed] [Google Scholar]
- 26.Hueston JL, Herren GP, Cueva JG, Buechner M, Lundquist EA, Goodman MB. The C. elegans EMA-like protein, ELP-1 is required for touch sensation and associates with microtubules and adhesion complexes. BMC Dev Biol. 2008;8:110. doi: 10.1186/1471-213X-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollmann M, Parwaresch R, Adam-Klages S, Kruse ML, Buck F, Heidebrecht HJ. Human EML4, a novel member of the EMAP family, is essential for microtubule formation. Exp Cell Res. 2006;312:3241–3251. doi: 10.1016/j.yexcr.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 28.Bechstedt S, Albert JT, Kreil DP, Muller-Reichert T, Gopfert MC, Howard J. A doublecortin containing microtubule-associated protein is implicated in mechanotransduction in Drosophila sensory cilia. Nat Commun. 2010;1:11. doi: 10.1038/ncomms1007. [DOI] [PMC free article] [PubMed] [Google Scholar]