

Abstract
Objective(s):
Lead (Pb) is a toxic metal inducing many destructive effects leading to a broad range of physiological, biochemical, and neurological dysfunctions in humans and laboratory animals.
Materials and Methods:
Here, we investigated the effect of chronic exposure to Pb (50 mg/l) on oxidative stress, hepatotoxicity, nephrotoxicity, and lipid profile of two different age groups of female rats treated with Pb from delivery until puberty period (40 days, Pb40) and post puberty period (65 days, Pb65).
Results:
Our results clearly show that the administration of Pb produces oxidative damage in liver and kidney, as strongly suggested by the significant increase in TBARS, decrease in total SH, and the alteration of SOD activity. Elevation in liver function biomarkers, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) and reduction in total protein (liver and plasma) and albumin are evidence of perturbations of liver synthetic function. In young Pb-treated group, Pb-induced nephropathy was more pronounced by the increase in the levels of creatinine, urea, and uric acid. However, hyperlipidemia was evident for both Pb-exposed groups leading to a potential risk for cardiovascular diseases and atherosclerosis.
Conclusion:
It is concluded that Pb induces metabolic and oxidative disturbances depending on the age of the animals, which are not negligible.
Keywords: Female, Hepatotoxicity Hyperlipidemia, Lead, Nephrotoxicity, Rat
Introduction
Environment pollution has become a major public concern. Water, air, and soil are all affected by pollution, whether anthropogenic (engine emissions of factories, sewage treatment plant, etc.) or natural (volcanic eruptions, erosion, etc.). On the other hand, population and industrial growth contributes to a considerable part for the contamination of the environment. Thus, the increase in the prevalence of certain diseases in developed and even developing countries is at the heart of social media and news (1).
Humans have been using Lead (Pb) since ancient times and the quantity of Pb used in the 20th century far surpasses the total consumption in all previous eras (2). Pb is a toxic metal to humans and animals and due to its persistency, it remains for long time in environment water, soil, dust, and in manufactured products containing Pb. Indeed, this environmental toxic metal enters the body through the digestive tract, mainly via intake of food and drinking water, or inhalation of airborne contaminated dust. Once absorbed, Pb diffuses rapidly through the bloodstream to various organs such as brain, liver, kidney, and to highly calcified tissues as teeth and bone.
The occupational exposure to Pb for the first time drew attention to its deleterious effects on workers and on serious consequences, particularly neuropsychiatric. Nowadays, acute occupational Pb poisoning is becoming a less important issue due to better working conditions and significant improvements in protective standards (3).
However, the developmental toxicity of Pb has become a significant area of research since children are much more sensitive than adults for learning impairment following low-level Pb exposure (4). Indeed, children are more vulnerable to Pb exposure for three main reasons: (i) young children are more at risk of ingesting environmental lead through normal mouthing behaviors (3), (ii) the average fractional gastrointestinal absorption of Pb is much greater in infants and young children than in adults (5), and (iii) the developing nervous system is thought to be far more vulnerable to the toxic effects of Pb than the mature brain (6).
Target organs in children are essentially the same as in adults, but some clinical manifestations of Pb effects are unique for the first aging group; furthermore, they can occur at levels much lower than in adults. Chronic low level of Pb exposure during this period reduces the IQ level significantly, and unfortunately, most of the neurobehavioral impacts are irreversible (7). Indeed, blood Pb levels of 10 µg/dl (equivalent to 0.48 µmol/l) or higher are considered toxic and result in neurological disorders, cognitive impairments, hypertension, and other disorders (8).
Lead-induced oxidative stress or disruption of prooxidant/antioxidant balance in blood and other soft tissues has been postulated to be the major mechanism of Pb associated tissue injury (9). It causes oxidative stress by inducing the generation of reactive oxygen species (ROS) (10), increasing the level of lipid peroxidation and thiobarbituric acid reactive substances (TBARS) (8, 10), and inhibiting the activity of many antioxidant enzymes, including glutathione (11, 12). Consequently, Pb alters the antioxidant defense system of cells, resulting in pathophysiological events in various body organs.
The liver and the kidney, being organs playing a vital part in the metabolism of Pb, are at special risk of damage. Indeed, liver which is responsible for maintaining the body’s metabolic homeostasis has been considered as the target organ for the toxic effects of Pb (13). Liver is the largest repository of Pb among soft tissues followed by kidney (10).
The absorbed Pb is conjugated in the liver and passed to the kidney, where a small quantity is excreted in urine and the rest is accumulated in various body organs and affects many biological activities at the molecular, cellular, and intercellular levels, which may result in morphological alterations that can remain even after Pb levels have fallen (14, 15).
Lipid disturbances have been classified as both principal and potential risk factors for cardiovascular diseases. Increased levels of total and LDL cholesterol were included with the principal risk factors for circulatory diseases. The new risk factors for cardiovascular diseases include an elevated level of triglycerides (TGs), the presence of small and dense LDL, and an increased concentration of lipoprotein (a) (Lpa). Currently, it is assumed that in persons exposed to Pb compounds, the lipid disturbances, representing risk factors for cardiovascular diseases, are manifested with a high frequency. Therefore, it can be concluded that Pb and its compounds induce pro-atherosclerotic effects in the lipid profile (16).
Based on the above considerations, this study was carried out to investigate the effects of chronic exposure to Pb on oxidative stress parameters in liver and kidneys of two different age groups of Wistar female rats treated with Pb from delivery until puberty period (young individuals) and post puberty period (adults). For this purpose, the concentrations of TBARS, total thiol group (SH), and SOD activity were measured in the liver and kidney of rats. Because excessive amounts of Pb may also cause nephropathy, liver dysfunction, and lipid profile disturbances, some relative metabolic parameters were determined to assess the level of toxicity in these animals.
Materials and Methods
Chemicals
Lead (II) acetate and phosphate ammonium (NH4)2 HPO4 were purchased from PROLABO (France). Butylated hydroxytoluene, 2-thiobarbituric acid (TBA), Coomassie
G250, 5,5’-dithiobis (2-nitrobenzoic acid) (DTNB), bovine serum albumin (BSA), Triton X-100, and the stock solution of Pb (1 x g Pb/l) were obtained from Sigma Chemicals (Germany). Concentrated pure acid nitric (HNO3, 69%) was obtained from Merck (Darmstadt, Germany). All other chemicals were of the highest grade available.
Determination of optimum dose
The choice of Pb dose was based on previous tests initiating with very high starting doses. The chosen dose of 50 mg Pb/l corresponds to an acceptable dose that does not cause any sign of toxicity until the end of the experimental period. Indeed, when the experimental protocol is established, the amount of administered Pb increases blood lead levels which can be detectable at bone level after 40 days of exposure.
Experimental animals
Experiments were started in female Wistar rats (200–220 g body weight) provided from society of pharmaceutical industries of Tunisia (SIPHAT, Tunis, Tunisia). After 1 week as adaptation period in a room with controlled temperature (22 °C ± 2 °C) and lighting (12-hr light–dark), they were mated with males.
A sperm-positive vaginal smear was taken to indicate the first day of pregnancy. From that moment, dams were housed individually in cages.
Beginning at lactational day 1, dams were given drinking water containing 50 mg/l of lead acetate until weaning. Control dams were given tap water. Thereafter, the offspring were exposed to the same solutions throughout the experiment, from weaning (day 21) until sacrifice. Female pups were housed in individually (n = 12–16) and then were sacrificed at 40 days of age (puberty, Pb40) and at 65 days of age (post puberty, Pb65) with their relative controls based on developmental stages given by Ronis et al (17).
During the experimental period, animals had access to food and water ad libitum; water consumption and weight gain were recorded every day. However, we were unable to determine milk intake for each animal per day; furthermore, it was not part of our objectives.
At the end of the treatment, animals were killed by decapitation without preliminary anesthesia, and whole blood was taken quickly from their neck. The liver and the right kidney were removed by transverse abdominal incision and kept frozen at -80 °C.
Blood lead determination
Pb concentration in whole blood samples (0.1 ml) was directly determined by graphite furnace atomic absorption spectroscopy method, after addition of matrix modifier (20 g/l (NH4)2 HPO4, 2% Triton X-100) without digestion (18). These measures were implemented using a ZEEnit 700-Analytik-Jena, Germany, equipped with deuterium and Zeeman background correction. Photometry was performed at a wavelength of 283.3 nm using a lead hollow cathode lamp. Calibration curve was obtained from the standard solutions prepared from a certified stock solution of Pb (1g Pb/l) and which corresponded to concentrations of 5, 10, 15, and 20 µg Pb/100 ml blood. Duplicate determinations were carried out for each sample and standards. Results were expressed as micrograms Pb per 100 ml of blood.
Hematological parameters
Hemoglobin was measured colorimetrically according to Drabkin and Austin’s method (19) through its transformation to cyanmethemoglobin under the action of potassium ferricyanide and potassium cyanide. Hemoglobin concentration was expressed in g/l.
Hematocrit measurements were carried out in capillary tubes centrifuged with HEMATOCRIT 20 Hettich for 15 min at 1000 rpm.
Glucose was measured by the glucose oxidase and peroxidase using quinoneimine as a chromogen. The amount of plasma glucose is related to amount of quinoneimine which is measured spectrophotometrically at 505 nm (20).
Hepatic markers of damage
Plasma activities of alanine aminotransferase (ALT) (EC 2.6.1.2) and aspartate aminotransferase (AST) (EC 2.6.1.1) were determined spectrophotometrically according to Bergmeyer et al (21). Changes in the absorbance were determined at 340 nm. Results were expressed in IU/l. Plasma alkaline phosphatase (ALP) (EC 3.1.3.1) activity was determined according to Price and Woodman (22). The absorbance was read at 405 nm, and the results were expressed in IU/l. Plasma albumin was determined according to Doumas et al (23). The reaction mixture was monitored spectrophotometrically at 628 nm. Results were expressed in g/l. All reagents and chemicals were obtained from Biomaghreb Laboratories, Tunis, Tunisia.
Renal markers of damage
Renal function was analyzed by determining creatinine (24), uric acid (25), and urea (26) levels in plasma. Samples, standards and controls, were each added to 1 ml reaction mixture and read spectrophotometrically at 492, 510, and 340 nm, respectively. All reagents and chemicals were obtained from Biomaghreb Laboratories, Tunis, Tunisia. Results were expressed in mg/l.
Oxidative stress parameters
TBARS concentration
TBARS were measured according to Oteiza et al (27) with slight modifications. A mixture of 150 µl of tissue homogenate, 0.5 ml of 30 g/l sodium dodecyl sulfate, 2 ml of 0.1 M HCL, 0.3 ml of 10 g/l phosphotungstic acid, and 1 ml of 7 g/l TBA was incubated in boiling water for 30 min. After cooling, 5 ml of 1-butanol was added. The organic layer was collected after centrifuging at 1000 g for 10 min at 4 °C. The absorbance was measured at 532 nm and compared with a standard curve constructed with known concentrations of 1,1,3,3-tetramethoxypropane. The data were expressed as nmol MDA/mg protein.
Total thiols (SH) level
An aliquot of supernatant (50 µl) was mixed with 1 ml of the Tris base (0.25 M)—EDTA (20 mM) buffer, pH 8.2, and the absorbance at 412 nm was measured; 20 µl of 10 mM DTNB was also added. After 15 min at ambient temperature, the absorbance was measured again with a DTNB blank. Results were expressed in [mM] (28).
SOD activity
SOD (EC 1.15.1.1) activity was estimated according to Mirsa and Fridovich (29). This method is based on the capacity of SOD in inhibiting autoxidation of epinephrine to adrenochrome. The color reaction was measured at 480 nm. One unit of enzyme was defined as the amount of enzyme required to inhibit the rate of autooxidation by 50% at 25 °C. Specific activity was defined as units per milligram protein (U/mg protein).
Protein assay
The protein content of liver and kidney supernatants and plasma was spectrophotometrically estimated according to the method of Biuret using BSA as standard (30).
Lipid status
Serum total cholesterol (TC), high density lipoprotein-cholesterol (HDL-Chol), low density lipoprotein-cholesterol (LDL-Chol), and TGs concentrations were determined using the corresponding diagnostic kits set by Randox Laboratories Ltd. (UK). In exploitation of lipid metabolism, we evaluated the cardiovascular risk factors as TC/HDL ratio and TG/HDL ratio. The atherogenic index (AI) was calculated as (TC–HDL)/HDL.
Statistical analysis
All results were expressed as mean±standard deviation. Comparisons between the groups were performed by one way ANOVA followed by Student’s t test. Differences were considered significant at P <0.01.
Results
Neither toxic signs nor changes in the behavior of the animals during the treatment were observed. Likewise, there were no specific signs attributable to the treatment with Pb, and mortality was not increased in any of the experimental groups over than in controls.
The amounts of metal ingested during the experimental periods (40 and 65 days, respectively) are shown in Table 1; these were calculated using this formula: [(ml water consumed/day) x metal dose]/rat weight (kg). This table also shows the concentrations of lead in the blood of experimental animals treated with Pb.
Table 1.
Lead consumption and blood lead levels in female rats treated with lead (50 mg/l) for 40 and 65 days
| Metal consumption (mg Pb/Kg b.w/day) | Blood lead level (µg/dl) | |||
|---|---|---|---|---|
| Control | Lead | Control | Lead | |
| 40 days | - | 11.26 ± 0.41 * | 1.5 ± 0.26 | 12.86 ± 0.98 * |
| 65 days | - | 8.83 ± 0.27 *,# | 1.42 ± 0.31 | 8.37 ± 0.63 *,# |
Values are means±SD for groups consisted of 12–16 rats. Statistically significant differences are indicated
P<0.01 compared with control group,
P<0.01 compared with (Pb40) group
Exposure to Pb from lactation had no effect on growth parameters (Table 2) of all treated animals when compared to their relative controls.
Table 2.
Growth parameters, absolute and relative liver and kidney weights in female rats treated with lead (50 mg/l) for 40 and 65 days
| Control 40 days | Lead 40 days | Control 65 days | Lead 65 days | |
|---|---|---|---|---|
| Growth parameters | ||||
| Initial body weight (g) | 5.75 ± 0.13 | 5.75 ± 0.15 | 5.56 ± 0.18 | 5.66 ± 0.10 |
| Final body weight (g) | 104.37 ± 2.10 | 103.96 ± 2.07 | 160 ± 3.43 | 167.67 ± 2.43 |
| B.W. gain (g) | 99.56 ± 2.02 | 98.21 ± 1.98 | 154.43 ± 3.48 | 160.9 ± 3.48 |
| Body surface area (g 0.7) | 26.03 ± 0.33 | 25.77 ± 0.37 | 34.78 ± 0.55 | 36.06 ± 0.39 |
| Food intake (g/Kg/day) | 14.07 ± 0.20 | 14.92 ± 0.37 | 11.97 ± 0.11 | 12.68 ± 0.18 |
| Fluid intake (ml/kg/day) | 22.60 ± 0.62 | 22.51 ± 0.82 | 17.86 ± 0.84 | 17.65 ± 0.55 |
| Energy intake (Kcal/day) | 44.10 ± 0.62 | 44.77 ± 1.12 | 35.91 ± 0.33 | 38.06 ± 0.55 |
| Feed efficiency (g/Kcal) | 2.30 ± 0.06 | 2.20 ± 0.05 | 4.45 ± 0.14 | 4.37 ± 0.08 |
| Organs weights (g) | ||||
| Liver weight | 6.31 ± 0.14 | 6.26 ± 0.14 | 6.87 ± 0.20 | 7.40 ± 0.20 a |
| Kidney weight | 0.62 ± 0.01 | 0.62 ± 0.01 | 0.64 ± 0.02 | 0.71 ± 0.01 b |
Values are mean±SD for groups consisted of 12–16 rats. Statistically significant differences are indicated
P <0.05,
P <0.01 compared with control group
Table 2 also shows the effect of Pb on the absolute (g) and relative (g/100 g b.w) weight of the liver and the kidney. At day 65, there was a significant increase of the both organs weight and relative weight when compared to controls.
Table 3 summarizes the data concerning the effects of Pb exposure on hematological parameters. Hemoglobin concentration was significantly reduced in all treated groups in comparison with controls. However, no significant changes were found in hematocrit value, glucose content, and plasma proteins level of all intoxicated animals at day 40 and day 65 of treatment.
Table 3.
Hematological parameters in female rats treated with lead (50 mg/l) for 40 and 65 days
| Control 40 days | Lead 40 days | Control 65 days | Lead 65 days | |
|---|---|---|---|---|
| Hemoglobin (g/l) | 108.12 ± 3.5 | 98.13 ± 1.9 a | 100.42 ± 3.56 | 88.98 ± 2.67 b |
| Hematocrit (%) | 40.25 ± 0.33 | 39.15 ± 0.39 | 42 ± 0.39 | 42.08 ± 0.32 |
| Glucose (g/l) | 1.51 ± 0.02 | 1.51 ± 0.02 | 1.29 ± 0.03 | 1.37 ± 0.02 |
| Proteins (g/l) | 52.42 ± 1.47 | 51.94 ± 2.2 | 55.24 ± 0.91 | 55.12 ± 0.75 |
Values are mean±SD for groups consisted of 12–16 rats. Statistically significant differences are indicated
P<0.05,
P<0.01 compared with control group
Liver function evaluation
Pb administration caused a significant increase in the activities of AST and ALP of both treated groups (Pb40 and Pb65), when compared with their relative controls. Furthermore, Pb administration caused a significant (P <0.05) increase in the activity of ALT, only in Pb65 group when compared to controls.
The data indicated also a significant (P<0.01) decrease in liver proteins level, in all treated groups together with a significant (P<0.05) decrease in albumin level in Pb65 group comparing to their relative controls (Table 4).
Table 4.
Hepatic and kidney markers of damage in female rats treated with lead (50 mg/l) for 40 and 65 days
| Control 40 days | Lead 40 days | Control 65 days | Lead 65 days | |
|---|---|---|---|---|
| Hepatic markers of damage | ||||
| ALT (IU/l) | 44.55 ± 0.98 | 44.23 ± 2.48 | 42 ± 1.49 | 51.72 ± 4.45 a |
| AST (IU/l) | 116 ± 6.86 | 132.87 ± 3.26 a | 107.07 ± 2.53 | 132.68 ± 4.61 c |
| ALP (IU/l) | 285 ± 12.26 | 411.58 ± 40.92 b | 558.67 ± 20.67 | 632.87 ± 31.58 a |
| Albumin (g/l) | 39.5 ± 1.03 | 37.38 ± 0.77 | 39.93 ± 0.64 | 37.72 ± 0.99 a |
| Proteins (g/100 g) | 9.23 ± 0.39 | 7.46 ± 0.46 b | 8.84 ± 0.28 | 7.29 ± 0.36 b |
| Kidney markers of damage | ||||
| Urea (mg/l) | 0.36 ± 0.01 | 0.45 ± 0.01 c | 0.29 ± 0.01 | 0.31 ± 0.01 |
| Creatinine (mg/l) | 4.26 ± 0.21 | 5.43 ± 0.18 c | 5.13 ± 0.19 | 6.39 ± 0.20 c |
| Uric acid (mg/l) | 8.56 ± 0.25 | 10.80 ± 0.33 c | 6.51 ± 0.59 | 5.97 ± 0.31 |
| Proteins (g/100 g) | 2.89 ± 0.21 | 2.68 ± 0.23 | 4.25 ± 0.17 | 4.01 ± 0.23 |
Values are mean±SD for groups consisted of 12–16 rats. Statistically significant differences are indicated
P<0.05;
P<0.01;
P<0.001 compared with control group
Kidney function evaluation
Treatment of rats with administered 50 mg Pb/l for 40 and 65 days caused an elevation in blood creatinine level (P<0.001). The data indicated also a marked elevation in blood urea level (P<0.001) only in Pb40 group and a significant (P<0.001) increase in uric acid level in the same group when compared to controls. Our results also show no significant differences in kidney proteins level in all treated groups in comparison with controls (Table 4).
Lipid profile
The results indicated that TGs, TC, and LDL-Chol concentrations were significantly increased following Pb intoxication in all treated groups when compared to their relative controls. Moreover, the data showed a significant (P<0.05) decrease in HDL-Chol level, only in Pb65 group.
Furthermore, Pb administration during 40 days and 65 days caused a significant increase in cardiovascular risk markers together with a significant (P<0.001) increase on the atherogenic index (AI) in all treated groups in comparison with their controls; this suggest the susceptibility of female rats to cardiovascular diseases and atherosclerosis risk (Table 5).
Table 5.
Plasma lipid status, cardiovascular risk markers, and atherogenic index in female rats treated with lead (50 mg/l) for 40 and 65 days
| Control 40 days | Lead 40 days | Control 65 days | Lead 65 days | |
|---|---|---|---|---|
| Triglycerides (g/l) | 0.88 ± 0.06 | 1.23 ± 0.07 b | 0.95 ± 0.09 | 1.41 ± 0.10 a |
| Cholesterol (g/l) | 0.52 ± 0.02 | 0.71 ± 0.02 c | 0.55 ± 0.01 | 0.62 ± 0.01 b |
| HDL-Chol (g/l) | 0.41 ± 0.01 | 0.40 ± 0.01 | 0.52 ± 0.01 | 0.41 ± 0.01 a |
| LDL-Chol (g/l) | 0.05 ± 0.01 | 0.18 ± 0.02 c | 0.09 ± 0.01 | 0.16 ± 0.01 b |
| LDL/HDL | 0.10 ± 0.04 | 0.47 ± 0.09 c | 0.16 ± 0.02 | 0.36 ± 0.05 c |
| Cholesterol/HDL | 1.36 ± 0.07 | 1.85 ± 0.17 b | 1.07 ± 0.03 | 1.31 ± 0.06 a |
| Triglycerides/HDL | 2.35 ± 0.21 | 3.28 ± 0.33 b | 1.65 ± 0.17 | 2.64 ± 0.41 b |
| AI | 0.33 ± 0.06 | 0.84 ± 0.15 c | 0.12 ± 0.02 | 0.51 ± 0.07 c |
Values are mean±SD for groups consisted of 12-16 rats. Statistically significant differences are indicated
P<0.05;
P<0.01;
P<0.001 compared with control group
Oxidative stress evaluation in liver and kidney
Chronic exposure to Pb increased significantly the TBARS level in the liver and the kidney homogenates of both treated groups (Pb40 and Pb65) in comparison with those measured in control animals (Figure 1).
Figure 1.
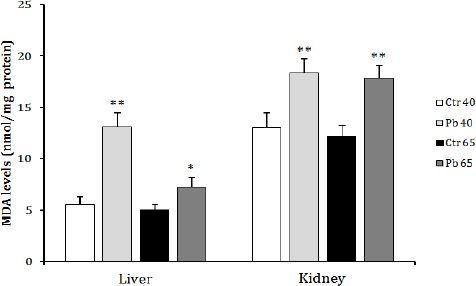
Effect of age dependent exposure to Pb (50 mg Pb/l) on liver and kidney malondialdehyde concentration (nmol MDA/mg proteins) in female rats. The animals were treated for a period of 40 days (puberty) and 65 days (post puberty). Values are means±SD for groups consisted of 12–16 rats. Statistically significant differences are indicated by * P<0.05, ** P<0.01 compared with control group
Total thiols (SH) levels were significantly (P<0.01) reduced in the liver of both treated groups (Pb40 and Pb65) when compared to controls (Figure 2).
Figure 2.
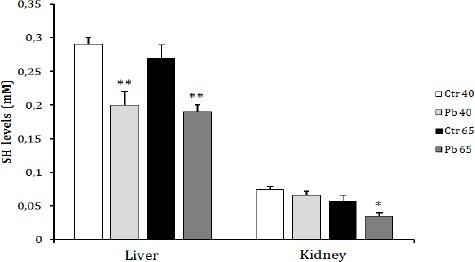
Effect of age dependent exposure to Pb (50 mg Pb/l) on liver and kidney total thiol group concentration (mM) in female rats. The animals were treated for a period of 40 days (puberty) and 65 days (post puberty). Values are means±SD for groups consisted of 12–16 rats. Statistically significant differences are indicated by *P<0.05, ** P<0.01 compared with control group
A non significant decrease in total SH level in the kidney was recorded in Pb40 group when compared to control. This decrease reached a significant level (P<0.05) in Pb65 group (Figure 2).
Data from Figure 3 show a marked increase in both liver (P<0.01) SOD activity after 40 days of Pb exposure and kidney (P<0.05) SOD activity after 65 days of exposure to the same metal when compared to their relative controls.
Figure 3.
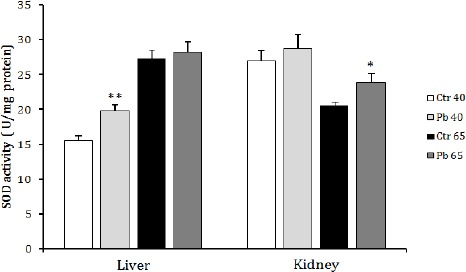
Effect of age dependent exposure to Pb (50 mg Pb/l) on liver and kidney superoxide dismutase activity (U/mg protein) in female rats. The animals were treated for a period of 40 days (puberty) and 65 days (post puberty). Values are means±SD for groups consisted of 2–16 rats. Statistically significant differences are indicated by *P<0.05, ** P<0.01 compared with control group
Discussion
In our study, no obvious influences on the general condition and appearance of the animals, feed consumption, and clinical observations were noted, which suggest that low-dose Pb has no apparent toxic effects on the function of most organs and growth parameters.
It is noted that the regulation of food intake and energy balance is controlled by hypothalamic neuropeptides and peripheral hormones (31). It is also well established that certain hypothalamic regions can control the food intake through neuropeptide Y (NPY), α-melanocyte-stimulating hormone (α-MSH), and coticotropin-releasing hormone (CRH) (32). In addition, peripheral hormones, especially insulin act by negative feedback on hypothalamus which causes inhibition of NPY release and stimulation of α-MSH and CRH (33, 34). The excess or deficit of some of these informational molecules would be responsible for an imbalance of food intake.
Based on this information, we can conclude that Pb doesn’t induce disturbance in mechanisms regulating food intake. Indeed, there are very few scientific studies seeking an association between Pb toxicity and food intake. However, some studies have highlighted the involvement of Pb in the disruption of body weight in animals exposed to this metal. The work of Ronis et al (17) showed that the treatment of pregnant rats at doses of Pb between 0.05% and 0.45% w/v causes disturbances in pups’ body weight at birth, and growth suppression during the pre-puberty and the puberty periods. The results of this study also showed that these observed changes were accompanied by a decrease in the plasma concentration of IGF (Insulin-like growth factor) and the rate of pituitary growth hormone (GH). From these data, it can be suggested that the observed decrease in body weight is due to the suppression of the growth of the animals following the disruption of the GH secretion.
Maternal Pb exposure has a significant influence on embryonic and fetal development, and has an important impact on pregnancy impairment outcomes including low birth weight, mental retardation, learning disabilities, and abnormal behavior (35). According to Gulson et al (36), Pb in mother’s milk contributes with about 36–80% of the infant Pb burden. Infant exposure to Pb is of concern because of the high susceptibility of the infant to toxic substances, and because there is no threshold for deleterious effects (37).
Our results showed an age-dependent decrease in blood Pb levels. We concluded that this difference between Pb40 and Pb65 groups could not be due to the administered amount of Pb, because all the experimental groups were exposed to the same dose of Pb throughout the experimental period.
Indeed, Varnai et al (38) explained that the gastrointestinal absorption in young animals is more important compared to adults. Furthermore, the gastrointestinal absorption of metal cations including Pb is much greater during lactation and pre-puberty periods because of (i) nutritional needs of young animals to grow and (ii) the involvement of the intestinal calcium-binding proteins (CaBP) in the transport of Pb (39), which make them more vulnerable to the deleterious effects of Pb poisoning.
The alterations in hematological parameters serve as the earliest indicators of toxic effects. In erythrocytes, Pb affects mainly the heme biosynthesis chain by inhibiting the key enzyme activity, δ-ALAD (delta aminolevulinic acid dehydratase) leading to the substrate δ-ALA accumulation (40). Anemia observed during Pb poisoning in this study is thought to result from inhibition of heme synthesis and a decreased life span of the erythrocytes (41).
Liver plays a central role in the detoxification process and along with kidney faces the threat of maximum exposure to xenobiotics and their metabolic by-products. During the hepatotoxic process, liver cell membrane will be damaged and several enzymes located in the hepatic cytosol, including ALT, AST, and ALP will be secreted into the blood (42). These serum enzymes are considered as markers of liver damage (43-45).
Indeed, in addition to oxidative stress, the hepatotoxicity induced by Pb may involve other pathways as endothelial cell injury (46), inflammation (47), and the transport pathway, since Pb ions mimic the action of other divalent cations, especially calcium (48).
In our study, Pb increased significantly ALT, AST, and ALP activities in treatment groups when compared with control rats. These changes pointed out the functional disorder of the liver. Several researchers have also reported perturbations in the activities of these enzymes during Pb poisoning (49-51).
Liver synthesizes plasma proteins, among them is albumin. Although a significant decrease in albumin level was only recorded in adult Pb-treated group (Pb 65), the total proteins level showed a significant decrease in all Pb-treated groups (Pb40 and Pb 65) indicating a perturbation in the synthetic function of the liver.
Kidney is a complex organ consisting of well-defined components that function in a highly coordinated manner. Several lines of evidence indicate that this organ has a crucial role in the toxicokinetics of Pb since it serves as a major organ of Pb excretion and a site of its accumulation (52, 53).
The toxic effects of Pb on the kidney appear to be primarily localized in the kidney tubule and are manifested by excessive urinary excretion of amino acids, glucose and phosphate, natriuresis, kaliuresis, and formation of nuclear inclusion bodies (54). These changes may be related to one or more factors, including increased blood Pb levels. Recent observations indicate that long-term exposure to considerably low Pb levels may increase risks of subclinical nephropathy, as indicated by biomarkers of renal dysfunction such as plasma creatinine, uric acid, and urea levels (55).
Our results showed that exposure to Pb increases significantly plasma creatinine levels in both treated groups (Pb40 and Pb65) comparing to their relative controls. Furthermore, a significant increase in urea and uric acid levels was only detected in Pb40 group comparing to controls. In fact, urea is the first acute renal marker, which increases when the kidney suffers any kind of injury; otherwise, creatinine is the most trustable renal marker. Its increase only occurs when the majority of renal function is lost (56). Indeed, clinical manifestations of renal impairment do not become evident until more than 50% of the nephrons are destroyed (57).
Few studies on humans and experimental animals have investigated the alteration of lipid metabolism following acute or chronic Pb exposure.
It has been shown that Pb accelerates lipid peroxidation and degradation of polyunsaturated membrane lipids and lipoproteins (58). Other studies have shown that chronic exposure to Pb causes alterations in the composition of red cell membrane fatty acids (59). All these observations are broadly suggestive of the involvement of Pb in lipid metabolism alteration.
Our results showed an increase in TGs, cholesterol, and LDL-chole levels. Similar observation of the relationships between Pb and hyperlipidemia was reported by other researches on laboratory animals (60, 61) and humans (62).
Indeed, enhanced cholesterogenesis observed in plasma of the Pb-treated animals may be attributed to a Pb-induced activation of 3-hydroxy-3- methylglutaryl coenzyme A (HMG CoA) reductase and HMG CoA synthase (the two rate-limiting enzymes in cholesterol synthesis) or it may be due to feedback inhibition (63). Furthermore, Pb has been shown to depress the activity of cytochrome P-450 (64). This can limit the biosynthesis of bile acids, which is the only significant route for elimination of cholesterol from the body.
On the other hand, increased de novo synthesis of cholesterol may occur following impaired feedback inhibition. Thus, the imbalance between cholesterol biosynthesis and elimination occurred by Pb, induced elevation of plasma TC concentration with simultaneous decrease in HDL-chole level, only in adult group Pb65.
The TC/HDL, TG/HDL, and LDL/HDL ratios are investigated to assess cardiovascular risks. An increase in these ratios indicates alteration in the cardiac system (65). It leads to elevated arterial pressure, degenerative changes of cardiac musculature, and decreased contractility (66). Increased LDL/HDL ratio which was observed in a previous study (67) indicates a linear positive association between blood Pb concentration and TC and LDL-chole levels.
It is well known that hyperlipedemia is the leading risk factor for atherosclerosis. Epidemiological investigation revealed a positive correlation between the severe degree of atherosclerosis and the concentrations of plasma cholesterol as well as LDL-chole. Numerous population studies have linked raised concentration of TC or LDL–chole in plasma with increased incidence of atherosclerotic events (68). In addition, the AI, defined as the ratio of TC–HDL and HDL, is believed to be an important risk factor of atherosclerosis. Our data clearly demonstrated that Pb has significantly increased this ratio.
Under physiological conditions, there is a balance between free radicals production and antioxidant defense mechanisms. This process involves mostly specific enzymes as SOD, CAT, and GPx and antiradical molecules that scavenge free radicals such as vitamins and thiol groups (SH).
Many studies have suggested the involvement of oxidative stress in some aspects of Pb toxicity (10, 43, 50, 69).
The most common group of indices used to assess oxidative stress is that of peroxidation products of lipids, usually polyunsaturated fatty acids, which are susceptible to free radicals attack (70). It is also known that the main Pb toxic effects are on the structure and cell membrane function. Many studies have showed that MDA, the most used end-product of lipid peroxidation, will be increased with Pb treatment (71-73). Our results also indicated that lipid peroxidation, determined by measuring the MDA levels, is amplified in liver and kidney after exposure to Pb.
Furthermore, it has been shown that the increase of the lipid peroxidation is generally accompanied by the decrease in the rate of SH groups (74), which is consistent with our results. Thus, it can be supposed that the Pb-induced decrease in total SH levels might result from their utilization to scavenge free radicals formed during exposure to Pb.
Different responses of SOD have been described in animals exposed to Pb including increases, decreases, or no changes in its activity (13, 74) depending on dose, length of treatment, and the target organ. In our experiment, Pb intoxication resulted in an increase of SOD activity in liver of Pb40 group and in kidney of Pb65 group comparing to their relative controls. As free radicals are the substrates of antioxidative enzymes, their overproduction may (i) stimulate the activities or biosynthesis of enzymatic proteins to adapt the increases of free radicals or (ii) override enzymatic activity and lead to the fall of its concentration (41) depending on target organ. However, hepatic and renal SOD activity does not show significant change in some of Pb treated groups when compared with their relative controls. This result is in agreement with the study undertaken by Patra et al (13) who recorded no significant change in the SOD activity in the liver, kidney, and brain of rats subjected to subchronic exposure to Pb. They hypothesized that the participation of iron in Fenton reaction in vivo leads to the production of more reactive hydroxyl radicals from superoxide radicals and hydrogen peroxide, which increase lipid peroxidation (13). This might be one of the reasons for significant alteration in lipid peroxidation and non significant changes in SOD activity.
Conclusion
During this study, we showed that Pb, the heavy metal widely spread in the environment and strongly used in the developing countries causes disturbances which are not negligible. Indeed, chronic exposure to Pb has resulted in significant liver and kidney metabolic disorders. Thus, increased levels of transaminases and alkaline phosphatase in plasma indicate cytolysis of liver cells. Reducing rate of albumin and liver proteins are indicative of liver synthetic function disruption. In kidneys, it has been shown that the glomerulus and the proximal tubule are the most vulnerable compartments of lead toxicity. Moreover, we noted that Pb induces dyslipidemia which is a major risk factor for the onset of cardiovascular diseases and atherosclerosis. Furthermore, it has been well established that chronic Pb exposure induces oxidative stress and many mechanisms are involved in the imbalance between pro and anti-oxidants.
Acknowledgment
This study has been supported by our laboratory, University of Sciences of Tunis, Tunisia.
References
- 1.Garnier R. Toxicitédu Plombet de ses derives. EMC-Toxicol Pathol. 2005;2:67–88. [Google Scholar]
- 2.El-Nekeety AA, El-Kady AA, Soliman MS, Hassan NS, Abdel-Wahab MA. Protective effect of Aquilegia vulgaris (L.) against lead acetate-induced oxidative stress in rats. Food Chem Toxicol. 2009;47:2209–2215. doi: 10.1016/j.fct.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 3.Lanphear BP, Hornung R, Ho M, Howard CR, Eberle S, Knauf K. Environmental lead exposure during early childhood. J Pediatr. 2002;140:40–47. doi: 10.1067/mpd.2002.120513. [DOI] [PubMed] [Google Scholar]
- 4.Davis JM, Svendsgaard DJ. Lead and child development. Nature. 1987;329:297–300. doi: 10.1038/329297a0. [DOI] [PubMed] [Google Scholar]
- 5.Bellinger DC. Lead. Pediatrics. 2004;113:1016–1022. [PubMed] [Google Scholar]
- 6.Lidsky TI, Schneider JS. Lead neurotoxicity in children: Basic mechanisms and clinical correlates. Brain. 2003;126:5–19. doi: 10.1093/brain/awg014. [DOI] [PubMed] [Google Scholar]
- 7.Fisher AM, Vessey JA. Preventing lead poisoning and its consequences. Ped Nursing. 1998;24:348–350. [PubMed] [Google Scholar]
- 8.Patrick L. Lead toxicity, a review of the literature. Part 1. Exposure, evaluation, and treatment. Altern Med Rev. 2006;11:2–22. [PubMed] [Google Scholar]
- 9.Flora SJ, Pande M, Mehta A. Beneficial effect of combined administration of some naturally occurring antioxidants (vitamins) and thiol chelators in the treatment of chronic lead intoxication. Chem Biol Interact. 2003;145:267–280. doi: 10.1016/s0009-2797(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 10.Kumar V, Abul KA, Nelson F, Richard NM. Robbins basic pathology. 8th ed. New Delhi, India: Elsevier; 2007. [Google Scholar]
- 11.Ashry KM, El-Sayed YS, Khamiss RM, El-Ashmawy IM. Oxidative stress and immunotoxic effects of lead and their amelioration with myrrh (Commiphora molmol) emulsion. Food Chem Toxicol. 2010;48:236–241. doi: 10.1016/j.fct.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Sidhu P, Nehru B. Lead intoxication: histological and oxidative damage in rat cerebrum and cerebellum. J Trace Elem Exp Med Biol. 2004;17:45–53. [Google Scholar]
- 13.Patra RC, Swarup D, Dwivedi SK. Antioxidant effects of α-tocopherol, ascorbic acid and L-methionine on lead induced oxidative stress to the liver, kidney and brain in rats. Toxicology. 2001;162:81–88. doi: 10.1016/s0300-483x(01)00345-6. [DOI] [PubMed] [Google Scholar]
- 14.Taib NT, Jarrar BM, Mubarak M. Ultrastructural alterations in hepatic tissues of white rats (Rattus norvegicus) induced by lead experimental toxicity. Saudi J Biol Sci. 2004;11:11–20. [Google Scholar]
- 15.Flora S, Flora G, Saxena G. Environmental occurrence, health effects and management of lead poisoning. In: Casas JS, Sordo J, editors. Lead: chemistry, analytical aspects, environmental impact and health effects. Amsterdam, Netherlands: Elsevier Science; 2006. pp. 158–228. [Google Scholar]
- 16.Poręba R, Powel G, Poręba M, Andrzejak R. Environmental and occupational exposure to lead as a potential risk factor for cardiovascular disease. Environ Toxicol Pharmacol. 2011;31:267–277. doi: 10.1016/j.etap.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Ronis MJJ, Badger TM, Shema SJ, Roberson PK. Endocrine mechanisms underlying the growth effects of developmental lead exposure in the rat. J Toxicol Environ Health A. 1998;54:101–120. doi: 10.1080/009841098158944. [DOI] [PubMed] [Google Scholar]
- 18.Bannon DI, Murashchik C, Zapf CR, Farfel MR, Chisoim JJ., Jr Graphite furnace atomic absorption spectroscopic measurement of blood lead in matrix-matched standards. Clin Chem. 1994;40:1730–1734. [PubMed] [Google Scholar]
- 19.Drabkin DL, Austin JH. Spectrophotometric constants for common hemoglobin derivatives in human, dog and rabbit blood. J Biol Chem. 1932;98:719–733. [Google Scholar]
- 20.Lott JA, Turner K. Evaluation of Trinder’s glucose oxidase method for measuring glucose in serum and urine. Clin Chem. 1975;21:1754–1760. [PubMed] [Google Scholar]
- 21.Bergmeyer HU, Scheibe P, Wahlefeld AW. Optimization of methods for aspartate aminotransferase and alanine aminotransferase. Clin Chem. 1978;24:58–73. [PubMed] [Google Scholar]
- 22.Price CP, Woodman DD. An improved autoanalyzer technique for the determination of serum alkaline phosphatase. Clin Chim Acta. 1971;35:265–271. doi: 10.1016/0009-8981(71)90192-6. [DOI] [PubMed] [Google Scholar]
- 23.Doumas BT, Natson WA, Biggs HG. Albumin standards and the measurement of serum albumin with bromocresol green. Clin Chim Acta. 1971;31:87–96. doi: 10.1016/0009-8981(71)90365-2. [DOI] [PubMed] [Google Scholar]
- 24.Larsen K. Creatinine assay by a reaction-kinetic principle. Clin Chim Acta. 1972;41:209–217. doi: 10.1016/0009-8981(72)90513-x. [DOI] [PubMed] [Google Scholar]
- 25.Fossati P, Prencipe L, Berti G. Use of 3, 5-dichloro-2- hydroxybenzenesulfonic acid/4-aminophenazone chromogenic system in direct enzyme asay of uric acid in serum and urine. Clin Chem. 1980;26:227–231. [PubMed] [Google Scholar]
- 26.Fawcett JK. A rapid and precise method for the determination of urea. J Clin Pathol. 1960;13:156–159. doi: 10.1136/jcp.13.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oteiza PI, Olin KL, Fraga CG, Keen CL. Zinc deficiency causes oxidative damage to proteins, lipids and DNA in rat testes. J Nutr. 1995;125:823–829. doi: 10.1093/jn/125.4.823. [DOI] [PubMed] [Google Scholar]
- 28.Miao-Lin H. Measurement of protein thiol groups and glutathione in plasma. Methods Enzymol. 1994;233:380–383. doi: 10.1016/s0076-6879(94)33044-1. [DOI] [PubMed] [Google Scholar]
- 29.Misra HP, Fridovich I. The role of superoxide anion in the autooxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247:3170–3175. [PubMed] [Google Scholar]
- 30.Gornall AG, Bardawill CJ, David MM. Determination of serum proteins by means of the Biuret reaction. J Biol Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- 31.Loviselli A, Secci G, Lai A, Velluzzi F. Mechanisms of regulation of the food intake: recent advances. Recenti Prog Med. 2007;98:1–6. [PubMed] [Google Scholar]
- 32.Lasram MM, Bini Douib I, Bouzid K, Annabi A, El Elj N, Dhouib H, et al. Abdelmoula J, Gharbi N. Effects of N-acetyl-L-Cysteine, in vivo, against pathological changes induced by malathion. Toxicol Mech Methods. 2014;24:294–306. doi: 10.3109/15376516.2014.886003. [DOI] [PubMed] [Google Scholar]
- 33.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 34.Weigle DS, Bukowski TR, Foster DC, Holderman S, Kramer JM, Lasser G, et al. Recombinant ob protein reduces feeding and body weight in the ob/ob mouse. J Clin Invest. 1995;96:2065–2070. doi: 10.1172/JCI118254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang BJ, Jang BJ, Son TG, Cho IH, Quan FS, Choe NH, et al. Ascorbic acid ameliorates oxidative damage induced by maternal low-level lead exposure in the hippocampus of rat pups during gestation and lactation. Food Chem Toxicol. 2012;50:104–108. doi: 10.1016/j.fct.2011.09.043. [DOI] [PubMed] [Google Scholar]
- 36.Gulson BL, Jameson CW, Mahaffey KR, Mizon KJ, Law AJ, Korsh MJ, et al. Relationships of lead in breast milk to lead in blood, urine, and diet of the infant and mother. Environ Health Perspect. 1998;106:667–674. doi: 10.1289/ehp.98106667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Needleman HL, Bellinger D. The health effect of low level exposure to lead. Annu Rev Public Health. 1991;12:111–140. doi: 10.1146/annurev.pu.12.050191.000551. [DOI] [PubMed] [Google Scholar]
- 38.Varnai VM, Piasek M, Blanusa M, Saric MM, Simic D, Kostial K. Calcium supplementation efficiently reduces lead absorption in suckling rats. Pharmacol Toxicol. 2001;89:326–330. doi: 10.1034/j.1600-0773.2001.d01-169.x. [DOI] [PubMed] [Google Scholar]
- 39.Fullmer CS, Edelstein S, Wasserman RH. Lead-binding properties of intestinal calcium-binding proteins. J Biol Chem. 1985;260:6816–6819. [PubMed] [Google Scholar]
- 40.Gurer-Orhan H, Handan US, Hilal O. Correlation between clinical indicators of lead poisoning and oxidative stress parameters in control and lead-exposed workers. Toxicology. 2004;195:147–154. doi: 10.1016/j.tox.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 41.Berrahal Annabi A, Nehdi A, Hajjaji N, Gharbi N, El-Fazâa S. Antioxidant enzymes activities and bilirubin level in adult rats treated with lead. C R Biol. 2007;330:581–588. doi: 10.1016/j.crvi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 42.Ncibi S, Othman MB, Akacha A, Krifi MN, Zourgui L. Opuntia ficus indica extract protects against chlorpyrifos-induced damage on mice liver. Food Chem Toxicol. 2008;46:797–802. doi: 10.1016/j.fct.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 43.Berrahal Annabi A, Lasram M, El Elj N, Kerkeni A, Gharbi N, El Fazâa S. Effect of age-dependent exposure to lead on hepatotoxicity and nephrotoxicity in male rats. Environ Toxicol. 2011;26:68–78. doi: 10.1002/tox.20530. [DOI] [PubMed] [Google Scholar]
- 44.Eraslan G, Kanbur M, Silici S. Effect of carbaryl on some biochemical changes in rats: the ameliorative effect of bee pollen. Food Chem Toxicol. 2009;47:86–91. doi: 10.1016/j.fct.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 45.Liju VB, Jeena K, Kuttan R. Acute and subchronic toxicity as well as mutagenic evaluation of essential oil from turmeric (Curcuma longa L) Food Chem Toxicol. 2013;53:52–61. doi: 10.1016/j.fct.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 46.Jarrar BM, Taib NT. Histological and histochemical alterations in the liver induced by lead chronic toxicity. Saudi J Biol Sci. 2012;19:203–210. doi: 10.1016/j.sjbs.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rikans LE, Yamano T. Mechanisms of cadmium mediated acute hepatotoxicity. J Biochem Mol Toxicol. 2000;14:110–117. doi: 10.1002/(sici)1099-0461(2000)14:2<110::aid-jbt7>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 48.Fotakis G, Timbrell JA. Role of trace elements in cadmium chloride uptake in hepatoma cell lines. Toxicol Lett. 2006;164:97–103. doi: 10.1016/j.toxlet.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 49.Dewanjee S, Das AK, Sahu R, Gangopadhyay M. Antidiabetic activity of Diospyros peregrina fruit: effect on hyperglycemia, hyperlipidemia and augmented oxidative stress in experimental type 2 diabetes. Food Chem Toxicol. 2009;47:2679–2685. doi: 10.1016/j.fct.2009.07.038. [DOI] [PubMed] [Google Scholar]
- 50.Mehana EE, Meki AMA, Fazili KM. Ameliorated effects of green tea extract on lead induced liver toxicity in rats. Exp Toxicol Path. 2012;64:291–295. doi: 10.1016/j.etp.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 51.Rahman S, Sultana S. Chemopreventive activity of glycyrrhizin on lead acetate mediated hepatic oxidative stress and its hyperproliferative activity in Wistar rats. Chem Biol Int. 2006;160:61–69. doi: 10.1016/j.cbi.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 52.Gargouri M, Magné C, Dauvergne X, Ksouri R, El Feki A, Giroux Metges MA, et al. Cytoprotective and antioxidant effects of the edible halophyte Sarcocornia perennis L. (swampfire) against lead-induced toxicity in renal cells. Ecotoxicol Environ Saft. 2013;95:44–51. doi: 10.1016/j.ecoenv.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 53.Liu CM, Ma JQ, Sun YZ. Quercetin protects the rat kidney against oxidative stress-mediated DNA damage and apoptosis induced by lead. Environ Toxicol Pharmacol. 2010;30:264–271. doi: 10.1016/j.etap.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 54.Jadhav SH, Sarkar SN, Patil RD, Tripathi HC. Effects of subchronic exposure via drinking water to a mixture of eight water-contaminating metals: a biochemical and histopathological study in male rats. Arch Environ Contam Toxicol. 2007;53:667–6677. doi: 10.1007/s00244-007-0031-0. [DOI] [PubMed] [Google Scholar]
- 55.Silbergeld EK. Facilitative mechanisms of lead as a carcinogen. Mutat Res. 2003;533:121–133. doi: 10.1016/j.mrfmmm.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 56.Borges LP, Brandao R, Godoi B, Nogueira CW, Zeni G. Oral administration of diphenyl diselenide protects against cadmium-induced liver damage in rats. Chem Biol Int. 2008;171:15–25. doi: 10.1016/j.cbi.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 57.Dioka CE, Orisakwe OE, Adeniyi FAA, Meludu SC. Liver and renal function tests in artisans occupationally exposed to lead in mechanic village in Nnewi, Nigeria. Int J Environ Res Public Health. 2004;1:21–25. doi: 10.3390/ijerph2004010021. [DOI] [PubMed] [Google Scholar]
- 58.Ribarov SR, Bernov LC, Benchev IC. The effect of lead on hemoglobin- catalysed lipid peroxidation. Biochim Biophys Acta. 1981;664:453–459. doi: 10.1016/0005-2760(81)90123-5. [DOI] [PubMed] [Google Scholar]
- 59.Osterode W, Ulberth F. Increased concentration of arachidonic acid in erythrocyte membrane in chronically lead-exposed men. J Toxicol Environ Health, Part A. 2000;59:87–95. doi: 10.1080/009841000156998. [DOI] [PubMed] [Google Scholar]
- 60.Ademuyiwa O, Agarwal R, Chandra R, Behari JR. Lead-induced phospholipidosis and cholesterogenesis in rat tissues. Chem Biol Interact. 2009;179:314–320. doi: 10.1016/j.cbi.2008.10.057. [DOI] [PubMed] [Google Scholar]
- 61.Newairy AA, Abdou HM. Protective role of flax lignans against lead acetate induced oxidativedamage and hyperlipidemia in rats. Food Chem Toxicol. 2009;47:813–818. doi: 10.1016/j.fct.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 62.Skoczyńska A, Gruber K, Belowska-Bień K, Mlynek V. Risk of cardiovascular diseases in lead-exposed workers of crystal glassworks, part I. Effect of lead on blood pressure and lipid metabolism. Med Pr. 2007;58:475–483. [PubMed] [Google Scholar]
- 63.Sawada H, Takami K, Asahi S. A toxicogenomic approach to drug-induced phospholipidosis: analysis of its induction mechanism and establishment of a novel in vitro screening system. Toxicol Sci. 2005;83:282–292. doi: 10.1093/toxsci/kfh264. [DOI] [PubMed] [Google Scholar]
- 64.Meredith PA, Campell BC, Goldberg A. The effect of industrial lead poisoning on cytochrome p-450 mediated phenazone hydroxylation. Eur J Clin Pharmacol. 1977;12:235–239. doi: 10.1007/BF00609867. [DOI] [PubMed] [Google Scholar]
- 65.Reaven GM. Importance of identifying the overweight patient who will benefit the most by losing weight. Ann Intern Med. 2003;138:420–423. doi: 10.7326/0003-4819-138-5-200303040-00012. [DOI] [PubMed] [Google Scholar]
- 66.Tsao DA, Yu HS, Chen JT, Ho CK, Chang HR. The change of beta adrenergic system in lead induced hypertension. Toxicol Appl Pharmacol. 2000;164:127–133. doi: 10.1006/taap.1999.8871. [DOI] [PubMed] [Google Scholar]
- 67.Ademuyiwa O, Ugbaja RN, Idumebor F, Adebawo O. Plasma lipid profiles and risk of cardiovascular disease in occupational lead exposure in Abeokuta, Nigeria. Lipids Health Dis. 2005;4:19. doi: 10.1186/1476-511X-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544–1568. doi: 10.1172/JCI107332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dotan Y, Lichtenberg D, Pinchuk I. Lipid peroxidation cannot be used as a universal criterion of oxidative stress. Prog Lipid Res. 2004;43:200–227. doi: 10.1016/j.plipres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 70.Lakshmi BVS, Sudhakar M, Aparna M. Protective potential of Black grapes against lead induced oxidative stress in rats. Environ Toxicol Pharmacol. 2013;35:361–368. doi: 10.1016/j.etap.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 71.Moneim AEA, Dkhil MA, Al-Quraishy S. The protective effect of flaxseed oil on lead acetate-induced renal toxicity in rats. J Hazard Mater. 2011;194:250–255. doi: 10.1016/j.jhazmat.2011.07.097. [DOI] [PubMed] [Google Scholar]
- 72.Xia D, Yu X, Liao S, Shao Q, Mou H, Ma W. Protective effect of Smilax glabra extract against lead-induced oxidative stress in rats. J Ethnopharmacol. 2010;130:414–420. doi: 10.1016/j.jep.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 73.Yegen B, Dedeoglu A, Aykac I, Oktay S, Yalcin AS. Effect of cold-restraint stress on glutathione and lipid peroxide levels in the liver and glandular stomach of rats. Pharm Res. 1990;221:45–48. doi: 10.1016/1043-6618(90)90742-v. [DOI] [PubMed] [Google Scholar]
- 74.El-Sokkary GH, Abdel-Rahman GH, Kamel ES. Melatonin protects against lead-induced hepatic and renal toxicity in male rats. Toxicology. 2005;213:25–33. doi: 10.1016/j.tox.2005.05.003. [DOI] [PubMed] [Google Scholar]