

Abstract
Context
The reconstruction of evolutionary scenarios for whole genomes in terms of genome rearrangements is a fundamental problem in evolutionary and comparative genomics. The DeCo algorithm, recently introduced by Bérard et al., computes parsimonious evolutionary scenarios for gene adjacencies, from pairs of reconciled gene trees. However, as for many combinatorial optimization algorithms, there can exist many co-optimal, or slightly sub-optimal, evolutionary scenarios that deserve to be considered.
Contribution
We extend the DeCo algorithm to sample evolutionary scenarios from the whole solution space under the Boltzmann distribution, and also to compute Boltzmann probabilities for specific ancestral adjacencies.
Results
We apply our algorithms to a dataset of mammalian gene trees and adjacencies, and observe a significant reduction of the number of syntenic conflicts observed in the resulting ancestral gene adjacencies.
Keywords: Synteny, Parsimony, Gene adjacency, Dynamic programming, Ensemble approach
Background
The reconstruction of the evolutionary history of genomic characters along a given species tree is a long-standing problem in computational biology. This problem has been well studied for several types of genomic characters, for which efficient algorithms exist to compute parsimonious evolutionary scenarios; classical examples include genes and genomes sequences [1], gene content [2], and gene family evolution [3,4]. Recently, Bérard et al. [5] extended the corpus of such results to syntenic characters. They introduced the notion of adjacency forest, that models the evolution of gene adjacencies within a phylogeny, motivated by the reconstruction of the architecture of ancestral genomes, and described an efficient dynamic programming (DP) algorithm, called DeCo, to compute parsimonious adjacency evolutionary histories. So far, DeCo is the only existing tractable model that considers the evolution of gene adjacencies within a general phylogenetic framework: other tractable models of genome rearrangements accounting for a given species phylogeny are either limited to single-copy genes and ignore gene-specific events [6], assume restrictions on the gene duplication events, such as considering only whole-genome duplication (see [7] and references there), or require a dated species phylogeny [8].
From a methodological point of view, most existing algorithms to reconstruct evolutionary scenarios along a species tree in a parsimony framework rely on dynamic-programming along this tree, whose introduction can be traced back to Sankoff in the 1970s (see [9] for a recent retrospective on this topic). Recently, several works considered more general approaches for such parsimony problems that either explore a wider range of values for combinatorial parameters of parsimonious models [10] or consider several alternate histories for a given instance, chosen for example from the set of all possible co-optimal scenarios or from the whole solution space, including suboptimal solutions (see [11-13] for examples of this approach for the gene tree/species tree reconciliation problem).
The present work follows the later approach and extends the DeCo DP scheme toward an exploration of the whole solution space of adjacency histories, under the Boltzmann probability distribution, that assigns a probability to each solution defined in terms of its parsimony score. This principle of exploring the solution space of a combinatorial optimization problem under the Boltzmann probability distribution is sometimes known as the "Boltzmann ensemble approach". It was initially introduced in the context of RNA folding, where the probability of any given conformation at the thermodynamic equilibrium follows a Boltzmann distribution, i.e. a conformation s is observed for a given RNA w with probability , where Ew,s is the free-energy of conformation s over w, k is the Boltzmann constant, T is the temperature, and is the partition function of w. This latter quantity can be seen as a renormalization factor, and is key in the study of RNA thermodynamics, but its computation involves summing over an exponential number of conformations compatible with the RNA sequence. A major paradigm shift occurred in RNA research when McCaskill [14] showed in 1990 how an efficient algorithm for the partition function could be adapted from a DP scheme for energy minimization through a simple change of algebra. This seminal work also introduced a variant of the inside-outside algorithm [15] for computing base-pairing probabilities.
While this Boltzmann ensemble approach has been used for a long time in RNA structure analysis, to the best of our knowledge it is not the case in comparative genomics, where exact probabilistic models have been favoured recently [16,17]. However, probabilistic models still pose computational challenges for large datasets, and so far a probabilistic model does not exist for gene adjacencies, which motivates our work. In the specific case of the DeCo model, the ability to explore alternative co-optimal or slightly sub-optimal solutions is crucial. Indeed, as DeCo models gene adjacencies, each ancestral gene can only be adjacent to at most two other genes, which is not considered in DeCo. However, the initial experiments using DeCo on mammalian gene trees resulted in hundreds of ancestral genes were involved in more than two ancestral gene adjacencies [5]. This raises the question of filtering inferred ancestral adjacencies to reduce the level of syntenic conflict, which can be done on the basis of their Boltzmann probabilities. We reason that some of the erroneously-predicted adjacencies may result from combinatorial optimization artifacts and that features of a gene adjacency parsimonious evolutionary scenario that are not robust to considering alternative equivalent, or slightly worse, solutions should be considered as dubious.
Methods
Models
A phylogeny is a rooted tree which represents the evolutionary relationships of a set of elements represented by its nodes: internal nodes are ancestors, leaves are extant elements, and edges represent direct descents between parents and children. We consider here three kinds of phylogenies (illustrated in Figure 1): species trees, reconciled gene trees and adjacencies trees/forests. Trees we consider are always rooted. For a tree T and a node x of T , we denote by T (x) the subtree rooted at x. If x is an internal node, we assume it has either one child, denoted by ax, or two children, denoted by ax and bx.
Figure 1.
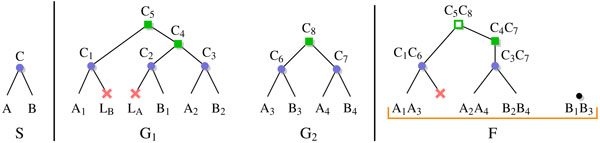
Example of an adjacency forest predicted from two reconciled gene trees. (Left) Species tree S, with two extant species A and B and an ancestral species C. (Middle) Two reconciled gene trees G1 and G2, with four extant genes in genome A, four extant genes in genome B and three ancestral genes in genome C. The set of extant adjacencies is (A1A3, B1B3, A2A4, B2B4) (Right) Parsimonious adjacency forest F composed of two adjacency trees. Blue dots are speciation nodes. Leaves are extant (species, genes, adjacencies), except when labelled with a red cross (gene loss). Green squares are (gene or adjacency) duplication nodes. Gene labels refer to the species of nodes. Every node of the adjacency tree is labelled by a couple of nodes from gene trees. Figure adapted from [5].
Species trees
A species tree S is a binary tree that describes the evolution of a set of related species, from a common ancestor (the root of the tree), through the mechanism of speciation. For our purpose, species are identified with genomes, and genes are arranged linearly or circularly along chromosomes.
Reconciled gene trees
A reconciled gene tree is also a binary tree that describes the evolution of a set of genes, called a gene family, through the evolutionary mechanisms of speciation, gene duplication and gene loss, within the given species tree S. Therefore, each node of a gene tree G represents a gene loss, an extant gene or an ancestral gene. Ancestral genes are represented by the internal nodes of G, while gene losses and extant genes are represented by the leaves of G.
We denote by s(g) ∈ S the species of a gene g ∈ G, and by e(g) the evolutionary event that leads to the creation of the two children ag and bg. If g is an internal node of G, then e(g) is a speciation (denoted by Spec) if the species pair {s(ag), s(bg)} equals the species pair {as(g), bs(g)}, or a gene duplication (GDup) if s(ag) = s(bg) = s(g). Finally, if g is a leaf, then e(g) indicates either a gene loss (GLoss) or an extant gene (Extant), in which case e(g) is not an evolutionary event.
Adjacency trees and forests
A gene adjacency is a pair of genes that appears consecutively along a chromosome. An adjacency tree represents the evolution of an ancestral adjacency through the evolutionary events of speciation, gene duplication, gene loss (these events, as described above, occur at the gene level and are modelled in the reconciled gene trees), and adjacency duplication (ADup), adjacency loss (ALoss) and adjacency break (ABreak), that are adjacency-specific events.
• The duplication of an adjacency {g1, g2} follows from the simultaneous duplication of both its genes g1 and g2 (with s(g1) = s(g2) and e(g1) = e(g2) = GDup), resulting in the creation of two distinct adjacencies each belonging to {ag1, bg1} × {ag2, bg2}.
• An adjacency may disappear due to several events, such as the loss of exactly one (gene loss) or both (adjacency loss) of its genes, or a genome rearrangement that breaks the contiguity between the two genes (adjacency break).
Finally, to model the complement of an adjacency break, i.e. the creation of adjacencies through a genome rearrangement, adjacency gain (AGain) events are also considered, and result in the creation of a new adjacency tree. It follows that the evolution of the adjacency between two genes can be described by a forest of adjacency trees, called an adjacency forest. In this forest, each node v belongs to a species denoted by s(v), and is associated to an evolutionary event e(v) ∈ {Spec, GDup, ADup} if g is an internal node, or {Extant, GLoss, ALoss, ABreak} if v is a leaf. Finally, adjacency gain events are associated to the roots of the trees of the adjacency forest. So in the same way that a gene tree G evolves within the species S, an adjacency forest F describing the evolution of the adjacency between two gene families G1 and G2 evolves within S, G1 and G2. We refer the reader to Figure 1 for an illustration.
Parsimony scores and the Boltzmann distribution
When considered in a parsimonious framework, the score of an adjacency forest F is the number of adjacency gains and breaks; other events are not considered as they are the by-products of evolutionary events already accounted for in the score of the reconciled gene trees G1 and G2. We denote by sa(F) the parsimony score of an adjacency forest F. Let be the set of all adjacency forests for G1 and G2, including both optimal and sub-optimal ones, where we assume that at least one extant adjacency is composed of extant genes from G1 and G2.
We define the Boltzmann factor of an adjacency forest F as
| (1) |
The partition function associated to two trees G1 and G2 is obtained as
| (2) |
where kT is an arbitrary constant. The partition function implicitly defines a Boltzmann probability distribution over , where the probability of an adjacency forest F is defined by:
| (3) |
By exponentially favouring adjacency forests with lower parsimony scores, the Boltzmann distribution provides an alternative way to probe the search space, which is heavily influenced by the choice of kT. Indeed, decreasing kT values will skew the Boltzmann distribution towards more parsimonious adjacency forests. Its limiting distributions are uniform over the whole search space (kT → +∞) or over the set of co-optimal forests (kT → 0) (see Figure 2 for an illustration).
Figure 2.
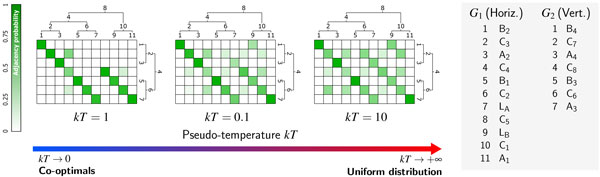
Temperature dependency of the adjacency matrix. Each value of the (pseudo) temperature parameter kT induces different Boltzmann probabilities for ancestral and extant adjacencies, allowing to probe the continuum between a parsimonious model (kT → 0) and the uniform distribution kt → ∞.
A Boltzmann probability distribution on the set of all adjacency forests for a given instance also implies a well defined notion of probability for features of adjacency forests. For example, one can associate a probability to a specific potential ancestral adjacency (i.e. adjacency between two genes from a given ancestral species) as the ratio of the sum of the probabilities of the adjacency forests that contain this adjacency with the partition function.
Algorithms
DeCo, the algorithm described in [5] to compute a parsimonious adjacency forest, is a DP scheme constrained by S, G1 and G2. We first present this algorithm, then describe how to extend it into an Boltzmann ensemble algorithm.
The DeCo DP scheme
Let G1 and G2 be two reconciled gene trees and g1 and g2 be two nodes, respectively of G1 and G2, such that s(g1) = s(g2). The DeCo algorithm computes, for every such pair of nodes g1 and g2, two quantities denoted by c1(g1, g2) and c0(g1, g2), that correspond respectively to the most parsimonious score of a parsimonious adjacency forest for the pairs of subtrees G(g1) and G(g2), under the hypothesis of a presence (c1) or absence (c0) of an ancestral adjacency between g1 and g2. As usual in DP along a species tree, the score of a parsimonious adjacency forest for G1 and G2 is given by min(c1(r1, r2), c0(r1, r2)) where r1 is the root of G1 and r2 the root of G2.
So, c1(g1, g2) and c0(g1, g2) can be computed as the minimum of a sum of the scores of adjacency gains or breaks and, more importantly, of terms of the form c1(x, y) and c0(x, y) with (x, y) ∈ {g1, ag1, bg1} × {g2, ag2, bg2} − (g1, g2), using the two combinatorial operator min and +.
(Un)-ambiguity of the DeCo DP scheme
As defined in [18], the ambiguity of a DP algorithm can be defined as follows: a DP explores a combinatorial solution space (here for DeCo, the space of all possible adjacency forests, including possible suboptimal solutions), that can be explicitly generated by replacing in the equations min by (the set-theoretic union operator) and + by the Cartesian product × between combinatorial sets. A DP algorithm is then unambiguous if the unions are disjoint, i.e. the sets provided as its arguments do not overlap.
We claim that the DeCo dynamic programming scheme is unambiguous. Indeed, computing c1(g1, g2) and c0(g1, g2) branches on disjoint subcases that each involve a different set of terms c1(x, y) and c0(x, y). The only case that deserves a closer attention is the case where e(g1) = e(g2) = GDup, as a simultaneous duplication can be obtained by two successive duplications. But in this case, the number of AGain events is different (see Figure 3), which ensures the pairwise difference of solutions.
Figure 3.

Partition function version of the DeCo dynamic programming scheme. This system of recurrences computes the α-rescaled partition function for two reconciled gene trees g1 and g2, using penalties AG and AB respectively for adjacency gains and breaks.
Stochastic backtrack algorithm through algebraic substitutions
As mentioned in [18], any unambiguous dynamic programming scheme can be adapted through algebraic changes to exhaustively generate the set of all adjacency forests, and also compute the corresponding partition function. To that purpose one simply needs to replace the arithmetic operators (min, +) with (Σ, ×), and to exponentiate any atomic cost into a (partial) Boltzmann factor e-C/kT (see Figure 3).
This precomputation allows us to sample adjacency forests under the Boltzmann distribution, by changing the deterministic backtrack used for maximum parsimony into a stochastic operation. Indeed, assume that the partition function version of the DeCo equation computes c1(g1, g2) (resp. c0(g1, g2)) as , where the ti denote the contribution to the partition function of one of the local alternatives within the DP scheme. The latter are typically computed recursively as combinations of atomic adjacency gain/break costs, and recursive terms of the form c1(x, y) and c0(x, y) with (x, y) ∈ {g1, ag1, bg1} × {g2, ag2, bg2} − {(g1, g2)}.
Then a (possibly non-parsimonious) random solution can be generated recursively for c1(g1, g2) (resp. c0(g1, g2)), by branching on some ti with probability ti/c1(g1, g2) (resp. ti/c0(g1, g2)), and proceed recursively on each occurrence of a recursive term within the alternative ti. The correctness of the algorithm, i.e. the fact that the random process generates each adjacency forests with Boltzmann probability, follows immediately from general considerations on unambiguous DP schemes [18].
The stochastic nature of the backtrack does not affect its worst-case complexity. This Boltzmann sampling algorithm, for an instance composed of two gene trees G1 and G2 of respective sizes (number of leaves) n1 and n2, has time complexity of for each backtrack.
Rescaling to avoid numerical overflows
The partition function values , handled during the computation, typically grow exponentially in the total number of nodes in G1 and G2, and may end up overflowing the floating point data type used within the DP tables. Following practice in RNA folding prediction [19], we address this issue by iteratively applying an homogeneous rescaling of these values during the computation, to keep the values found in the DP table asymptotically close to 1, while still allowing for analysis of the Boltzmann distribution.
To that purpose, one introduces a rescaling factor α which is applied, as a multiplicative term, to some of the DP rules. A rescaling is homogeneous for a pair of (sub)trees (G1(g1), G2(g2)) (abridged into (g1, g2) from now) when the number of occurrences of α, encountered during the generation of a given solution F, only depends on (g1, g2) and not on specific features of F. Let us denote by κg1, g2 the number of occurrences of α for (g1, g2), then the rescaled contribution of a given solution F is now , while the rescaled partition function, computed by the modified DP scheme, is given by
| (4) |
A direct execution of the stochastic backtrack algorithm then returns each forest F with probability
| (5) |
In other words, the introduction of the rescaling does not induce any bias in the stochastic sampling, i.e. the sampling still follows a Boltzmann distribution.
On the other hand, α can be used to constrain the values to avoid numerical overflows. For instance, setting yields . Furthermore, if the rescaling terms are regularly distributed during the execution of the DP scheme, then the intermediate values c0|1(g1, g2) also typically remain close to 1, thereby avoiding numerical over/underflows. In practice, is the end product of the computation, and thus cannot be used to determine a suitable value for α. However, any value that avoids numerical over/underflow can be used, so DeClone accepts as input a prescribed value for α. Note also that α can also be typically inferred from a partial computation, based on the first occurrence of an under/overflow in the DP matrices. To apply these concepts in the context of the DeCo DP scheme, we are left to find an homogeneous rescaling.
Fortunately, we observe that the number of recursive calls , where ≠ GDup and ≠ GDup, is provably constant within the solutions generated from any call c0|1(g1, g2). For the sake of simplicity, we assume here that calls of the form c0|1(g1, g2), where e(g1) = GLoss (resp. e(g2) = GLoss), are expanded into calls c0|1(g1, ag2) and c0|1(g1, bg2) (resp. c0|1(ag1, g2) and c0|1(bg1, g2)), unless g2 (resp. g1) is also a leaf. From this observation that can be tediously verified by induction, we adapt the DP scheme as illustrated by Figure 3.
Inside-Outside algorithm
While the sampling algorithm described above provides a flexible, easy to implement, approach to analyze the Boltzmann distribution, it only allows for the computation of estimates for properties of interest (for example the occurrence of a specific ancestral adjacency in evolutionary scenarios), whose accuracy may critically depend on the number of samples, the - a priori unknown - variance of the underlying distribution, or other factors. However, whenever the property of interest, in conjunction with the DP scheme, fulfills certain technical conditions [18], it is possible to compute its expectation exactly in polynomial time, by transforming the DP scheme using a variant of the inside-outside algorithm.
More precisely, our objective is to compute the probabilities associated with each of the left-hand-side (LHS) to right-hand-side (RHS) transitions in the DP recurrence. Let us denote by l → r an LHS/RHS transition, such that
| (6) |
and by Fl→r the set of forests whose production borrows the l → r transition. The Boltzmann probability of (l → r) is then defined as
| (7) |
Since is known, it is sufficient to compute the numerator of the above fraction, i.e. the total Boltzmann factor of the forests that feature (l → r). On the other hand, the number of forests in typically grows exponentially on n1 + n2, so one must find an efficient strategy for computing this summation.
The principle of the inside-outside algorithm [15] is to decompose each of the executions, associated with a forest in , into: a) an inside part, generated from the recursive calls in the RHS r; and b) an outside part, which denotes the context in which the LHS l appears, i.e. an execution of the DP scheme which features a recursive call to l, and is truncated at that point. Let us remark that the inside and outside parts are independent, i. e. any inside part can be combined with any outside part to form a valid execution of the DP scheme, and the score of the associated forest is simply obtained by summing the scores of its two parts. Thus, the total Boltzmann factor of the forests , can be decomposed as
where Cr denotes the constant score increment in the RHS, and is the outside partition function, i.e. the total Boltzmann factor of all outside parts that are truncated at l. This term can be computed in by inverting the DP scheme of Figure 3 in a purely generic, yet quite technical, fashion [18]. To limit the risk of mistakes in the derivation/implementation of DP equations for d0|1(g1, g2), we implemented an ad hoc parser, based on the inversion principle described by Ponty and Saule [18].
Once the probabilities P (l → r) are known, it is possible to determine the probability of an (ancestral) adjacency (g1, g2) by simply summing over the probabilities of transitions that infer such an adjacency, i. e. that feature a recursive call of the form c1(g1, g2) within their RHS. Iterating this over all (g1, g2) pairs, one obtains an adjacency matrix, as shown in Figure 2.
Results and discussion
Data
We re-analyzed a dataset described in [5] composed of 5, 039 reconciled gene trees and 50, 389 extant gene adjacencies, forming 6, 074 DeCo instances, with genes taken from 36 mammalian genomes from the Ensembl database in 2012. In [5], these data were analyzed using the DeCo algorithm that computed a single parsimonious adjacency forest per instance. All together, these adjacency forests defined 112, 188 (resp. 96, 482) ancestral and extant genes (resp. adjacencies), where, by "ancestral adjacency", we mean adjacency that involves two genes g1 and g2 whose descendants in their respective gene trees satisfy that they do not belong to the same species s(g1) (equal to s(g2)), i.e. g1 and g2 are pre-speciation genes, that were not duplicated within their species (this choice is motivated by the fact that the reconstruction of ancestral genomes considers pre-speciation genomes. More important, we can observe 5, 817 ancestral genes participating to three or more ancestral adjacencies, which represent a significant level of syntenic conflict (close to 5%), as a gene can only be adjacent to at most two neighboring genes along a chromosome.
DeCo scores, solution space
Unlike reconciled gene trees, whose mutation cost can be high, most adjacency forests have a relatively low cost, with only 32 instances leading to forest of score 5 or above, while the average number of parsimonious syntenic events (adjacency gain and break) is 1.25. This illustrates the fact that syntenic events, that are due to genome rearrangements, are rare evolutionary events, which suggests that parsimony is a relevant criterion for such characters, and that robustness of syntenic characters with respect to the whole solution space should be assessed in terms of optimal or slightly suboptimal evolutionary scenarios.
Boltzmann sampling and exact Boltzmann probabilities
For each instance, we sampled 1, 000 adjacency forests under the Boltzmann distribution, for three values of kT, 0.001, 0.1, 0.5, and recorded the frequency of all observed ancestral adjacencies. Then for the same values of kT, we computed the exact Boltzmann probability of all potential ancestral adjacencies using the inside-outside algorithm. The result observed were very similar whether sampling or exact probabilities were considered. However, the time required to compute exact Boltzmann probabilities is polynomial, so the exact Boltzmann approach based on the inside-outside algorithm should naturally be favoured in applications. In consequence, we discuss only the case of exact Boltzmann probabilities below.
The main difference between the three values of kT is that, with kT = 0.5, non-optimal adjacency forests have a higher Boltzmann probability in the Boltzmann distribution, while kT = 0.1 skews the distribution toward optimal adjacency forests and slightly suboptimal ones, and kT = 0.01 ensures that the probability of sub-optimal adjacency forests is extremely low and almost does not contribute to the partition function. We then looked at the numbers of ancestral adjacencies, genes and syntenic conflicts from ancestral adjacencies in terms of Boltzmann probability. Table 1 below summarizes the obtained results.
Table 1.
Characteristics of ancestral genes and adjacencies from observed ancestral adjacencies filtered by Boltzmann probability (leftmost column), with different kT values.
| Ancestral genes | Ancestral adjacencies | Syntenic conflicts | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Proba. | kT = 0.5 | kT = 0.1 | kT = 0.01 | kT = 0.5 | kT = 0.1 | kT = 0.01 | kT = 0.5 | kT = 0.1 | kT = 0.01 |
| ≥ 0.1 | 122,593 | 120,495 | 120,531 | 137,133 | 116,671 | 116,873 | 31,415 | 14,655 | 14,863 |
| ≥ 0.2 | 121,896 | 119,792 | 119,864 | 130,928 | 113,835 | 114,109 | 26,415 | 12,631 | 12,871 |
| ≥ 0.3 | 120,869 | 118,871 | 118,913 | 122,073 | 110,472 | 110,676 | 19,729 | 10,259 | 10,471 |
| ≥ 0.4 | 118,240 | 117,883 | 117,990 | 111,224 | 107,492 | 107,845 | 11,569 | 8,439 | 8,738 |
| ≥ 0.5 | 113,023 | 116,476 | 116,810 | 100,265 | 103,963 | 105,015 | 5,658 | 6,578 | 7,373 |
| ≥ 0.6 | 104,970 | 114,699 | 114,902 | 88,943 | 100,064 | 100,511 | 3,044 | 5,081 | 5,387 |
| ≥ 0.7 | 92,647 | 112,834 | 112,924 | 75,063 | 96,546 | 96,789 | 1,363 | 4,104 | 4,301 |
| ≥ 0.8 | 75,408 | 110,272 | 110,503 | 58,665 | 92,181 | 92,621 | 492 | 3,290 | 3,496 |
| ≥ 0.9 | 45,753 | 107,769 | 107,863 | 34,005 | 88,132 | 88,373 | 65 | 2,717 | 2,886 |
| = 1 | 13 | 16,470 | 106,903 | 7 | 10,798 | 87,048 | 0 | 0 | 2,690 |
The difference observed between the results with different values of kT supports that parsimony is an appropriate criterion for looking at gene adjacency evolution. Indeed, in the results obtained with kT = 0.5, that gives a higher probability to non-optimal adjacency forests, it appears that the number of conserved ancestral adjacencies drops sharply after probability 0.6, showing that very few ancestral adjacencies appear with high probability. However, with kT = 0.1 and kT = 0.01, by taking a high probability threshold (starting at a threshold of 0.6), we reduce significantly the number of syntenic conflicts while maintaining a relatively similar number of ancestral genes than the experiments described in [5]; this observation illustrates the potential of the ensemble approach compared to the classical dynamic approach that relies on a single arbitrary optimal solution. Next, the experiment with kT = 0.01 that considers only co-optimal scenarios (the probability of non-optimal scenarios falls under the numerical precision) shows that, despite conserving only ancestral adjacencies with maximal support in terms of Boltzmann probability, a significant number of syntenic conflicts remains. We conjecture that this is due to errors in the considered reconciled gene trees, and it would be interesting to see if the information about highly supported conflicting adjacencies can be used to correct reconciled gene tree.
Conclusions
The main contribution of our work is an extension of the DeCo dynamic programming scheme to consider adjacency forests in a probabilistic framework, under the Boltzmann distribution. The application of our algorithms on a mammalian genes dataset, together with a simple threshold-based, approach to filter ancestral adjacencies, proved to be effective to reduce significantly the number of syntenic conflicts, illustrating the interest of the ensemble approach. This preliminary work raises several questions and can be extended along several lines. Among them, we can cite two of immediate interest. First, given the Boltzmann probabilities of the adjacency gains and breaks associated to ancestral adjacencies, we could use them to compute a Maximum Expected Accuracy adjacency forest, which is a parsimonious adjacency forest in a scoring model where each event is weighted by Boltzmann probability (see [20] for an example of this approach for RNA secondary structures). This would provide a unique evolutionary scenario per instance. Next, we considered here an evolutionary model based on speciation, duplication and loss. A natural extension would be to include the event of lateral gene transfer in the model. Efficient reconciliation algorithms exist for several variants of this model [3,4], together with an extension of DeCo, called DeCoLT [21]. DeCoLT is also based on dynamic programming, and it is likely that the techniques we developed in the present work also apply to this algorithm.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors participated in all aspects of the study.
Acknowledgements
J.P.P.Z. visit to Simon Fraser University was funded by the São Paulo Research Foundation (FAPESP).
Declarations
The publication charges this article were funded by the Simon Fraser University (SFU) Open Access fund.
This article has been published as part of BMC Bioinformatics Volume 16 Supplement 19, 2015: Brazilian Symposium on Bioinformatics 2014. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcbioinformatics/supplements/16/S19
References
- Liberles DA. Ancestral Sequence Reconstruction. Oxford University Press, Oxford, UK; 2007. [Google Scholar]
- Csürös M. RECOMB-CG. Lecture Notes in Computer Science. Vol. 5267. Springer, Berlin, Germany; 2008. Ancestral reconstruction by asymmetric Wagner parsimony over continuous characters and squared parsimony over distributions; pp. 72–86. doi:10.1007/978-3-540-87989-3_6. [Google Scholar]
- Bansal MS, Alm EJ, Kellis M. Efficient algorithms for the reconciliation problem with gene duplication, horizontal transfer and loss. Bioinformatics. 2012;28(12):283–291. doi: 10.1093/bioinformatics/bts225. doi:10.1093/bioinformatics/bts225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon J-P, Scornavacca C, Gorbunov KY, Szöllosi GJ, Ranwez V, Berry V. RECOMB-CG. Lecture Notes in Computer Science. Vol. 6398. Springer, Berlin, Germany; 2010. An efficient algorithm for gene/species trees parsimonious reconciliation with losses, duplications and transfers; pp. 93–108. doi:10.1007/978-3-642-16181-0_9. [Google Scholar]
- Bérard S, Gallien C, Boussau B, Szöllosi GJ, Daubin V, Tannier E. Evolution of gene neighborhoods within reconciled phylogenies. Bioinformatics. 2012;28(18):382–388. doi: 10.1093/bioinformatics/bts374. doi:10.1093/bioinformatics/bts374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biller P, Feijão P, Meidanis J. Rearrangement-based phylogeny using the single-cut-or-join operation. IEEE/ACM Trans. Comput. Biology Bioinform. 2013;10(1):122–134. doi: 10.1109/TCBB.2012.168. doi:10.1109/TCBB.2012.168. [DOI] [PubMed] [Google Scholar]
- Gagnon Y, Blanchette M, El-Mabrouk N. A flexible ancestral genome reconstruction method based on gapped adjacencies. BMC Bioinformatics. 2012;13(S-19):4. doi: 10.1186/1471-2105-13-S19-S4. doi:10.1186/1471-2105-13-S19-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Ratan A, Raney BJ, Suh BB, Zhang L, Miller W, Haussler D. DUPCAR: reconstructing contiguous ancestral regions with duplications. Journal of Computational Biology. 2008;15(8):1007–1027. doi: 10.1089/cmb.2008.0069. doi:10.1089/cmb.2008.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csürös M. Models and Algorithms for Genome Evolution. Springer, Berlin, Germany; 2013. How to infer ancestral genome features by parsimony: Dynamic programming over an evolutionary tree; pp. 29–45. doi:10.1007/978-1-4471-5298-9_3. [Google Scholar]
- Libeskind-Hadas R, Wu Y-C, Bansal MS, Kellis M. Pareto-optimal phylogenetic tree reconciliation. Bioinformatics. 2014;30(12):87–95. doi: 10.1093/bioinformatics/btu289. doi:10.1093/bioinformatics/btu289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal MS, Alm EJ, Kellis M. Reconciliation revisited: Handling multiple optima when reconciling with duplication, transfer, and loss. Journal of Computational Biology. 2013;20(10):738–754. doi: 10.1089/cmb.2013.0073. doi:10.1089/cmb.2013.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scornavacca C, Paprotny W, Berry V, Ranwez V. Representing a set of reconciliations in a compact way. J. Bioinformatics and Computational Biology. 2013;11(2) doi: 10.1142/S0219720012500254. doi:10.1142/S0219720012500254. [DOI] [PubMed] [Google Scholar]
- Doyon J-P, Hamel S, Chauve C. An efficient method for exploring the space of gene tree/species tree reconciliations in a probabilistic framework. IEEE/ACM Trans. Comput. Biology Bioinform. 2012;9(1):26–39. doi: 10.1109/TCBB.2011.64. doi:10.1109/TCBB.2011.64. [DOI] [PubMed] [Google Scholar]
- McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29(6-7):1105–1119. doi: 10.1002/bip.360290621. doi:10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- Baker JK. Trainable grammars for speech recognition. The Journal of the Acoustical Society of America. 1979;65(S1):132–132. doi:10.1121/1.2017061. [Google Scholar]
- Arvestad L, Lagergren J, Sennblad B. The gene evolution model and computing its associated probabilities. J. ACM. 2009;56(2) doi:10.1145/1502793.1502796. [Google Scholar]
- Mahmudi O, Sjöstrand J, Sennblad B, Lagergren J. Genome-wide probabilistic reconciliation analysis across vertebrates. BMC Bioinformatics. 2013;14(S-15):10. doi: 10.1186/1471-2105-14-S15-S10. doi:10.1186/1471-2105-14-S15-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponty Y, Saule C. In: Algorithms in Bioinformatics (Proceedings of WABI'11). Lecture Notes in Computer Science. Przytycka, T., Sagot, M.-F, editor. Vol. 6833. Springer, Berlin Heidelberg, Germany; 2011. A combinatorial framework for designing (pseudoknotted) RNA algorithms; pp. 250–269. doi:10.1007/978-3-642-23038-7_22. [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie/Chemical Monthly. 1994;125(2):167–188. doi:10.1007/BF00818163. [Google Scholar]
- Clote P, Lou F, Lorenz WA. Maximum expected accuracy structural neighbors of an RNA secondary structure. BMC Bioinformatics. 2012;13(S-5):6. doi: 10.1186/1471-2105-13-S5-S6. doi:10.1186/1471-2105-13-S5-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson M, Szöllosi GJ, Daubin V, Tannier E. Lateral gene transfer, rearrangement, reconciliation. BMC Bioinformatics. 2013;14(S-15):4. doi: 10.1186/1471-2105-14-S15-S4. doi:10.1186/1471-2105-14-S15-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]