

Abstract
Both selective estrogen receptor modulators and aromatase inhibitors are widely used for the treatment of breast cancer. Compounds with both aromatase inhibitory and estrogen receptor modulatory activities could have special advantages for treatment of breast cancer. Our previous efforts led to the discovery of norendoxifen as the first compound with dual aromatase inhibitory and estrogen receptor binding activities. To optimize its efficacy and aromatase selectivity versus other cytochrome P450 enzymes, a series of structurally related norendoxifen analogues were designed and synthesized. The most potent compound, 4'-hydroxynorendoxifen (10), displayed elevated inhibitory potency against aromatase and enhanced affinity for estrogen receptors when compared to norendoxifen. The selectivity of 10 for aromatase versus other cytochrome P450 enzymes was also superior to norendoxifen. 4'-Hydroxynorendoxifen is therefore an interesting lead for further development to obtain new anticancer agents of potential value for the treatment of breast cancer.
Introduction
Breast cancer is the most frequently diagnosed cancer in women. It is estimated that one out of eight women in the United States (about 12%) will develop invasive breast cancer during the course of her life.1 Breast cancer is second only to lung cancer as the cause of cancer-related deaths in women.2 Most breast cancer tumors are estrogen receptor (ER) positive, making them suitable for antiestrogen therapy. The selective estrogen receptor modulator (SERM) tamoxifen (1, Figure 1) has been widely used for treatment of ER-positive breast cancer, with efficacy documented in various randomized clinic trials.3, 4 However, the use of tamoxifen is limited by intrinsic and acquired drug resistance, and long-term tamoxifen treatment also increases the occurrence of endometrial cancer.3, 5, 6 For postmenopausal breast cancer patients, aromatase inhibitors (AIs), e.g. letrozole (3) and anastrozole (4), have been used to inhibit estrogen biosynthesis. Several comparative clinical trials have demonstrated that AIs are superior to tamoxifen in the treatment of postmenopausal women with ER-positive breast cancer.7-11 Unfortunately, since AIs nonselectively deplete estrogen in the whole body, they inevitably lead to severe musculoskeletal pain, reduction of bone density, and increased frequency of fractures and cardiovascular events.12-16
Figure 1.

The structures of the selective estrogen receptor modulator tamoxifen, its active metabolite 4-hydroxytamoxifen, and the aromatase inhibitors letrozole and anastrozole.
One possible strategy to improve patient compliance and treatment outcomes is to develop agents with dual AI and SERM activities. One of the potential benefits of a dual AI/SERM agent is improved efficacy. It is possible that the AI and SERM activities could act synergistically to inhibit breast tumor growth. According to Brodie et al., a combination of the aromatase inhibitor letrozole and the estrogen receptor antagonist fulvestrant was more effective than either letrozole or fulvestrant alone in suppressing breast tumor growth, especially in delaying the development of tumor resistance.17 For this reason, dual AI/SERM agents are expected to have efficacy superior to tamoxifen and conventional aromatase inhibitors. A second advantage of dual AI/SERM agents could be fewer side effects. The SERM activity of a dual AI/SERM agent may stimulate estrogen receptors in non-cancerous tissues and ameliorate the side effects (e.g. osteoporosis, musculoskeletal pain, and cardiovascular events) caused by estrogen depletion. This hypothesis is supported by the ATAC (anastrozole, tamoxifen, alone or in combination) trial, which demonstrated that a combination of the AI anastrozole and the SERM tamoxifen resulted in fewer bone fractures than when anastrozole was used alone.9, 18 Although the ATAC trial showed no superior therapeutic effect of the tamoxifen plus anastrozole combination over anastrozole alone,9 this result may not be generalizable to all SERM and AI combinations or to dual AI/SERM agents. Of note, the serum concentrations of anastrozole were lower in the combination arm of the ATAC study, and this fact challenges the generalized conclusion that all SERM/AI combinations would inevitably produce no additional benefit relative to an AI alone.
Norendoxifen (5, Figure 2) is a metabolite of tamoxifen. In 2012, we reported that norendoxifen is a potent aromatase inhibitor and that it has good selectivity for aromatase versus other cytochrome P450 enzymes.19 In 2013, we disclosed the first synthesis of (E,Z)-norendoxifen (5). Biological testing results confirmed the aromatase inhibitory activity of (E,Z)-norendoxifen and further established high affinity for both ER-α and ER-β.20 Next, the E- and Z-norendoxifen isomers (E-5 and Z-5) were prepared via stereoselective synthetic routes. The biological testing results revealed that E-norendoxifen is the more potent aromatase inhibitor, while Z-norendoxifen has higher binding affinity for both ER-α and ER-β.
Figure 2.

The structures and biological activities of (E,Z)-, Z- and E-norendoxifen.
The potent aromatase inhibitory activity together with the high binding affinity for both ER-α and ER-β support the further utilization of norendoxifen as a lead compound for the development of possible dual AI/SERM agents. Moreover, the fact that millions of patients have already been exposed to norendoxifen as a metabolite of tamoxifen supports a high safety profile expected for norendoxifen and related compounds. In this report, a series of norendoxifen analogues were designed, synthesized and tested with the aim to optimize the efficacy against both aromatase and ER, explore the structure-activity relationships, and improve the aromatase selectivity versus other cytochrome P450 enzymes.
Results and Discussion
Structure-based Optimization
To assist rational drug design, the interaction of E-norendoxifen with aromatase was investigated. A hypothetical binding model of E-norendoxifen in the crystal structure of the active site of aromatase (PDB code: 3s7921) obtained by molecular docking is displayed in Figure 3a.20 Since this model is consistent with the structure-activity relationships of previously published analogues,20 it was used to guide further structure-based optimization.
Figure 3.

The hypothetical binding model of E-norendoxifen in the active site of aromatase (a) and Z-norendoxifen in the active site of estrogen receptor-α (b), and the molecular modifications proposed for structure-based optimization (c).
To investigate the interaction of Z-norendoxifen with ER-α, a hypothetical binding model of Z-norendoxifen in the ligand-binding site of ER-α was obtained by mutating the crystal structure of the 4-hydroxytamoxifen (2, Figure 1) complex with ER-α (PDB code: 3ert22), followed by full energy minimization using Amber 10 (Figure 3b). Considering the high structural similarity between Z-norendoxifen and 4-hydroxytamoxifen, the model proposed in Figure 3b is a logical one to use for further structure-based optimization. Since the ligand binding pocket of ER-β is nearly identical to ER-α (only two residues were different: Met421 and Leu384 in ER-α correspond to Ile and Met in ER-β, respectively), one might expect that structure modifications of norendoxifen would result in similar effects on binding to both ER-α and ER-β. This notion is generally supported by the ER-α and ER-β binding data listed in Figure 2. Therefore, molecular modeling of the binding of Z-norendoxifen to ER-β was not performed.
Based on the hypothetical binding models shown in Figure 3a-b, a series of structurally related norendoxifen analogues were designed and the optimization strategies are summarized in Figure 3c. Detailed rationales for the proposed modifications are described below.
(1) Incorporating a hydroxyl group in the para position of the “A” ring
For aromatase, attaching a hydroxyl group in this position was proposed in order to increase aromatase inhibitory activity of E-norendoxifen by forming a new hydrogen bond with the carbonyl group of Ile133 or the guanidine group of Arg115 (see Figure 3 for ring labeling). For ER, this hydroxyl group would increase affinity of Z-norendoxifen by hydrogen bonding to the imidazole ring of His524.
(2) Replacing the “B” ring hydroxyl group with hydrogen or an amino group
The modeling results indicate that the “B” ring para hydroxyl group of Z-norendoxifen is crucial for ER binding by forming bifurcated hydrogen bonds with Glu353 and Arg394. This hydroxyl group is also thought to be important for aromatase inhibitory activity of E-norendoxifen because it hydrogen bonds to the backbone carbonyl group of Leu372. The importance of this hydroxyl group might be verified by preparing analogues with the hydroxyl group replaced by hydrogen or an amino group.
(3) Introducing iron-coordinating groups or various alkyl groups in the location of the ethyl side chain
An iron-coordinating group (usually imidazole or 1,2,4-triazole) is present in most aromatase inhibitors, [e.g. letrozole (3) and anastrozole (4)], and it is a crucial pharmacophore for aromatase inhibitory activity.23 According to the hypothetical binding model of E-norendoxifen to aromatase, the ethyl side chain is pointing toward the heme iron, with the distance from the methyl carbon to heme iron being 3.9 Å. Introducing functional groups (−CN, imidazole or 1,2,4-triazole) that could coordinate to the heme iron is therefore expected to improve aromatase inhibitory activity. Besides iron-coordinating groups, various alkyl groups were also introduced in this position to investigate their effects on binding to aromatase and ER.
(4) Replacing the side chain terminal amino group with different substituents, including hydrazine, hydroxyl, carboxyl, and halogens
Based on the modeling results, the terminal amino group is essential for both the aromatase inhibitory activity of E-norendoxifen and the ER binding affinity of Z-norendoxifen because it forms a salt bridge with Asp309 of aromatase and Asp351 of ER. Replacing this amino group with different substituents would be useful to confirm the importance of a positively charged group in this position for the biological activity.
(5) Replacing the ether oxygen with a methylene group
According to the hypothetical binding model of E-norendoxifen with aromatase, the ether oxygen forms a hydrogen bond with the side chain of Ser478. The contribution of this hydrogen bond to the aromatase inhibitory activity could be determined by eliminating the ether functionality. For ER, the ether oxygen is proposed to have no direct interaction with surrounding residues, and therefore replacement of the ether oxygen with a methylene group would not be expected to affect ER binding. A potential benefit of replacing the ether oxygen with a methylene group would be to increase metabolic stability in vivo by preventing oxidative O-dealkylation.
Chemistry and Biological Activity
Synthesis of (E,Z)-Norendoxifen Analogues with Different Substituents on the “A” Ring
To synthesize “A” ring hydroxylated (E,Z)-norendoxifen 10, 4,4'-dihydroxybenzophenone (6) was first alkylated with 2-iodoacetamide (Scheme 1). The benzophenone 7 and propiophenone 824 were reacted under McMurry cross-coupling conditions to provide the product 9 with E/Z ratio 4.5:1. The product 9 quickly isomerized to a 1:1 mixture of E and Z isomers when dissolved in chloroform and kept at room temperature overnight. In the last step, deprotection of the pivaloyl group and reduction of the amide group were finished in one pot to afford the product 10 as a 1:1 mixture of E and Z isomers.
Scheme 1.

Synthesis of 10a
aReagents and conditions: (a) ICH2CONH2, Cs2CO3, DMF, 37%; (b) Zn, TiCl4, THF, 55%; (c) LiAlH4, AlCl3, THF, 70%.
Compound 14a was designed by attaching a fluorine atom to the “A” ring of norendoxifen. The symmetrical compound 14b was designed by replacing the benzene “A” ring with an ethyl group. To prepare 14a-b, 4,4'-dihydroxybenzophenone (6) and ketones 11a-b were reacted under McMurry cross-coupling reaction conditions to provide the diphenols 12a-b (Scheme 2). Treatment of the diphenols 12a-b with one equivalent of 2-iodoacetamide in the presence of potassium carbonate afforded the mono-alkylated products 13a and 13b in 40% and 47% yields, respectively. Then the amide group was reduced with LiAlH4 to provide the desired products 14a-b in good yield, with 14a being obtained as an isomeric mixture.
Scheme 2.

Synthesis of 14a-ba
aReagents and conditions: (a) Zn, TiCl4, THF, 91-96%; (b) ICH2CONH2, K2CO3, acetone, 40-47%; (c) LiAlH4, AlCl3, THF, 44-76%.
The biological testing results for compounds 10 and 14a-b are summarized in Table 1. According to the testing results, introducing a hydroxyl group in the para position of the “A” ring of 10 increased both aromatase inhibitory activity and ER binding affinity. This supported the expectation that a hydroxyl group in this position would benefit aromatase inhibitory activity of E-norendoxifen via hydrogen bonding with Ile133 or Arg115 and enhance ER binding affinity of Z-norendoxifen by forming an additional hydrogen bond with His524. Incorporating a fluorine atom in this position (14a) decreased aromatase inhibitory activity but only had a minor negative effect on ER binding. Replacement of the “A” ring with an ethyl group (14b) decreased both aromatase inhibitory activity and ER binding affinity. Letrozole was used as a positive control for aromatase inhibition and it displayed an IC50 of 5.3 nM.25
Table 1.

| ||||
|---|---|---|---|---|
| Compound | R | Aromatase (IC50, nM) | ER-α (EC50, nM) | ER-β (EC50, nM) |
| Norendoxifen | -H | 102 ± 33 | 27.0 ± 4.8 | 35.2 ± 16.8 |
| 10 | -OH | 45.0 ± 3.0 | 15.0 ± 3.3 | 9.5 ± 0.2 |
| 14a | -F | 1020 ± 300 | 87.8 ± 5.8 | 49.2 ± 13.2 |
| 14b | 1320 ± 1 | 277 ± 61 | 202 ± 21 | |
The values are mean values of at least three experiments.
Compound 14b has no E or Z isomers, while the rest of the compounds were tested as 1:1 mixtures of E and Z isomers.
Synthesis of Norendoxifen Analogues with Different Substituents on the “B” Ring
To validate the importance of the “B” ring hydroxyl group, an analogue 18 without this hydroxyl group was synthesized. Starting from 4-hydroxybenzophenone (15), the McMurry cross-coupling reaction with propiophenone provided the phenol 16 (Scheme 3). The phenolic hydroxyl group was alkylated with 2-iodoacetamide and followed by LiAlH4 reduction of the amide 17 to afford the product 18.
Scheme 3.

Synthesis of 18a
aReagents and conditions: (a) propiophenone, Zn, TiCl4, THF, 98%; (b) ICH2CONH2, acetone, K2CO3, 73%; (c) LiAlH4, AlCl3, THF, 63%.
To explore the effect of replacing the “B” ring hydroxy group with an amino group, compounds 25a and 25b were prepared as shown in Scheme 4. The benzophenone 21 was prepared via Friedel-Crafts reaction of benzoyl chloride 19 and anisole 20 as described by Davies et al.26 The methoxy group of 21 was subsequently cleaved and the phenol was alkylated with dibromoethane. Treatment of 23 with propiophenone or 8 under McMurry cross-coupling reaction conditions directly provided the anilines 24a and 24b. In the last step, the side chain amination and removal of the pivalate group were finished in one-pot by treating 24a and 24b with ammonium hydroxide to afford the products 25a and 25b in good yield.
Scheme 4.

Synthesis of 25a-ba
aReagents and conditions: (a) AlCl3, CH2Cl2, 55%; (b) HBr, AcOH, 94%; (c) dibromoethane, K2CO3, acetone, 68%; (d) propiophenone or 8, Zn, TiCl4, THF, 53-55%; (e) NH4OH, NaI, THF, 52-56%.
The biological testing results for compound 18, 25a and 25b are summarized in Table 2. According to the testing results, removing the para hydroxyl group in the “B” ring of norendoxifen to produce 18 decreased aromatase inhibitory activity significantly and resulted in complete loss of the binding affinity to both ER-α and ER-β. Replacing the hydroxyl group of norendoxifen with an amino group in 25a improved aromatase inhibitory activity but decreased ER binding affinity. These results are consistent with the proposal that this hydroxyl group binds to aromatase by hydrogen bonding with Leu372 and binds to ER via bifurcated hydrogen bonds with Glu353 and Arg394. Compound 25b displayed elevated potency against both aromatase and ER when compared with 25a. This fact is consistent with the results with norendoxifen and 10 showing that incorporating a hydroxyl in the para position of the “A” ring benefits both aromatase inhibitory activity and ER binding affinity.
Table 2.

| |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Aromatase (IC50, nM) | ER-α (EC50, nM)b | ER-β (EC50, nM)b |
| Norendoxifen | -H | -OH | 102 ± 33 | 27.0 ± 4.8 | 35.2 ± 16.8 |
| 18 | -H | -H | 913 ± 100 | 0% competition | 0% competition |
| 25a | -H | -NH2 | 53.1 ± 1.8 | 274 ± 44 | 182 ± 82 |
| 25b | -OH | -NH2 | 48.1 ± 8.1 | 144 ± 62 | 49.9 ± 9.2 |
The values are mean values of at least three experiments.
Percent ER competition was determined at the concentration of 100,000 nM for each compound.
Compound 25a were tested as 2.5:1 mixture of E and Z isomers, and the rest of the compounds were tested as 1:1 mixtures of E and Z isomers.
Synthesis of Norendoxifen Analogues with Different Replacements for the Ethyl Side Chain
The synthetic route for compounds 29a-e is outlined in Scheme 5. The McMurry cross-coupling reaction of 4,4'-dihydroxybenzophenone 6 and ketones 26a-e provided the diphenols 27a-e. The diphenols 27a-e were treated with one equivalent of 2-iodoacetamide in the presence of potassium carbonate to provide the mono-alkylated products 28a-e as the major products. Further reduction of the amides 28a-e with LiAlH4 afforded the analogues 29a-e in good yield.
Scheme 5.

Synthesis of 29a-ea
aReagents and conditions: (a) Zn, TiCl4, THF, 65-96%; (b) ICH2CONH2, acetone, K2CO3, 27-39%; (c) LiAlH4, AlCl3, THF, 54-82%.
Compound 32 was prepared via a slightly revised route (Scheme 6). 4,4'-Dihydroxybenzophenone 6 was first mono-alkylated with dibromoethane to provide 30 in good yield. The product 30 underwent McMurry cross-coupling reaction with benzaldehyde to afford 31. In the last step, the analogue 32 was obtained by amination of the bromide 31 with ammonium hydroxide.
Scheme 6.

Synthesis of 32a
aReagents and conditions: (a) dibromoethane, acetone, H2O, K2CO3, 36%; (b) benzaldehyde, Zn, TiCl4, THF, 57%; (c) NH4OH, NaI, THF, 75%.
To synthesize the conformationally restricted norendoxifen analogue 36, 4,4'-dihydroxybenzophenone (6) and ketone 33 underwent the McMurry cross-coupling reaction to afford the diphenol 34 (Scheme 7). The diphenol 34 was mono-alkylated with 2-iodoacetamide followed by LiAlH4 reduction to afford the product 36 in good yield.
Scheme 7.

Synthesis of 36a
aReagents and conditions: (a) Zn, TiCl4, THF, 85%; (b) ICH2CONH2, K2CO3, acetone, 40%; (c) LiAlH4, AlCl3, THF, 66%.
In order to make norendoxifen analogues with an iron-coordinating group in the location of the ethyl side chain, the diphenol 27a was treated with one equivalent of methyl chloromethyl ether to provide the mono-protected product 37 in 41% yield (Scheme 8). Treatment of the phenol 37 with 3827 in the presence of cesium carbonate converted 37 to 39 in an isolated yield of 72%. Compound 39 underwent a series of sequential reactions including bromination with NBS, alkylation of KCN, and deprotection of MOM and Boc groups with HCl to afford the product 40 in very good yield. The product 41 with an imidazole group was also obtained via a similar sequence of reactions (including bromination with NBS, alkylation of the anion derived from imidazole and deprotection of the MOM and Boc group with HCl).
Scheme 8.

Synthesis of 40-41a
aReagents and conditions: (a) NaH, MOMCl, THF, 41%; (b) Cs2CO3, DMF, 50 °C, 72%; (c) NBS, CCl4; (d) KCN, THF, H2O; (e) imidazole, NaH, THF; (f) MeOH, HCl, 64-81% in 3 steps.
To prepare 1,2,4-triazole-containing analogues, the intermediate 39 was first brominated with one equivalent of NBS and then treated with 1,2,4-triazole in the presence of NaH (Scheme 9). As expected, this provided a mixture of two isomers 42 and 43, which could be separated by silica gel column chromatography. Intermediate 42 was treated with HCl to remove the MOM and Boc groups and this afforded the product 44 as a 1:1 mixture of E and Z isomers. Treatment of compound 43 with HCl also provided a 1:1 mixture of E and Z isomers. Surprisingly, these two isomers Z-45 and E-45 could be separated by silica gel chromatography. The stereochemistries of Z-45 and E-45 were confirmed by NMR spectroscopy as described in the stereochemistry determination section.
Scheme 9.

Synthesis of 44, Z-45 and E-45a
aReagents and conditions: (a) NBS, CCl4; (b) NaH, 1,2,4-triazole, THF; (c) MeOH, HCl, 7-31% in 3 steps.
The biological testing results for compound 29a-e, 32, 36, 40-41, 44, E-45 and Z-45 are summarized in Table 3. According to the testing results, replacement of the ethyl group with different alkyl groups (29a-e, 32) had different effects on aromatase, ER-α and ER-β. The ethyl group (norendoxifen) and chloroethyl group (29b, this group is also presents in the SERM toremifene) were optimal for aromatase inhibitory activity, while only the ethyl group was optimal for ER-α binding affinity. ER-β appeared to tolerate different alkyl groups in this position, because norendoxifen, compound 32, and compounds 29a-c displayed very similar potencies against ER-β. According to the hypothetical binding mode of Z-norendoxifen with ER-α (Figure 3b), the ethyl side chain is situated in a hydrophobic pocket surrounded by Met388, Met421 and Leu428. In ER-β, the Met421 was replaced with an Ile residue, which could account for the better tolerance of ER-β with different alkyl groups. Replacement of the ethyl group with a benzene ring in compound 29e decreased both aromatase inhibitory activity and ER binding affinity. Similarly, the cyclized, conformationally restricted analogue 36 of norendoxifen also showed decreased aromatase inhibitory activity and ER binding affinity. The incorporation of iron-coordinating groups in the location of the ethyl group led to the very potent aromatase inhibitors 40 and 41, but it also markedly decreased the binding affinity with ER. The increase of aromatase inhibitory activity might be due to the coordination of the iron by the nitrile group (40) or imidazole group (41). Interestingly, the 1,2,4-triazole analogue 44 displayed good aromatase inhibitory activity and moderate ER binding affinities, while the 1,3,4-triazole analogues E-45 and Z-45 showed moderate aromatase inhibitory activity and good ER binding affinities.
Table 3.
The Aromatase Inhibitory Activity and Estrogen Receptor Binding Affinities of 29a-e, 32, 36, 40-41, 44, E-45 and Z-45a,b,c

| ||||
|---|---|---|---|---|
| Compound | R | Aromatase (IC50, nM) | ER-α (EC50, nM)b | ER-β (EC50, nM)b |
| Norendoxifen | -CH2CH3 | 102 ± 33 | 27.0 ± 4.8 | 35.2 ± 16.8 |
| 32 | -H | 6290 ± 2160 | 1050 ± 270 | 19.5 ± 11.0 |
| 29a | -CH3 | 337 ± 13 | 675 ± 458 | 27.8 ± 18.7 |
| 29b | -CH2CH2Cl | 143 ± 2 | 265 ± 7 | 52.0 ± 22.0 |
| 29c | -CH2CH2CH3 | 261 ± 40 | 384 ± 161 | 36.5 ± 8.9 |
| 29d | -CH(CH3)2 | 328 ± 53 | 54% competition | 68% competition |
| 29e | -Ph | 1930 ± 160 | 653 ± 102 | 478 ± 97 |
| 40 | -CH2CN | 48.9 ± 12.4 | 48% competition | 64% competition |
| 41 |
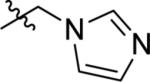
|
16.9 ± 0.8 | 59% competition | 74% competition |
| 44 |
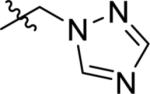
|
174 ± 10 | 290 ± 194 | 519 ± 345 |
| E-45 |
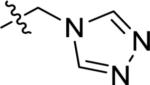
|
715 ± 10 | 56.5 ± 11.0 | 47.7 ± 25.4 |
| Z-45 |
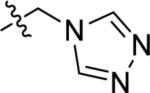
|
699 ± 63 | 176 ± 54 | 57.5 ± 33.4 |
| 36 | 493 ± 29 | 3180 ± 240 | 1350 ± 550 | |
The values are mean values of at least three experiments.
Percent ER competition was determined at the concentration of 100,000 nM for each compound.
Compound 29e has no E or Z isomers, compound E-45 and Z-45 were tested as pure E or Z isomers, while the rest of the compounds were tested as 1:1 mixtures of E and Z isomers.
Synthesis of Norendoxifen Analogues with Different Replacements of the Aminoethoxyl Side Chain
To make the hydroxy analogue 48 and carboxy analogue 49, the diphenol 4620 was first treated with ethyl 2-iodoacetate in the presence of potassium carbonate to provide the mono-alkylated product 47. Reduction of the ester group with LiAlH4 provided the hydroxy analogue 48. Hydrolysis of the ester group with KOH afforded the carboxy compound 49 (Scheme 10).
Scheme 10.

Synthesis of 48 and 49a
aReagents and conditions: (a) ICH2CO2Et, acetone, K2CO3, 23%; (b) LiAlH4, THF, 65%; (3) KOH, THF, H2O, 97%.
To make the bromo analogue 50 and the hydrazine analogue 51, the benzophenone 30 was reacted with propiophenone under McMurry cross-coupling reaction conditions to directly afford the product 50 in good yield (Scheme 11). Then, the bromine 50 was reacted with hydrazine to yield the product 51. Unfortunately, the product 51 was quite unstable and it decomposed in methanol at room temperature.
Scheme 11.

Synthesis of 50a
aReagents and conditions: (a) propiophenone, Zn, TiCl4, THF, 87%; (b) N2H4·H2O, THF.
The analogues 55a and 55b were prepared starting from benzophenone 52,20 which was alkylated with 1-bromo-2-chloroethane or dibromopropane to provide 53a and 53b (Scheme 12). Treatment of benzophenones 53a and 53b with propiophenone under McMurry cross-coupling conditions afforded 54a and 54b. Then, the pivalate group was removed and the side chain of the compound derived from 54b was aminated to yield analogues 55a and 55b.
Scheme 12.

Synthesis of 55a-ba
aReagents and conditions: (a) 1-bromo-2-chloroethane, acetone, K2CO3, 49%; (b) dibromopropane, CH3CN, Cs2CO3, 79%; (c) propiophenone, Zn, TiCl4, THF, 96%; (d) LiAlH4, THF, 58%; (e) NH4OH, NaI, THF, 54%.
The biological testing results for compounds 48-50 and 55a-b are summarized in Table 4. Replacement of the terminal amino group with a hydroxyl group, carboxyl group, or halogens in analogues 48-50 and 55a significantly impaired both aromatase inhibitory activity and ER binding affinity. This demonstrated the importance of a positively charged group in this position for interaction with Asp309 of aromatase and Asp351 of ER. Extending the aminoethoxyl side chain to the aminopropoxyl side chain in 55b had no influence on the binding affinity with ER, but decreased aromatase inhibitory activity significantly.
Table 4.
The Aromatase Inhibitory Activity and Estrogen Receptor Binding Affinities of 48-50 and 55a-ba,b,c,d

| ||||
|---|---|---|---|---|
| Entry | R | Aromatase (IC50, nM)b | ER-α (EC50, nM)c | ER-β (EC50, nM)c |
| Norendoxifen | -CH2NH2 | 102 ± 33 | 27.0 ± 4.8 | 35.2 ± 16.8 |
| 48 | - CH2OH | 30500 ± 2900 | 62% competition | 56% competition |
| 49 | -COOH | 3680 ± 190 | 55% competition | 57% competition |
| 50 | -CH2Br | 20% inhibition | 60% competition | 60% competition |
| 55a | -CH2Cl | 27% inhibition | 62% competition | 40% competition |
| 55b | -CH2CH2NH2 | 1240 ± 180 | 34.9 ± 11.4 | 21.3 ± 5.5 |
The values are mean values of at least three experiments.
Percent aromatase inhibition was determined at the concentration of 50,000 nM for each compound.
Percent ER competition was determined at the concentration of 100,000 nM for each compound.
All compounds were tested as 1:1 mixtures of E and Z isomers.
Synthesis of Norendoxifen Analogues in which the Ether Oxygen is Replaced with a Methylene Group
Compounds 61a-b were designed by replacing the ether oxygen with a methylene group and the synthetic route is outlined in Scheme 13. The benzophenone 57, which was prepared via Friedel-Crafts reaction of the commercially available benzoyl chloride 56 and anisole 20, was hydrolyzed and the product 58 underwent the McMurry cross-coupling reaction with propiophenone or 8 to afford 59a28 and 59b. A Heck coupling reaction of the bromides 59a-b with acrylamide provided 60a and 60b. The double bond in the side chain was selectively reduced by catalytic hydrogenation with Rh(PPh3)3Cl. The pivalate group was cleaved and the amide group was reduced in one pot with LiAlH4 to provide analogues 61a and 61b.
Scheme 13.

Synthesis of analogues 61a-ba
aReagents and conditions: (a) AlCl3, DCM, 89%; (b) HBr, AcOH, 89%; (c) propiophenone or 8, Zn, TiCl4, THF, 60-61%; (d) acrylamide, Pd(PPh3)4, TEA, DMF; (e) Rh(PPh3)3Cl, H2, MeOH; (f) LiAlH4, AlCl3, THF, 18-34% in 3 steps.
The biological testing results for compounds 61a-b are summarized in Table 5. Replacement of the ether oxygen with a methylene group in 61a decreased aromatase inhibitory activity when compared with norendoxifen but had no significant effect on ER binding. The decrease of aromatase inhibitory activity might be due to the loss of the hydrogen bonding of the ether oxygen with Ser478. Interestingly, compound 61b displayed an improved aromatase inhibitory activity but a decreased ER binding affinity when compared with 61a.
Table 5.

| |||||
|---|---|---|---|---|---|
| Entry | R | X | Aromatase (IC50, nM) | ER-α (EC50, nM) | ER-β (EC50, nM) |
| Norendoxifen | -H | -O- | 102 ± 33 | 27.0 ± 4.8 | 35.2 ± 16.8 |
| 61a | -H | -CH2- | 7340 ± 450 | 27.1 ± 15.0 | 13.3 ± 1.4 |
| 61b | -OH | -CH2- | 491 ± 35 | 1920 ± 620 | 293 ± 187 |
The values are mean values of at least three experiments.
All compounds were tested as 1:1 mixtures of E and Z isomers.
Summary of the Structure-Activity Relationships
In sum, 25 structurally related norendoxifen analogues have been synthesized and biologically tested. The structure-activity relationships are summarized in Figure 4. Generally, the structure-activity relationships are consistent with the molecular modeling predictions and this supports the reliability of utilizing the molecular modeling results for further lead optimization.
Figure 4.

Summary of structure-activity relationships for the prepared norendoxifen analogues.
aIn compound 61b, the -OH group is unfavorable for ER binding affinity.
The Inhibitory Activities of 4'-Hydroxynorendoxifen (10) Against Other Cytochrome P450 Enzymes
Among the prepared norendoxifen analogues, the 4'-hydroxynorendoxifen (10) displayed improved potency against both aromatase and ER when compared with norendoxifen. Interestingly, our recent research confirmed that 4'-hydroxynorendoxifen is an active metabolite of tamoxifen.29 The presence of 4'-hydroxynorendoxifen in human plasma after tamoxifen treatment suggests that it might contribute to the clinical effects of tamoxifen in breast cancer patients. Considering its potent AI activity and the high binding affinity for both ER-α and ER-β, 4'-hydroxynorendoxifen was selected for further evaluation. To investigate the likelihood of drug interactions, the inhibitory activities of 4'-hydroxynorendoxifen against major cytochrome P450 enzymes were evaluated (Table 6). According to the testing results, 4'-hydroxynorendoxifen (Ki 20.0 nM) is 4-fold more potent than norendoxifen (Ki 77 nM) against aromatase (CYP19). 4'-Hydroxynorendoxifen also potently inhibits CYP1A2 (Ki 56 nM), while it only displays moderate or weak inhibitory activities toward other cytochrome P450 enzymes (including CYP3A5, CYP3A4, CYP2D6, CYP2A6, Ki from 423-1640 nM). 4'-Hydroxynorendoxifen showed elevated selectivity (3-fold) toward CYP19 versus CYP1A2 when compared with norendoxifen (1-fold). 4'-Hydroxynorendoxifen also displayed a good selectivity (> 21-fold) toward CYP19 versus all other CYP P450 enzymes tested.
Table 6.
The Inhibitory Activities of Norendoxifen and 4′-Hydroxynorendoxifen against Major Cytochrome P450 Enzymes.a

| ||||
|---|---|---|---|---|
| Cytochrome P450 Enzymes | Norendoxifena | 4′-Hydroxynorendoxifen | ||
| IC50 (nM) | Ki (nM) | IC50 (nM) | Ki (nM) | |
| Aromatase (CYP19) | 102 ± 33 | 77.0 ± 9.5 | 45.0 ± 3.0 | 20.0 ± 4.0 |
| Recombinant CYP1A2 | 207 ± 26 | 76.0 ± 3.0 | 207 ± 56 | 56.0 ± 5.0 |
| Recombinant CYP3A5 | 723 ± 27 | 829 ± 62 | 556 ± 52 | 855 ± 14 |
| Recombinant CYP3A4 | 285 ± 81 | 375 ± 6 | 1880 ± 340 | 1640 ± 90 |
| Recombinant CYP2D6 | No inhibition | No inhibition | 3600 ± 400 | 423 ± 124 |
| Recombinant CYP2A6 | 6370 ± 980 | 2180 ± 260 | 6730 ± 660 | 710 ± 212 |
The testing results for norendoxifen were previously published.30
Attempted Synthesis of E- and Z-4'-Hydroxynorendoxifen
Based on previous testing results, E- and Z-norendoxifen displayed distinct biological activities against aromatase and ER.20 The synthesis of the pure E and Z isomers of 4'-hydroxynorendoxifen was therefore attempted to provide material for biological testing.
In order to prepare the pure Z isomer of 4'-hydroxynorendoxifen, the propiophenone 8 and benzophenone 52 were first reacted under McMurry cross-coupling conditions and this afforded the product 62 with E/Z ratio > 10:1 (Scheme 14). Further trituration of 62 with methanol provided the pure E isomer of 62 in 78% yield. Although partial isomerization was observed during the alkylation of 62 with 2-iodoacetamide, the pure E isomer of 63 was obtained with 56% yield. The stereochemistry of 63 was confirmed by NMR spectroscopy as described in the stereochemistry determination section. In the last step, facile isomerization happened during the LiAlH4 reduction of 63 and the workup procedures, which resulted in an E/Z ratio at 1:3 in the product 64.
Scheme 14.

Synthesis of 64a
aReagents and conditions: (a) Zn, TiCl4, THF, 78%; (b) ICH2CONH2, K2CO3, acetone, H2O, 56%; (c) LiAlH4, AlCl3, THF, 2 h, 53%.
In order to prepare the pure E isomer of 4'-hydroxynorendoxifen, the amide 9 (prepared in Scheme 1, E/Z = 4.5:1) was reduced with LiAlH4. Facile isomerization was still observed and the product 65 was obtained with E/Z ratio 3:2 (Scheme 15).
Scheme 15.

Synthesis of 65a
aReagents and conditions: (a) LiAlH4, AlCl3, THF, 70%.
Previously, by using a similar route, we were able to prepare Z-norendoxifen with E/Z ratio 1:10.20 Here, in the case of 4'-hydroxynorendoxifen, the additional hydroxy group seemed to accelerate the rate of E/Z isomerization. The fast E/Z isomerization not only challenged the synthesis of the pure E and Z isomers of 4'-hydroxynorendoxifen, it also made their biological activity testing difficult. Considering these factors, further efforts to prepare pure E- and Z-4'-hydroxynorendoxfen were not attempted.
Stereochemistry Determination of Compound Z-45, E-45 and 63
The Z and E geometries of compounds Z-45, E-45 and 63 were confirmed by 1H-1H COSY and NOE NMR spectroscopy as previous described for stereochemistry determination of norendoxifen20 and 4-hydroxytoremifene.31 The 1H-1H COSY and NOE correlations used for the stereochemistry determination are summarized in Figure 5, and the detailed assignment procedures for each molecule are described below.
Figure 5.

The 1H-1H COSY and NOE correlations used for the stereochemistry determination of compound Z-45, E-45 and 63.
For compound Z-45, Hb was first assigned by the presence of NOE correlation with Ha, and then Hc was assigned by COSY correlation with Hb. The Z geometry of Z-45 was confirmed by the NOE correlation between Hc and Hd. For compound E-45, Hb was assigned by the NOE correlation with Ha, and then Hc was assigned by COSY correlation with Hb. Strong NOE correlations were observed both for Hd-He and Hd-Hf, and no NOE correlation was found between Hd and Hc. Those facts confirmed the E geometry of E-45. For compound 63, Hb was assigned by the NOE correlation with Ha. Hc was assigned by COSY correlation with Hb. Strong NOE correlations were observed both for Hd-He and Hd-Hf, and no NOE correlation was found between Hd and Hc. These facts confirmed the E geometry of 63.
Molecular Dynamics Simulations
The transcriptional activity of ER upon ligand binding depends on the specific ER conformation the binding ligand can trigger, especially with respect to the position of helix-12 (H-12, residues 536-544).22, 32 When 4-hydroxytamoxifen (the biologically active form of tamoxifen) binds to ER-α, the dimethylaminoethoxy side chain projects out and prevents the reorientation of H-12 that is required for coactivator recruitment, thereby locking ER-α in the antagonistic conformation.22 This specific antagonistic conformation (Figure 7b) is reported to be the origin of the selective estrogen receptor modulatory activities of 4-hydroxytamoxifen.33, 34
Figure 7.

(a) Mass-weighted RMSD of helix-12 (H-12) during molecular dynamics simulations. (b) Overlap of the averaged structure of Z-norendoxifen (cyan)-ER-α complex (H-12 colored in blue) with the crystal structure of 4-hydroxytamoxifen (pink)-ER-α complex (H-12 colored in magenta).
Considering the structural similarity of Z-norendoxifen and 4-hydroxytamoxifen, a molecular dynamics (MD) simulation study was performed to explore whether Z-norendoxifen could trigger and stabilize ER-α in a similar antagonistic conformation as 4-hydroxytamoxifen. The MD simulation started with the hypothetical binding mode of Z-norendoxifen in the ligand binding site of ER-α (as shown in Figure 3b) obtained by modifying the crystal structure of the 4-hydroxytamoxifen-ER-α complex (PDB ID 3ert), and the total simulation time was 2 ns. According to the RMSD during the course of simulations, the whole system reached an equilibrium state at approximately 1 ns. The RMSD of ER-α finally stabilized at approximately 2.2 Å, and the RMSD of Z-norendoxifen was approximately 0.3 Å (Figure 6a). The distance profiles for the key interactions between Z-norendoxifen and ER-α were also monitored (in Figure 6c). The simulation results showed that the salt bridge interaction with Asp 351, and the hydrogen bonds with Glu353 and Arg394 were well maintained during the whole simulations. Although small fluctuations were observed for the hydrogen bond with the crystal water molecule, this hydrogen bond was also present for more than 70% of the total molecular dynamics simulation time. The low RMSD together with the stable interaction distance profiles indicate that the binding mode of Z-norendoxifen in the ligand binding site of ER-α is quite stable and reliable.
Figure 6.

(a) Mass-weighted RMSD of ER-α and Z-norendoxifen during simulations. (b) The interaction diagram of Z-norendoxifen in the ligand-binding site of ER-α. (c) Time dependence of key interaction distances between Z-norendoxifen and ER-α during simulations.
To investigate the effect of Z-norendoxifen binding on the repositioning of H-12, two different RMSDs were calculated for H-12 (residues 536-544, Figure 7a). The first RMSD was calculated by fitting H-12 itself, which accounts for the conformational change of H-12 itself. The second RMSD of H-12 was calculated by fitting residues 306-535 of ER-α. The second RSMD accounts for the conformational change of H-12 plus the translocation and rotation of H-12 relative to the rest of ER-α. According to the results, the RMSD of H-12 by fitting H-12 itself stayed at 1.0 Å, which indicates only small conformational changes occurred on H-12. The RMSD of H-12 obtained by fitting the non-H-12 part of ER-α stayed at 1.5 Å, which is only 0.5 Å larger than that of H-12 itself. The small difference between the two RMSDs indicates that H-12 remains predominantly in the same position on the ER-α surface during the whole simulation and the translocation and rotation of H-12 relative to the other parts of ER-α is very small. An averaged structure of Z-norendoxifen-ER-α complex was obtained by averaging 100 snapshots in the last 1 ns of the molecular dynamics trajectory. The averaged structure was overlapped with the crystal structure of 4-hydroxytamoxifen-ER-α complex (PDB ID 3ert, in Figure 7b). The overlap clearly shows that the H-12 of Z-norendoxifen-ER-α complex is maintained in the same location as H-12 of the 4-hydroxytamoxifen-ER-α complex. These results demonstrate that Z-norendoxifen can stabilize H-12 in the same antagonistic position as 4-hydroxytamoxifen. Therefore, according to the molecular dynamics simulation, Z-norendoxifen is likely to display a similar ER modulation effect as 4-hydroxytamoxifen.
Antagonism of Transcriptional Activity
Neither affinity for the estrogen receptor nor the presence of a protonated amino group at the end of the side chain capable of hydrogen bonding to the Asp351 carboxylate of the estrogen receptor guarantee any estrogenic or antiestrogenic pharmacological activity of the ligand. Changes in the structure of the ligand can be expected to influence the pharmacology, and the type of activity can also be influenced by mutation of Asp351, as it has been documented that the Asp351Tyr mutation converts raloxifene from being an antiestrogen to being an estrogen35 and the Asp351Gly mutation converts 4-hydroxytamoxifen from being estrogenic to being antiestrogenic.36 It is therefore important to investigate estrogen receptor ligands in functional assays that monitor the effects of ligand binding on mRNA levels. In the present case, to assess the abilities of the norendoxifen analogues to antagonize β-estradiol (E2) transcriptional activity in MCF-7 cells, 18 of the analogues were tested at a concentration of 1 µM in minimum essential media (MEM) supplemented with 10% charcoal stripped fetal bovine serum (FBS). The reason for choosing these 18 compounds is that they have relatively high ER binding affinities against ER-α or ER-β or both. The progesterone receptor expression is commonly used to assess estrogenic or antiestrogenic activity.37 As shown in Figure 8, the presence of 10 nM E2 was able to significantly increase the mRNA expression of the progesterone receptor (PGR) gene compared to the control, which contained 0.1% methanol (vehicle) in the MEM supplemented with 10% charcoal stripped FBS. The PGR mRNA expression level with 10 nM E2 stimulation alone was considered as 100% PGR mRNA expression. Endoxifen was used as a positive control at a concentration of 1 [.proportional]M and it can antagonize the PGR mRNA expression in the presence of 10 nM E2 to 10% expression level compared to 10 nM E2 stimulation alone, which is consistent with the published result.37 Among the 18 test compounds, norendoxifen, 10, 14a, 44, E-45, Z-45, 55b, 61a and 61b were able to antagonize the PGR mRNA expression level to 32%, 14%, 42%, 47%, 47%, 44%, 33%, 16%, 6%, respectively. All of the test compounds have shown statistically significant differences compared to the control stimulated by E2 alone. The antiestrogenic effects of the compounds as monitored by reduction of estradiol-stimulated mRNA levels correlate weakly with estrogen receptor affinities. For example, compounds 10 (14% mRNA level) and 61a (16% mRNA level) both have relatively high affinities for ER-α (15 and 27 nM EC50 values, respectively), but 61b (6% mRNA level) actually reduced mRNA level more than any other compound tested (Figure 8) but had low affinity for ER-α (1920 nM EC50 value). This may reflect the fact that affinities for the receptors don't necessarily translate directly into biological effects in functional assays, such as antagonism of the progesterone receptor expression.
Conclusion
A series of structurally related norendoxifen analogues were designed by molecular modeling using a structure-based drug design approach. These analogues were synthesized and evaluated pharmacologically. Most of them displayed potent aromatase inhibitory activity and also showed high estrogen receptor binding affinities. The structure-activity relationships obtained were generally consistent with the molecular modeling predictions. According to molecular dynamics simulations, Z-norendoxifen can stabilize helix H-12 of ER-α in the same antagonistic conformation as 4-hydroxytamoxifen, and Z-norendoxifen is likely to display the same ER-α modulatory activity as 4-hydroxytamoxifen. 4'-Hydroxynorendoxifen (10) displayed elevated potency against aromatase, higher affinity for ER-α and ER-β, and was a more potent antagonist of estradiol-stimulated progesterone receptor mRNA expression in MCF-7 cells when compared to norendoxifen. The aromatase selectivity versus other cytochrome P450 enzymes for 4'-hydroxynorendoxifen was also superior to norendoxifen. These results suggested that the tamoxifen metabolite 4'-hydroxynorendoxifen is a promising candidate for further development toward breast cancer treatment.
Experimental Section
General
Melting points were determined using capillary tubes with a Mel-Temp apparatus and are uncorrected. The nuclear magnetic resonance (1H and 13C NMR) spectra were recorded using a Bruker ARX300 spectrometer (300 MHz) with a QNP probe or a Bruker DRX-2 spectrometer (500 MHz) with a BBO probe. High-resolution mass spectra were recorded on a double-focusing sector mass spectrometer with magnetic and electrostatic mass analyzers. The purities of biologically tested compounds are ≥ 95% as determined by HPLC or elemental analyses. For elemental analyses, the observed percentages differ less than 0.40% from the calculated values. For purities estimated by HPLC, the major peak accounted for ≥ 95% of the combined total peak area when monitored by a UV detector at 254 nm. The HPLC analyses were performed on a Waters 1525 binary HPLC pump/Waters 2487 dual λ absorbance detector system using a 5 μm C18 reversed phase column. Cytochrome P450 (CYP) inhibitor screening kits for aromatase (CYP19) inhibition studies were purchased from BD Biosciences (San Jose, CA). Estrogen receptor α and β competitor assay kits were purchased from Invitrogen (Carlsbad, CA).
General Procedure for the McMurry Cross-Coupling Reaction
Zinc powder (653 mg, 10 mmol) was suspended in dry THF (8 mL) and the mixture was cooled to 0 °C. Then, TiCl4 (0.55 mL, 5 mmol) was added dropwise under argon. When the addition was complete, the mixture was warmed to room temperature and heated to reflux for 2 h. After cooling down, a solution of the corresponding benzophenone (1 mmol) and ketone (3 mmol) in dry THF (8 mL) was added, and the mixture was heated at reflux in the dark for 3 h. After being cooled to room temperature, THF was evaporated. The residue was dissolved in saturated NH4Cl aqueous solution (20 mL) and extracted with ethyl acetate (20 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography to provide the McMurry cross-coupling product.
General Procedure for the Mono-alkylation of Diphenol with 2-Iodoacetamide
A suspension of the corresponding diphenol (1 mmol) and K2CO3 (415 mg, 3 mmol) in acetone (6 mL) was heated to reflux for 10 min. Then, a solution of 2-iodoacetamide (240 mg, 1.3 mmol) in acetone (6 mL) was added in small portions over 3 h and the mixture was stirred at reflux for 1 h. After cooling down, the solvent was evaporated and the residue was dissolved in saturated NH4Cl aqueous solution (30 mL) and extracted with ethyl acetate (30 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography to provide the mono-alkylated product.
General Procedure for the Amide Reduction with LiAlH4
A suspension of AlCl3 (400 mg, 3 mmol) and LiAlH4 (380 mg, 10 mmol) in dry THF (10 mL) was stirred under argon and cooled to 0 °C. A solution of the corresponding amide (1 mmol) in dry THF (10 mL) was added. The mixture was warmed to room temperature and stirred under argon overnight. The reaction was quenched with H2O (1 mL) and the THF was evaporated. The residue was dissolved in saturated NH4Cl aqueous solution (25 mL) and extracted with ethyl acetate (25 mL × 5). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography to provide the reduction product.
2-(4-(4-Hydroxybenzoyl)phenoxy)acetamide (7)
A suspension of 6 (1.00 g, 4.67 mmol) and Cs2CO3 (4.04 g, 12.4 mmol) in DMF (10 mL) was heated to 80 °C. A solution of 2-iodoacetamide (970 mg, 5.24 mmol) in DMF (6 mL) was added in small portions over 3 h and the mixture was stirred at 80 °C overnight. After cooling down, the solvent was evaporated and the residue was dissolved in saturated NH4Cl aqueous solution (45 mL) and extracted with ethyl acetate (30 mL × 6). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (1-9 hexanes-ethyl acetate) to provide the product 7 as white solid (462 mg, 37%): mp 203-206 °C. 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.73-7.70 (m, 2 H), 7.65-7.63 (m, 2 H), 7.00-6.97 (m, 2 H), 6.84-6.82 (m, 2 H), 4.51 (s, 2 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 195.5, 171.4, 161.6, 160.3, 132.6, 132.1, 131.8, 128.8, 114.9, 114.0, 66.5; ESIMS m/z (relative intensity) 294 (MNa+, 100); HRESIMS m/z calcd for C15H14NO4 (MH+) 272.0923, found 272.0930.
4-(1-(4-(2-Amino-2-oxoethoxy)phenyl)-1-(4-hydroxyphenyl)but-1-en-2-yl)phenyl Pivalate (9)
Intermediate 7 (90 mg, 0.33 mmol) and 8 (221 mg, 0.943 mmol) were reacted according to the general McMurry cross-coupling reaction procedure. The product was further purified by silica gel column chromatography (1:2 hexanes-ethyl acetate) to afford the product 9 as a white solid (85.5 mg, 55%): mp 189-192 °C. NMR shows a nearly 4.5:1 mixture of E and Z isomers. The mixture isomerized to be a 1:1 mixtue of E and Z isomers when dissolved in CDCl3 and kept at room temperature overnight. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.15-7.12 (m, 2 H), 7.09-7.05 (m, 4 H), 7.02-6.99 (m, 2 H), 6.90-6.87 (m, 2 H), 6.83-6.74 (m, 8 H), 6.66-6.63 (m, 2 H), 6.58-6.55 (m, 2 H), 6.46-6.43 (m, 2 H), 4.46 (s, 2 H), 4.31 (s, 2 H), 2.47-2.41 (m, 4 H), 1.29 (s, 18 H), 0.91-0.86 (m, 6 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 179.1, 173.8, 157.4, 157.0, 156.6, 156.2, 150.4, 141.6, 141.5, 141.3, 139.7, 139.0, 138.6, 136.2, 135.8, 133.5, 133.3, 132.1, 132.0, 131.9, 122.1, 116.2, 115.6, 114.9, 68.2, 68.0, 40.3, 30.2, 28.2, 14.7; MALDIMS m/z (relative intensity) 473 (M+, 100); HRESIMS m/z calcd for C29H32NO5 (MH+) 474.2281, found 474.2287.
4,4'-(1-(4-(2-Aminoethoxy)phenyl)but-1-ene-1,2-diyl)diphenol (10)
The amide 9 (84.0 mg, 0.177 mmol) was reacted with LiAlH4 according the general procedure for amide reduction with LiAlH4, and the product was further purified by silica gel column chromatography (9:1 dichloromethane-methanol) to provide the product 10 as a pale yellow oil (46.6 mg, 70%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.07 (m, 2 H), 6.97 (m, 2 H), 6.90-6.86 (m, 6 H), 6.75-6.72 (m, 4 H), 6.64 (m, 2 H), 6.58-6.54 (m, 6 H), 6.41 (m, 2 H), 3.98 (t, J = 5.3 Hz, 2 H), 3.84 (t, J = 5.3 Hz, 2 H), 2.99 (t, J = 5.3 Hz, 2 H), 2.90 (t, J = 5.3 Hz, 2 H), 2.40 (m, 4 H), 0.88 (m, 6 H); 13C NMR (75 MHz, methanol-d4) δ 158.8, 157.9, 157.2, 156.6, 156.2, 141.9, 141.6, 138.9, 138.4, 138.1, 136.7, 136.4, 135.0, 133.1, 132.0, 131.7, 115.9, 115.7, 115.2, 114.4, 69.5, 69.2, 41.6, 41.5, 29.8, 14.1; EIMS m/z (relative intensity) 375 (M+, 100); HRESIMS m/z calcd for C24H26NO3 (MH+) 376.1913, found 376.1897. Anal. Calcd for C24H25NO3·0.8 MeOH: C, 74.26; H, 7.09; N, 3.49. Found: C, 74.15; H, 6.93; N, 3.45.
4,4'-(2-(4-Fluorophenyl)but-1-ene-1,1-diyl)diphenol (12a)
The starting material 6 (1.11 g, 5.18 mmol) and 11a (2.24 g, 14.7 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (3:1 hexanes-ethyl acetate) to provide the product 12a as a white solid (1.67 g, 96%): mp 197-199 °C. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.04-6.98 (m, 4 H), 6.82-6.73 (m, 4 H), 6.65-6.61 (m, 2 H), 6.45-6.41 (m, 2 H), 2.46-2.38 (m, 2 H), 0.86 (t, J = 7.3 Hz, 3 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 156.8, 155.9, 140.6, 140.1, 136.6, 136.2, 133.3, 132.6, 132.5, 131.9, 116.1, 116.0, 115.7, 115.5, 30.2, 14.7; EIMS m/z (relative intensity) 334 (M+, 100); HREIMS m/z calcd for C22H19FO2 (M+) 334.1364, found 334.1366.
4,4'-(2-Ethylbut-1-ene-1,1-diyl)diphenol38
The starting material 6 (1.05 g, 4.90 mmol) and pentan-3-one (11b, 1.28 g, 14.8 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (4:1 hexanes-ethyl acetate) to provide the product 12b as a white solid (1.19 g, 91%): mp 160-162 °C (lit.38 163 °C). 1H NMR (300 MHz, acetone-d6) δ 6.94 (m, 4 H), 6.74 (m, 4 H), 2.12 (q, J = 7.5 Hz, 4 H), 0.98 (t, J = 7.5 Hz, 6 H).
2-(4-(2-(4-Fluorophenyl)-1-(4-hydroxyphenyl)but-1-en-1-yl)phenoxy)acetamide (13a)
The diphenol 12a (870 mg, 2.60 mmol) was reacted with 2-iodoacetamide according to the general mono-alkylation procedure for diphenols, and the product was purified by silica gel column chromatography (1:2 hexanes-ethyl acetate) to provide the product 13a as a white solid (410 mg, 40%): mp 160-162 °C. The NMR spectrum shows a nearly 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.15-7.11 (m, 2 H), 7.06-6.98 (m, 6 H), 6.90-6.88 (m, 2 H), 6.83-6.75 (m, 8 H), 6.65-6.61 (m, 2 H), 6.58-6.55 (m, 2 H), 6.45-6.42 (m, 2 H), 4.46 (s, 2 H), 4.31 (s, 2 H), 2.45-2.39 (m, 4 H), 0.87 (t, J = 7.3 Hz, 6 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 173.9, 164.1, 160.9, 157.5, 157.0, 156.7, 156.2, 141.5, 141.2, 139.8, 139.7, 138.9, 138.6, 136.1, 135.8, 133.4, 133.3, 132.6, 132.5, 132.0, 131.8, 116.2, 116.0, 115.7, 115.6, 114.9, 68.2, 68.0, 30.2, 14.6; negative ion ESIMS m/z (relative intensity) 390 [(M - H+)−, 100]; negative ion HRESIMS m/z calcd for C24H21FNO3 (M - H+)− 390.1506, found 390.1519.
2-(4-(2-Ethyl-1-(4-hydroxyphenyl)but-1-en-1-yl)phenoxy)acetamide (13b)
The diphenol 12b (295 mg, 1.10 mmol) was reacted with 2-iodoacetamide according to the general mono-alkylation procedure for diphenols, and the product was purified by silica gel column chromatography (2:3 hexanes-ethyl acetate) to provide the product 13b as a white solid (168 mg, 47%): mp 167-169 °C. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.04-7.01 (m, 2 H), 6.92-6.88 (m, 2 H), 6.83-6.79 (m, 2 H), 6.69-6.66 (m, 2 H), 4.41 (s, 2 H), 2.15-2.06 (m, 4 H), 0.98-0.92 (m, 6 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 174.0, 156.9, 156.3, 143.0, 139.2, 137.5, 136.4, 131.8, 131.6, 116.0, 115.4, 68.2, 25.6, 14.4; negative ion ESIMS m/z (relative intensity) 324 [(M - H+)−, 100]; HRESIMS m/z calcd for C20H23NO3Na (MNa+) 348.1576, found 348.1562.
4-(1-(4-(2-Aminoethoxy)phenyl)-2-(4-fluorophenyl)but-1-en-1-yl)phenol (14a)
The amide 13a (218 mg, 0.557 mmol) was reacted with LiAlH4 according to the general procedure for amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 14a as a colorless oil (160 mg, 76%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.11-6.99 (m, 8 H), 6.91-6.73 (m, 10 H), 6.67-6.63 (m, 2 H), 6.57-6.54 (m, 2 H), 6.45-6.42 (m, 2 H), 3.98 (t, J = 5.1 Hz, 2 H), 3.82 (t, J = 5.1 Hz, 2 H), 2.98 (t, J = 5.1 Hz, 2 H), 2.88 (t, J = 5.1 Hz, 2 H), 2.47-2.40 (m, 4 H), 0.89 (t, J = 7.3 Hz, 6 H); 13C NMR (75 MHz, methanol-d4) δ 164.2, 160.9, 159.1, 158.3, 157.6, 156.7, 140.9, 140.7, 140.2, 137.7, 137.4, 136.0, 135.7, 133.1, 132.7, 132.6, 131.6, 116.0, 115.8, 115.5, 115.4, 115.2, 114.5, 70.3, 70.0, 41.9, 41.8, 29.9, 14.1; ESIMS m/z (relative intensity) 378 (MH+, 100); HRESIMS m/z calcd for C24H25FNO2 (MH+) 378.1870, found 378.1867. Anal. Calcd for C24H24FNO2: C, 76.37; H, 6.41; N, 3.71. Found: C, 76.04; H, 6.54; N, 3.70.
4-(1-(4-(2-Aminoethoxy)phenyl)-2-ethylbut-1-en-1-yl)phenol (14b)
The amide 13b (70 mg, 0.200 mmol) was reacted with LiAlH4 according to the general procedure of amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 14b as a white solid (27.4 mg, 44%): mp 138-140 °C. 1H NMR (300 MHz, methanol-d4) δ 7.00 (m, 2 H), 6.90 (m, 2 H), 6.83 (m, 2 H), 6.67 (m, 2 H), 3.97 (t, J = 5.2 Hz, 2 H), 2.97 (t, J = 5.2 Hz, 2 H), 2.13 (m, 4 H), 0.99 (m, 6 H); 13C NMR (75 MHz, CDCl3) δ 158.6, 156.7, 142.2, 138.2, 138.0, 136.3, 131.3, 115.7, 115.0, 70.1, 41.8, 25.4, 13.7; ESIMS m/z (relative intensity) 312 (MH+, 100); HRESIMS m/z calcd for C20H26NO2 (MH+) 312.1964, found 312.1963. Anal. Calcd for C20H25NO2·0.5 MeOH: C, 75.20; H, 8.31; N, 4.28. Found: C, 75.02; H, 8.28; N, 4.23.
4-(1,2-Diphenylbut-1-en-1-yl)phenol (16)39
The starting material 15 (1.01 g, 5.10 mmol) and propiophenone (2.16 g, 16.1 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (85:15 hexanes-ethyl acetate) to provide the product 16 as a yellow oil (1.54 g, 98%). The NMR spectrum shows a 5:1 mixture of E and Z isomers. The mixture quickly isomerized to be a 1:1 mixtue of E and Z isomers when dissolved in CDCl3 and kept at room temperature overnight. 1H NMR (300 MHz, CDCl3) δ 7.37-7.33 (m, 2 H), 7.27-7.24 (m, 4 H), 7.19-7.09 (m, 12 H), 7.02-6.99 (m, 2 H), 6.90-6.87 (m, 2 H), 6.83-6.80 (m, 2 H), 6.76-6.73 (m, 2 H), 6.49-6.46 (m, 2 H), 2.53-2.46 (m, 4 H), 0.98-0.91 (m, 6 H).
2-(4-(1,2-Diphenylbut-1-en-1-yl)phenoxy)acetamide (17)
A mixture of 16 (935 mg, 3.11 mmol), 2-iodoacetamide (1.21 g, 6.54 mmol) and K2CO3 (1.92 g, 13.9 mmol) was dissolved in acetone (16 mL). The suspension was heated at reflux and stirred for 4 h. After cooling down, the acetone was carefully evaporated, and the residue was dissolved in water (30 mL) and extracted with ethyl acetate (30 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (45:55 hexanes-ethyl acetate) to provide the product 17 as a white solid (815 mg, 73%): mp 131-134 °C. The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, CDCl3) δ 7.38-7.33 (m, 2 H), 7.25-7.10 (m, 16 H), 7.02-6.99 (m, 2 H), 6.92-6.81 (m, 6 H), 6.58-6.55 (m, 2 H), 4.52 (s, 2 H), 4.36 (s, 2 H), 2.51-2.43 (m, 4 H), 0.97-0.90 (m, 6 H); 13C NMR (75 MHz, CDCl3) δ 171.1, 155.8, 155.0, 143.5, 143.0, 142.3, 142.2, 142.1, 142.0, 137.9, 137.8, 137.5, 137.0, 132.1, 130.9, 130.7, 129.6, 129.4, 128.2, 127.9, 127.8, 127.3, 126.6, 126.1, 125.8, 114.3, 113.5, 67.1, 66.9, 29.0, 13.5; ESIMS m/z (relative intensity) 380 (MNa+, 100); HRESIMS m/z calcd for C24H23NO2Na (MNa+) 380.1627, found 380.1624.
2-(4-(1,2-Diphenylbut-1-en-1-yl)phenoxy)ethan-1-amine (18)40
The amide 17 (540 mg, 1.51 mmol) was reacted with LiAlH4 according to the general procedure of amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 18 as a pale yellow oil (328 mg, 63%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.33-7.30 (m, 2 H), 7.27-7.18 (m, 4 H), 7.14-7.05 (m, 12 H), 6.96-6.91 (m, 4 H), 6.85-6.83 (m, 2 H), 6.77-6.74 (m, 2 H), 6.58-6.55 (m, 2 H), 4.02 (t, J = 5.3 Hz, 2 H), 3.85 (t, J = 5.3 Hz, 2 H), 3.00 (t, J = 5.3 Hz, 2 H), 2.90 (t, J = 5.3 Hz, 2 H), 2.50-2.40 (m, 4 H), 0.93-0.86 (m, 6 H). Anal. Calcd for C24H25NO: C, 83.93; H, 7.34; N, 4.08. Found: C, 83.53; H, 7.41; N, 3.98.
(4-Methoxyphenyl)(4-nitrophenyl)methanone (21)26
A solution of 19 (2.04 g, 11.0 mmol) and 20 (6 mL, 32.9 mmol) in dry dichloromethane (12 mL) was stirred in an ice bath. Aluminium chloride (1.50 g, 11.2 mmol) was added. The mixture was kept at 0 °C for 1 h, and then warmed to room temperature and stirred overnight. The reaction mixture was poured into ice-water (40 mL), stirred for 15 min and extracted with dichloromethane (40 mL × 3). The organic layers were combined and dried over Na2SO4. Solvent was evaporated and the residue was washed with hexanes (30 mL) and recrystalized from hexanes (44 mL) – chloroform (22 mL) to provide the product 21 as gray needless (1.55 g, 55%): mp 120-122 °C. 1H NMR (300 MHz, CDCl3) δ 8.34-8.31 (m, 2 H), 7.90-7.86 (m, 2 H), 7.82-7.79 (m, 2 H), 7.01-6.97 (m, 2 H), 3.91 (s, 3 H).
(4-Hydroxyphenyl)(4-nitrophenyl)methanone (22)41
A solution of 21 (1.44 g, 5.60 mmol) in HBr (48% v/v, 15 mL) and glacial acetic acid (15 mL) was heated at reflux for 9 h. After cooling down, solvent was carefully removed in vacuo. The residue was dissolved in water (50 mL) and extracted with ethyl acetate (50 mL × 4). The organic layers were combined and dried over Na2SO4. Evaporation of the solvent provided the product 22 as a yellow solid (1.28 g, 94%): mp 194-197 °C (lit.41 198-200 °C). 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 8.32-8.29 (m, 2 H), 7.85-7.82 (m, 2 H), 7.71-7.67 (m, 2 H), 6.89-6.85 (m, 2 H).
(4-(2-Bromoethoxy)phenyl)(4-nitrophenyl)methanone (23)
A suspension of 22 (693 mg, 2.85 mmol) and K2CO3 (1.53 g, 11.0 mmol) in dibromomethane (5 mL), water (1 mL) and acetone (6 mL) was heated at reflux for 4 h. After cooling down, the solvent was evaporated and the residue was dissolved in water (40 mL) and extracted with ethyl acetate (40 mL × 3). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (80:20 hexanes-ethyl acetate) to provide the product 23 as a white solid (681 mg, 68%): mp 132-134 °C. 1H NMR (300 MHz, CDCl3) δ 8.35-8.31 (m, 2 H), 7.90-7.86 (m, 2 H), 7.84-7.79 (m, 2 H), 7.03-6.98 (m, 2 H), 4.39 (t, J = 6.2 Hz, 2 H), 3.68 (t, J = 6.2 Hz, 2 H); 13C NMR (75 MHz, CDCl3) δ 193.4, 162.4, 149.5, 143.5, 132.7, 130.3, 129.5, 123.5, 114.5, 67.9, 28.5; EIMS m/z (relative intensity) 349 (M+, 74), 109 (100); HREIMS m/z calcd for C15H12NO4Br (M+) 348.9944, found 348.9937.
4-(1-(4-(2-Bromoethoxy)phenyl)-2-phenylbut-1-en-1-yl)aniline (24a)
The benzophenone 23 (410 mg, 1.17 mmol) and propiophenone (590 mg, 4.40 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (3:1 hexanes-ethyl acetate) to afford the product 24a as a brown glass (260 mg, 53%). The NMR spectrum shows a nearly 3:1 mixture of E and Z isomers. 1H NMR (300 MHz, CDCl3) δ 7.20 -7.08 (m, 8.9 H), 7.04-7.01 (m, 0.7 H, isomer 1), 6.91-6.86 (m, 2 H, isomer 2), 6.80-6.77 (m, 0.7 H, isomer 1), 6.71-6.68 (m, 0.7 H, isomer 1), 6.65-6.62 (m, 2 H, isomer 2), 6.56-6.52 (m, 0.7 H, isomer 1), 6.38-6.34 (m, 2 H, isomer 2), 4.31 (t, J = 6.2 Hz, 2 H, isomer 2), 4.15 (t, J = 6.2 Hz, 0.7 H, isomer 1), 3.68 (t, J = 6.2 Hz, 2 H, isomer 2), 3.55 (t, J = 6.2 Hz, 0.7 H, isomer 1), 2.52-2.42 (m, 2.7 H), 0.95-0.89 (m, 4.2 H); EIMS m/z (relative intensity) 421 (M+, 100); HRESIMS m/z calcd for C24H25NOBr (MH+) 422.1119, found 422.1124.
4-(1-(4-Aminophenyl)-1-(4-(2-bromoethoxy)phenyl)but-1-en-2-yl)phenyl Pivalate (24b)
The benzophenone 23 (250 mg, 0.714 mmol) and 8 (523 mg, 2.23 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (3:1 hexanes-ethyl acetate) to afford the product 24b as a brown oil (207 mg, 55%). NMR shows a nearly 2.5:1 mixture of E and Z isomers. 1H NMR (300 MHz, CDCl3) δ 7.18-7.09 (m, 5 H), 7.04-7.01 (m, 0.8 H, isomer 1), 6.90-6.86 (m, 5 H), 6.81-6.78 (m, 0.8 H, isomer 1), 6.73-6.70 (m, 0.8 H, isomer 1), 6.68-6.65 (m, 2 H, isomer 2), 6.59-6.56 (m, 0.8 H, isomer 1), 6.43-6.40 (m, 2 H, isomer 2), 4.31 (t, J = 6.2 Hz, 2 H, isomer 2), 4.16 (t, J = 6.2 Hz, 0.8 H, isomer 1), 3.66 (t, J = 6.2 Hz, 2 H, isomer 2), 3.55 (t, J = 6.2 Hz, 0.8 H, isomer 1), 2.51-2.42 (m, 2.8 H), 1.35 (s, 12.6 H), 0.96-0.91 (m, 4.2 H); 13C NMR (75 MHz,, CDCl3) δ 177.1, 156.7, 149.0, 142.9, 140.0, 139.6, 138.2, 137.1, 134.2, 132.1, 131.9, 130.7, 130.5, 120.8, 115.4, 114.9, 114.2, 113.5, 67.8, 67.5, 39.0, 29.7, 29.3, 29.0, 27.1, 13.7; ESIMS m/z (relative intensity) 522 (MH+, 100); HRESIMS m/z calcd for C29H32BrNO3 (MH+) 522.1644, found 522.1647.
4-(1-(4-(2-Aminoethoxy)phenyl)-2-phenylbut-1-en-1-yl)aniline (25a)
A mixture of 24a (177 mg, 0.419 mmol) and NaI (244 mg, 1.62 mmol) was dissolved with DMF (10 mL), and then ammonium hydroxide aqueous solution (30%, 8 mL) was added. The solution was heated to 65 °C and stirred in a sealed tube for 24 h. After cooling down, the mixture was diluted with distilled water (30 mL) and extracted with ethyl acetate (40 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (9:1 dichloromethane-methanol) to provide the product 25a as a colorless oil (77.4 mg, 52%). The NMR spectrum shows a 2.5:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.14-7.02 (m, 9 H), 6.95-6.89 (m, 2.8 H), 6.77-6.74 (m, 0.8 H, isomer 1), 6.71-6.68 (m, 0.8 H, isomer 1), 6.60-6.52 (m, 2.8 H), 6.36-6.32 (m, 2 H, isomer 2), 4.00 (t, J = 5.2 Hz, 2 H, isomer 2), 3.82 (t, J = 5.2 Hz, 0.8 H, isomer 1), 2.99 (t, J = 5.2 Hz, 2 H, isomer 2), 2.89 (t, J = 5.2 Hz, 0.8 H, isomer 1), 2.53-2.41 (m, 2.8 H), 0.92-0.86 (m, 4.2 H); ESIMS m/z (relative intensity) 359 (MH+, 100); HRESIMS m/z calcd for C24H27N2O (MH+) 359.2123, found 359.2116. Anal. Calcd for C24H26N2O·0.6 MeOH: C, 78.23; H, 7.58; N, 7.42. Found: C, 78.24; H, 7.39; N, 7.39.
4-(1-(4-(2-Aminoethoxy)phenyl)-1-(4-aminophenyl)but-1-en-2-yl)phenol (25b)
A mixture of 24b (181 mg, 0.346 mmol) and NaI (281 mg, 1.87 mmol) was dissolved with THF (5 mL), and then ammonium hydroxide aqueous solution (30%, 8 mL) was added. The solution was heated to 100 °C and stirred in a sealed tube for 24 h. After cooling down, THF was evaporated, and the aqueous solution was extracted with ethyl acetate (20 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (9:1 dichloromethane-methanol) to provide the product 25b as a yellow glass (72.9 mg, 56%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.10-7.07 (m, 2 H), 6.93-6.86 (m, 8 H), 6.77-6.74 (m, 2 H), 6.70-6.66 (m, 2 H), 6.60-6.54 (m, 8 H), 6.39-6.36 (m, 2 H), 4.00 (t, J = 5.1 Hz, 2 H), 3.86 (t, J = 5.1 Hz, 2 H), 3.01 (t, J = 5.1 Hz, 2 H), 2.93 (t, J = 5.1 Hz, 2 H), 2.48-2.36 (m, 4 H), 0.91-0.86 (m, 6 H); 13C NMR (75 MHz,, methanol-d4) δ 158.8, 157.9, 156.6, 147.1, 146.0, 141.5, 141.1, 139.0, 138.5, 138.2, 135.5, 135.1, 133.2, 132.8, 132.0, 131.7, 131.3, 116.3, 115.7, 115.1, 114.3, 69.8, 69.5, 41.7, 41.6, 30.8, 14.2; ESIMS m/z (relative intensity) 375 (MH+, 100); HRESIMS m/z calcd for C24H27N2O2 (MH+) 375.2072, found 375.2070. HPLC purity 96.7% (C-18 reverse phase, MeOH-H2O, 90:10).
4,4'-(2-Phenylprop-1-ene-1,1-diyl)diphenol (27a)42
The starting material 6 (1.35 g, 6.30 mmol) and acetophenone (26a, 2.14 g, 17.8 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (2:1 hexanes-ethyl acetate) to provide the product 27a as a white solid (1.74 g, 91%): mp 227-229 °C (lit42 135 °C). 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.07-7.06 (m, 4 H), 7.01-6.99 (m, 3 H), 6.74-6.72 (m, 2 H), 6.65-6.63 (m, 2 H), 6.42-6.40 (m, 2 H), 2.05 (s, 3 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 155.1, 154.4, 144.6, 138.6, 135.3, 135.0, 133.4, 131.9, 131.1, 129.1, 127.6, 125.5, 114.5, 113.9, 23.0.
4,4'-(4-Chloro-2-phenylbut-1-ene-1,1-diyl)diphenol (27b)
The starting material 6 (1.05 g, 4.9 mmol) and 26b (2.36 g, 14.0 mmol) were reacted according the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (2:1 hexanes-ethyl acetate) to afford the product 27b as a yellow glass (1.66 g, 96%). 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.13-7.06 (m, 7 H), 6.79-6.76 (m, 2 H), 6.68-6.64 (m, 2 H), 6.43-6.40 (m, 2 H), 3.37 (t, J = 7.5 Hz, 2 H), 2.90 (t, J = 7.5 Hz, 2 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 157.1, 156.2, 143.2, 142.9, 135.9, 135.7, 135.6, 133.2, 131.9, 130.9, 129.4, 127.5, 116.3, 115.5, 44.2, 40.0; EIMS m/z (relative intensity) 350 (M+, 100); HREIMS m/z calcd for C22H19O2Cl (M+) 350.1068, found 350.1056.
4,4'-(2-Phenylpent-1-ene-1,1-diyl)diphenol (27c)42
The starting material 6 (1.19 g, 5.55 mmol) and 26c (2.57 g, 17.3 mmol) were reacted according the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (2:1 hexanes-ethyl acetate) to afford the product 27c as a white solid (1.70 g, 93%): mp 194-196 °C (lit35 142-145 °C). 1H NMR (300 MHz, methanol-d4) δ 7.14-6.99 (m, 7 H), 6.78-6.74 (m, 2 H), 6.67-6.63 (m, 2 H), 6.42-6.38 (m, 2 H), 2.43-2.38 (m, 2 H), 1.34-1.27 (m, 2 H), 0.79 (t, J = 7.3 Hz, 3 H); 13C NMR (75 MHz, methanol-d4) δ 157.1, 156.3, 144.5, 140.3, 136.6, 136.2, 133.1, 131.7, 130.8, 128.8, 126.8, 115.8, 115.1, 39.0, 23.2, 14.6.
4,4'-(3-Methyl-2-phenylbut-1-ene-1,1-diyl)diphenol (27d)43
The starting material 6 (1.12 g, 5.22 mmol) and 26d (2.30 g, 15.52 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was further purified by silica gel column chromatography (2:1 hexanes-ethyl acetate) to afford the product 27d as a white solid (1.63 g, 95%): mp 136-139 °C (lit43 138 °C). 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.10-7.07 (m, 2 H), 7.04-6.99 (m, 5 H), 6.76-6.73 (m, 2 H), 6.67-6.64 (m, 2 H), 6.36-6.33 (m, 2 H), 3.00-2.96 (m, 1 H), 0.87 (s, 3 H), 0.86 (s, 3 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 154.9, 153.9, 144.7, 140.0, 138.1, 134.8, 134.7, 131.0, 130.7, 130.2, 126.8, 125.4, 114.7, 113.7, 31.4, 21.4.
4,4'-(2,2-Diphenylethene-1,1-diyl)diphenol (27e)44
The starting material 6 (0.99 g, 4.6 mmol) and 26e (2.40 g, 13.2 mmol) were reacted according the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (9:1 hexanes-ethyl acetate) to provide the product 27e as a white solid (1.1 g, 65%): mp 220-222 °C (lit37 222.9-223.7 °C). 1H NMR (300 MHz, acetone-d6) δ 7.10-7.05 (m, 6 H), 7.01-6.98 (m, 4 H), 6.84-6.81 (m, 4 H), 6.58-6.54 (m, 4 H); 13C NMR (75 MHz acetone-d6) δ 156.6, 145.3, 141.6, 139.1, 135.8, 133.1, 131.8, 128.2, 126.5, 115.1.
2-(4-(1-(4-Hydroxyphenyl)-2-phenylprop-1-en-1-yl)phenoxy)acetamide (28a)
The diphenol 27a (200 mg, 0.66 mmol) was reacted with 2-iodoacetamide according the general mono-alkylation procedure for diphenols, and the product was purified by silica gel column chromatography (1:2 hexanes-ethyl acetate) to provide the product 28a as a white solid (93.2 mg, 39%): mp 137-140 °C. The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.15-6.99 (m, 14 H), 6.88-6.85 (m, 2 H), 6.79-6.74 (m, 4 H), 6.65-6.62 (m, 2 H), 6.55-6.52 (m, 2 H), 6.45-6.42 (m, 2 H), 4.45 (s, 2 H), 4.30 (s, 2 H), 2.08 (s, 3 H), 2.05 (s, 3 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 173.7, 157.2, 156.8, 156.5, 145.8, 139.6, 139.2, 138.9, 136.3, 136.0, 135.7, 133.6, 133.4, 132.8, 132.6, 130.6, 129.2, 127.3, 116.2, 115.6, 115.5, 114.8, 68.2, 68.0, 24.6, 24.5; EIMS m/z (relative intensity) 359 (M+, 100); HRESIMS m/z calcd for C23H21NO3Na (MNa+) 382.1419, found 382.1430.
2-(4-(4-Chloro-1-(4-hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)acetamide (28b)
The diphenol 27b (710 mg, 2.02 mmol) was reacted with 2-iodoacetamide according the general mono-alkylation procedure of diphenols, and the product was purified by silica gel column chromatography (1:2 hexanes-ethyl acetate) to provide the product 28b as a yellow glass (280 mg, 34%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.22-7.19 (m, 2 H), 7.13-7.05 (m, 12 H), 6.91-6.88 (m, 2 H), 6.79-6.76 (m, 4 H), 6.65-6.62 (m, 2 H), 6.55-6.52 (m, 2 H), 6.44-6.40 (m, 2 H), 4.46 (s, 2 H), 4.29 (s, 2 H), 3.38-3.34 (m, 4 H), 2.92-2.86 (m, 4 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 173.6, 157.5, 142.6, 138.3, 138.2, 136.2, 135.4, 135.3, 133.3, 133.1, 132.2, 131.9, 130.9, 129.5, 127.8, 116.4, 115.8, 115.6, 114.9, 68.2, 68.0, 44.2, 39.9; ESIMS m/z (relative intensity) 430 (MNa+, 66), 339 (100); HRESIMS m/z calcd for C24H22NO3ClNa (MNa+) 430.1186, found 430.1180.
2-(4-(1-(4-Hydroxyphenyl)-2-phenylpent-1-en-1-yl)phenoxy)acetamide (28c)
The diphenol 27c (940 mg, 2.84 mmol) was reacted with 2-iodoacetamide according to the general mono-alkylation procedure of diphenols, and the product was purified by silica gel column chromatography (1:2 hexanes-ethyl acetate) to provide the product 28c as a colorless glass (432 mg, 39%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.15-6.99 (m, 14 H), 6.92-6.88 (m, 2 H), 6.79-6.74 (m, 4 H), 6.65-6.62 (m, 2 H), 6.56-6.53 (m, 2 H), 6.42-6.39 (m, 2 H), 4.47 (s, 2 H), 4.30 (s, 2 H), 2.41-2.32 (m, 4 H), 1.32-1.22 (m, 4 H), 0.79-0.73 (m, 6 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 176.4, 159.9, 159.4, 159.0, 158.5, 146.7, 143.9, 143.5, 142.0, 141.6, 141.3, 138.8, 138.5, 135.9, 135.7, 134.6, 134.4, 133.4, 131.5, 129.6, 118.6, 118.0, 117.9, 117.2, 70.6, 70.4, 41.7, 25.8, 17.6; CIMS m/z (relative intensity) 388 (MH+, 100); HRESIMS m/z calcd for C25H25NO3Na (MNa+) 410.1732, found 410.1733.
2-(4-(1-(4-Hydroxyphenyl)-3-methyl-2-phenylbut-1-enyl)phenoxy)acetamide (28d)
The diphenol 27d (865 mg, 2.62 mmol) was reacted with 2-iodoacetamide according to the general mono-alkylation procedure of diphenols, and the product was purified by silica gel column chromatography (1:2 hexanes-ethyl acetate) to provide the product 28d as a white solid (341 mg, 34%): mp 205-206 °C. The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.18-7.00 (m, 14 H), 6.90-6.87 (m, 2 H), 6.80-6.75 (m, 4 H), 6.68-6.65 (m, 2 H), 6.50-6.47 (m, 2 H), 6.40-6.36 (m, 2 H), 4.45 (s, 2 H), 4.25 (s, 2 H), 3.05-2.92 (m, 2 H), 0.90-0.87 (m, 12 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 173.8, 157.2, 156.7, 156.2, 155.7, 147.1, 146.8, 141.2, 139.2, 138.7, 135.9, 132.7, 132.5, 132.1, 131.9, 131.7, 128.4, 127.1, 116.3, 115.7, 115.4, 114.6, 68.2, 68.0, 33.1, 33.0, 22.9; CIMS m/z (relative intensity) 388 (MH+, 100); HRESIMS m/z calcd for C25H25NO3Na (MNa+) 410.1732, found 410.1727.
2-(4-(1-(4-Hydroxyphenyl)-2,2-diphenylvinyl)phenoxy)acetamide (28e)
The diphenol 27e (350 mg, 0.96 mmol) was reacted with 2-iodoacetamide according to the general mono-alkylation procedure of diphenols, and the product was purified by silica gel column chromatography (2:3 hexanes-ethyl acetate) to provide the product 28e as a colorless glass (110 mg, 27%). 1H NMR (300 MHz, CDCl3) δ 7.12-6.97 (m, 12 H), 6.88-6.85 (m, 2 H), 6.65-6.56 (m, 4 H), 4.42 (s, 2 H); 13C NMR (75 MHz, CDCl3) δ 171.8, 155.4, 154.7, 144.0, 139.7, 139.6, 138.1, 135.7, 132.8, 132.7, 131.3, 127.7, 126.2, 114.7, 113.7, 66.8; ESIMS m/z (relative intensity) 444 (MNa+, 100); HRESIMS m/z calcd for C28H23NO3Na (MNa+) 444.1576, found 444.1569.
4-(1-(4-(2-Aminoethoxy)phenyl)-2-phenylprop-1-en-1-yl)phenol (29a)
The amide 28a (87.0 mg, 0.242 mmol) was reacted with LiAlH4 according the general procedure of amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 29a as a white solid (45.1 mg, 54%): mp 216-218 °C. The NMR spectrum showed a 1:1 mixture of E and Z isomers. 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.10-7.05 (m, 10 H), 7.02-6.98 (m, 4 H), 6.84-6.82 (m, 2 H), 6.74-6.72 (m, 4 H), 6.64-6.62 (m, 2 H), 6.51-6.49 (m, 2 H), 6.42-6.40 (m, 2 H), 4.00 (t, J = 5.0 Hz, 2 H), 3.85 (t, J = 5.0 Hz, 2 H), 3.04 (t, J = 5.0 Hz, 2 H), 2.95 (t, J = 5.0 Hz, 2 H), 2.06 (s, 3 H), 2.05 (s, 3 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 156.9, 156.3, 155.3, 154.6, 144.4, 138.3, 136.7, 136.4, 134.9, 134.7, 134.1, 133.8, 131.9, 131.8, 131.1, 131.0, 129.1, 127.6, 125.6, 114.6, 114.0, 113.7, 113.0, 68.1, 67.8, 40.4, 40.3, 23.1, 23.0; ESIMS m/z (relative intensity) 346 (MH+, 100); HRESIMS m/z calcd for C23H24NO2 (MH+) 346.1807, found 346.1803. Anal. Calcd for C23H23NO2·0.5 MeOH: C, 78.09; H, 6.97; N, 3.88. Found: C, 77.71; H, 6.72; N, 4.07.
4-(1-(4-(2-Aminoethoxy)phenyl)-4-chloro-2-phenylbut-1-en-1-yl)phenol (29b)
The amide 28b (240 mg, 0.59 mmol) was reacted with LiAlH4 according the general procedure of amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 29b as a colorless oil (138.7 mg, 60%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.19-7.06 (m, 14 H), 6.90-6.87 (m, 2 H), 6.80-6.74 (m, 4 H), 6.68-6.65 (m, 2 H), 6.53-6.50 (m, 2 H), 6.44-6.40 (m, 2 H), 3.98 (t, J = 5.1 Hz, 2 H), 3.80 (t, J = 5.1 Hz, 2 H), 3.39-3.34 (m, 4 H), 2.99 (t, J = 5.1 Hz, 2 H), 2.92-2.86 (m, 6 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 161.6, 160.8, 159.9, 159.1, 145.5, 145.2, 139.6, 139.5, 138.6, 138.4, 138.0, 137.9, 135.6, 134.4, 133.3, 131.8, 130.1, 118.8, 117.9, 117.0, 72.8, 72.5, 46.4, 44.5, 44.4, 42.4; ESIMS m/z (relative intensity) 394 (MH+, 100); HRESIMS m/z calcd for C24H25NO2Cl (MH+) 394.1574, found 394.1575. Anal. Calcd for C24H24NO2Cl: C, 73.18; H, 6.14; N, 3.56. Found: C, 73.41; H, 6.41; N, 3.44.
4-(1-(4-(2-Aminoethoxy)phenyl)-2-phenylpent-1-en-1-yl)phenol (29c)
The amide 28c (316 mg, 0.82 mmol) was reacted with LiAlH4 according the general procedure of amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 29c as a white solid (238 mg, 78%): mp 100-105 °C. The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.10-6.98 (m, 14 H), 6.85-6.82 (m, 2 H), 6.75-6.71 (m, 4 H), 6.64-6.61 (m, 2 H), 6.49-6.47 (m, 2 H), 6.41-6.39 (m, 2 H), 3.98 (t, J = 5.1 Hz, 2 H), 3.82 (t, J = 5.1 Hz, 2 H), 3.01 (t, J = 5.1 Hz, 2 H), 2.91 (t, J = 5.1 Hz, 2 H), 2.38-2.32 (m, 4 H), 1.29-1.24 (m, 4 H), 0.76-0.74 (m, 6 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 157.1, 156.2, 155.3, 154.4, 142.9, 142.8, 139.6, 139.3, 138.2, 136.7, 136.3, 135.0, 134.7, 131.8, 130.5, 129.4, 127.5, 125.6, 114.6, 113.9, 113.7, 113.0, 109.9, 68.6, 68.3, 40.6, 40.4, 37.7, 21.9, 13.8; ESIMS m/z (relative intensity) 374 (MH+, 100); HRESIMS m/z calcd for C25H28NO2 (MH+) 374.2120, found 374.2121. Anal. Calcd for C25H27NO2·0.4 MeOH: C, 78.97; H, 7.46; N, 3.63. Found: C, 78.73; H, 7.12; N, 3.77.
4-(1-(4-(2-Aminoethoxy)phenyl)-3-methyl-2-phenylbut-1-enyl)phenol (29d)
The amide 28d (206 mg, 0.532 mmol) was reacted with LiAlH4 according the general procedure of amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 29d as a colorless oil (163 mg, 82%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.15-7.01 (m, 14 H), 6.86-6.83 (m, 2 H), 6.79-6.74 (m, 4 H), 6.69-6.66 (m, 2 H), 6.45-6.43 (m, 2 H), 6.39-6.36 (m, 2 H), 3.93 (t, J = 5.1 Hz, 2 H), 3.72 (t, J = 5.1 Hz, 2 H), 3.05-2.95 (m, 2 H), 2.96 (t, J = 5.1 Hz, 2 H), 2.82 (t, J = 5.1 Hz, 2 H), 0.90-0.86 (m, 12 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 157.0, 156.0, 155.2, 154.2, 145.2, 145.0, 139.8, 137.9, 137.8, 136.1, 136.0, 134.5, 134.4, 131.0, 130.7, 130.2, 126.9, 125.5, 114.8, 113.9, 113.8, 112.9, 68.9, 68.6, 40.7, 40.5, 31.5, 31.4, 21.5; APCIMS m/z (relative intensity) 374 (MH+, 100); HRESIMS m/z calcd for C25H28NO2 (MH+) 374.2120, found 374.2120. Anal. Calcd for C25H27NO2·0.1 MeOH: C, 80.03; H, 7.33; N, 3.72. Found: C, 79.97; H, 7.00; N, 3.81.
4-(1-(4-(2-Aminoethoxy)phenyl)-2,2-diphenylvinyl)phenol (29e)
The amide 28e (101 mg, 0.24 mmol) was reacted with LiAlH4 according the general procedure of amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 29e as a white solid (62.6 mg, 64%): mp 164-166 °C. 1H NMR (300 MHz, CDCl3 and methanol-d4) δ 7.04-6.94 (m, 10 H), 6.91-6.88 (m, 2 H), 6.81-6.78 (m, 2 H), 6.60-6.58 (m, 2 H), 6.52-6.49 (m, 2 H), 3.89 (t, J = 5.0 Hz, 2 H), 2.96 (t, J = 5.0 Hz, 2 H); 13C NMR (75 MHz, CDCl3 and methanol-d4) δ 158.4, 156.7, 145.7, 141.7, 140.4, 138.3, 136.6, 134.0, 132.7, 128.9, 127.3, 115.8, 114.8, 70.0, 42.0; ESIMS m/z (relative intensity) 408 (MH+, 100); HRESIMS m/z calcd for C28H26NO2 (MH+) 408.1964, found 408.1953. Anal. Calcd for C28H25NO2·0.6 MeOH: C, 80.50; H, 6.47; N, 3.28. Found: C, 80.28; H, 6.17; N, 3.02.
(4-(2-Bromoethoxy)phenyl)(4-hydroxyphenyl)methanone (30)45
A solution of 6 (2.38 g, 11.1 mmol) and K2CO3 (3.02 g, 21.8 mmol) in 1,2-dibromoethane (15 mL), acetone (30 mL) and water (4 mL) was heated to reflux for 4 h. The solvent was evaporated and the residue was dissolved with saturated NH4Cl aqueous solution (50 mL) and extracted with ethyl acetate (50 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (3:2 hexanes-ethyl acetate) to provide the product 30 as a white solid (1.29 g, 36%): mp 118-119 °C (lit45 139-142 °C). 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.69-7.66 (m, 2 H), 7.63-7.60 (m, 2 H), 6.91-6.89 (m, 2 H), 6.83-6.81 (m, 2 H), 4.29 (t, J = 6 Hz, 2 H), 3.60 (t, J = 6 Hz, 2 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 195.5, 161.4, 161.3, 132.5, 132.1, 131.0, 128.9, 114.9, 113.9, 67.8, 28.7.
4-(1-(4-(2-Bromoethoxy)phenyl)-2-phenylvinyl)phenol (31)
The benzophenone 30 (200 mg, 0.623 mmol) and benzaldehyde (212 mg, 2.00 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (4:1 hexanes-ethyl acetate) to afford the product 31 as a yellow oil (141 mg, 57%). The NMR spectrum shows a 2:1 mixture of E and Z isomers. The mixture quickly isomerized to be a 1:1 mixtue of E and Z isomers when dissolved in CDCl3 and kept at room temperature overnight. 1H NMR (300 MHz, CDCl3) δ 7.29-7.26 (m, 2 H), 7.22-7.19 (m, 2 H), 7.18-7.03 (m, 14 H), 6.88-6.84 (m, 6 H), 6.81-6.76 (m, 4 H), 4.34-4.28 (m, 4 H), 3.69-3.62 (m, 4 H); 13C NMR (75 MHz, CDCl3) δ 157.6, 157.4, 155.4, 155.0, 141.5, 137.7, 137.1, 136.3, 133.5, 132.6, 131.7, 129.4, 129.0, 128.9, 128.0, 126.5, 126.4, 126.3, 115.5. 115.0, 114.7, 114.4, 67.9, 67.8, 29.2, 29.1; EIMS m/z (relative intensity) 394 (M+, 16), 107 (100); negative ion HRESIMS m/z calcd for C22H18O2Br (M - H+)− 393.0490, found 393.0493.
4-(1-(4-(2-Aminoethoxy)phenyl)-2-phenylvinyl)phenol (32)
A mixture of 31 (141 mg, 0.356 mmol) and NaI (224 mg, 1.49 mmol) was dissolved with THF (4 mL), and then ammonium hydroxide aqueous solution (30%, 5 mL) was added. The solution was heated to 70 °C and stirred in a sealed tube for 24 h. After cooling down, THF was carefully evaporated, and the aqueous layer was extracted with ethyl acetate (15 mL × 3). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (9:1 dichloromethane-methanol). The purified product was dissolved in DMSO (2 mL) and then diluted with water (12 mL). The solid was collected by filtration and dried in vacuo to provide the product 32 as a white solid (88.4 mg, 75%): mp 75-79 °C. The NMR spectrum shows a nearly 1:1 mixture of E and Z isomers. 1H NMR (500 MHz, DMSO-d6) δ 7.18-7.16 (m, 2 H), 7.11-7.00 (m, 8 H), 6.98-6.94 (m, 6 H), 6.90-6.84 (m, 6 H), 6.83-6.81 (m, 2 H), 6.73-6.71 (m, 2 H), 6.69-6.67 (m, 2 H), 3.89-3.87 (m, 4 H), 2.87-2.83 (m, 4 H); 13C NMR (125 MHz, DMSO-d6) δ 158.6, 158.3, 157.6, 157.1, 141.8, 137.9, 135.8, 134.0, 132.5, 131.3, 131.2, 130.7, 129.4, 129.3, 128.8, 128.7, 128.3, 128.2, 126.7, 126.6, 125.5, 125.0, 116.0, 115.4, 115.0, 114.5, 70.5, 41.2; ESIMS m/z (relative intensity) 332 (MH+, 100); HRESIMS m/z calcd for C22H22NO2 (MH+) 332.1651, found 332.1649. Anal. Calcd for C22H21NO2·0.1 H2O: C, 79.30; H, 6.41; N, 4.20. Found: C, 79.18; H, 6.45; N, 3.98.
4,4'-((2,3-Dihydro-1H-inden-1-ylidene)methylene)diphenol (34)46
The starting material6 (1.24 g, 5.78 mmol) and ketone 33 (2.18 g, 16.5 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was further purified by silica gel column chromatography (2:1 hexanes-ethyl acetate) to afford the product 34 as a yellow oil (1.54 g, 85%). 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.14-7.11 (m, 1 H), 7.07-7.02 (m, 2 H), 7.00-6.93 (m, 3 H), 6.78-6.68 (m, 5 H), 6.46-6.43 (m, 1 H), 2.85 (s, 4 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 157.1, 156.6, 148.7, 143.2, 140.1, 137.0, 136.1, 135.9, 132.4, 131.8, 128.1, 126.7, 126.3, 126.1, 116.7, 115.9, 35.7, 31.9; APCIMS m/z (relative intensity) 315 (MH+, 100).
2-(4-((2,3-Dihydro-1H-inden-1-ylidene)(4-hydroxyphenyl)methyl)phenoxy)acetamide (35)
The diphenol 34 (970 mg, 3.08 mmol) was reacted with 2-iodoacetamide according to the general mono-alkylation procedure of diphenols, and the product was purified by silica gel column chromatography (1:2 hexanes-ethyl acetate) to provide the product 35 as a yellow glass (463 mg, 40%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.16-7.08 (m, 6 H), 7.03-6.94 (m, 6 H), 6.87-6.69 (m, 10 H), 6.48-6.46 (m, 1 H), 6.39-6.36 (m, 1 H), 4.45 (s, 2 H), 4.42 (s, 2 H), 2.85-2.83 (m, 8 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 173.8, 157.7, 157.2, 148.8, 142.9, 141.0, 140.7, 139.3, 138.3, 136.5, 135.5, 132.7, 132.4, 131.9, 131.8, 128.4, 126.8, 126.4, 126.2, 116.8, 116.1, 116.0, 115.3, 68.1, 35.8, 35.6, 31.9; EIMS m/z (relative intensity) 371 (M+, 100); HRESIMS m/z calcd for C24H21NO3Na (MNa+) 394.1419, found 394.1432.
4-((4-(2-Aminoethoxy)phenyl)(2,3-dihydro-1H-inden-1-ylidene)methyl)phenol (36)
The amide 35 (291 mg, 0.783 mmol) was reacted with LiAlH4 according the general procedure of amide reduction with LiAlH4, and the product was purified by silica gel column chromatography (1:9 methanol-dichloromethane) to provide the product 36 as a yellow glass (186 mg, 66%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.15-7.11 (m, 4 H), 7.07-7.02 (m, 4 H), 6.99-6.95 (m, 4 H), 6.84-6.82 (m, 2 H), 6.81-6.79 (m, 2 H), 6.76-6.57 (m, 6 H), 6.46-6.44 (m, 1 H), 6.40-6.38 (m, 1 H), 3.99 (t, J = 5.1 Hz, 2 H), 3.96 (t, J = 5.1 Hz, 2 H), 3.01 (t, J = 5.1 Hz, 2 H), 2.98 (t, J = 5.1 Hz, 2 H), 2.87-2.85 (m, 8 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 157.6, 157.1, 155.8, 155.3, 147.3, 147.2, 141.5, 139.0, 138.8, 136.7, 135.7, 135.1, 134.2, 134.1, 130.9, 130.8, 130.2, 126.7, 126.6, 125.1, 124.8, 124.6, 124.5, 124.4, 115.2, 114.3, 114.2, 113.5, 68.8, 40.6, 40.5, 34.2, 34.1, 30.3; EIMS m/z (relative intensity) 357 (M+, 96), 314 (100); HREIMS m/z calcd for C24H24NO2 (MH+) 358.1807, found 358.1801. Anal. Calcd for C24H23NO2: C, 80.64; H, 6.49; N, 3.92. Found: C, 80.29; H, 6.44; N, 3.86.
4-(1-(4-(Methoxymethoxy)phenyl)-2-phenylprop-1-en-1-yl)phenol (37)
A solution of 27a (247 mg, 0.817 mmol) and NaH (31 mg, 95%, 1.23 mmol) in dry THF (4 mL) was stirred under argon for 10 min, and then methyl chloromethyl ether (0.07 mL, 0.92 mmol) was added. The mixture was stirred at room temperature overnight and quenched with saturated NaHCO3 aqueous solution (3 mL). The solvent was evaporated, and the residue was dissolved with water (15 mL) and extracted with ethyl acetate (15 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (85:15 hexanes-ethyl acetate) to provide the product 37 as a pale yellow oil (115 mg, 41%). The NMR spectrum showed a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, CDCl3) δ 7.28-7.09 (m, 14 H), 7.04-7.01 (m, 2 H), 6.82-6.78 (m, 4 H), 6.76-6.73 (m, 2 H), 6.71-6.68 (m, 2 H), 6.49-6.46 (m, 2 H), 5.21 (s, 2 H), 5.07 (s, 2 H), 3.53 (s, 3 H), 3.43 (s, 3 H), 2.15 (s, 6 H); 13C NMR (75 MHz, CDCl3) δ 155.8, 155.1, 154.3, 153.6, 144.3, 138.2, 137.4, 137.1, 136.1, 135.8, 134.6, 132.2, 132.0, 131.4, 131.2, 129.3, 127.9, 127.3, 127.2, 127.1, 126.0, 115.7, 115.1, 114.9, 114.3, 94.5, 94.4, 56.1, 56.0, 23.5, 23.4; EIMS m/z (relative intensity) 346 (M+, 100); HREIMS m/z calcd for C23H22O3 (M+) 346.1563, found 346.1558.
tert-Butyl (2-(4-(1-(4-(Methoxymethoxy)phenyl)-2-phenylprop-1-en-1-yl)phenoxy)ethyl)carbamate (39)
A suspension of 37 (112 mg, 0.323 mmol), 38 (196 mg, 0.621 mmol) and Cs2CO3 (254 mg, 0.78 mmol) in dry DMF (2 mL) was stirred at 50 °C under argon overnight. After cooling down, DMF was removed in vacuo. The residue was dissolved with water (15 mL) and extracted with ethyl acetate (15 mL × 4). The organic layers were combined, washed with 1 M KOH solution (20 mL) and brine (20 mL), and dried over Na2SO4. The solvent was evaporated and the residue was further purified by silica gel column chromatography (85:15 hexanes-ethyl acetate) to provide the product 39 as a transparent oil (114 mg, 72%). The NMR spectrum showed a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, CDCl3) δ 7.25-7.10 (m, 14 H), 7.03-7.00 (m, 2 H), 6.89-6.86 (m, 2 H), 6.81-6.78 (m, 4 H), 6.70-6.67 (m, 2 H), 6.57-6.54 (m, 2 H), 5.20 (s, 2 H), 5.06 (s, 2 H), 4.04 (t, J = 5.0 Hz, 2 H), 3.89 (t, J = 5.0 Hz, 2 H), 3.56 (t, J = 5.0 Hz, 2 H), 3.52 (s, 3 H), 3.47 (t, J = 5.0 Hz, 2 H), 3.43 (s, 3 H), 2.15 (s, 3 H), 2.14 (s, 3 H), 1.47 (s, 9 H), 1.44 (s, 9 H); 13C NMR (75 MHz, CDCl3) δ 157.2, 156.5, 155.9, 155.2, 144.3, 138.1, 137.3, 136.9, 136.5, 136.1, 134.6, 132.1, 132.0, 131.2, 131.1, 129.3, 127.9, 127.4, 127.1, 127.0, 126.0, 115.7, 115.1, 113.9, 113.2, 94.5, 94.4, 79.5, 67.1, 66.8, 56.0, 55.9, 40.1, 28.4, 23.4; ESIMS m/z (relative intensity) 512 (MNa+, 100); HRESIMS m/z calcd for C30H35NO5Na (MNa+) 512.2413, found 512.2394.
4-(4-(2-Aminoethoxy)phenyl)-4-(4-hydroxyphenyl)-3-phenylbut-3-enenitrile (40)
A solution of 39 (216 mg, 0.441 mmol) and N-bromosuccinimide (79.1 mg, 0.445 mmol) in CCl4 (6 mL) was heated at reflux under argon for 3 h. After cooling down, the solid was removed by filtration and the filtrate was concentrated in vacuo. The residue was dissolved in THF (6 mL) and a solution of KCN (97.5 mg, 1.50 mmol) in water (1.5 mL) was added. The mixture was stirred at room temperature overnight. The solvent was evaporated, and the residue was dissolved with water (15 mL) and extracted with ethyl acetate (15 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (7:3 hexanes-ethyl acetate). The purified product was dissolved with methanol (3 mL) and dichloromethane (3 mL), and then concentrated HCl (1.2 mL) was added. The mixture was stirred at room temperature overnight. The solvent was removed, and the residue was dissolved with water (20 mL), neutralized with NaHCO3 to pH = 7, and extracted with ethyl acetate (20 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (9:1 dichloromethane-methanol) to provide the product 40 as a red glass (132 mg, 81%). The NMR spectrum showed a 1:1 mixture of E and Z isomers. 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.18-7.09 (m, 12 H), 7.07-7.05 (m, 2 H), 6.91-6.89 (m, 2 H), 6.82-6.80 (m, 2 H), 6.77-6.75 (m, 2 H), 6.67-6.65 (m, 2 H), 6.54-6.52 (m, 2 H), 6.46-6.44 (m, 2 H), 3.98 (t, J = 5.1 Hz, 2 H), 3.81 (t, J = 5.1 Hz, 2 H), 3.51 (s, 2 H), 3.48 (s, 2 H), 3.00 (t, J = 5.1 Hz, 2 H), 2.90 (t, J = 5.1 Hz, 2 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 158.2, 157.3, 156.7, 155.8, 144.1, 144.0, 139.9, 134.6, 134.2, 132.9, 132.5, 131.8, 130.5, 129.3, 128.2, 127.0, 126.3, 126.1, 118.4, 115.3, 114.4, 114.3, 113.3, 69.0, 68.7, 40.6, 40.5, 25.2; ESIMS m/z (relative intensity) 371 (MH+, 100); HRESIMS m/z calcd for C24H23N2O2 (MH+) 371.1759, found 371.1756. Anal. Calcd for C24H22N2O2·0.3 MeOH: C, 76.79; H, 6.15; N, 7.37. Found: C, 76.67; H, 5.97; N, 7.44.
4-(1-(4-(2-Aminoethoxy)phenyl)-3-(1H-imidazol-1-yl)-2-phenylprop-1-en-1-yl)phenol (41)
A solution of 39 (195 mg, 0.398 mmol) and N-bromosuccinimide (71.3 mg, 0.401 mmol) in dry CCl4 (6 mL) was heated at reflux under argon for 3 h. After cooling down, the solid was removed by filtration and the filtrate was concentrated in vacuo. The residue was dissolved in dry THF (6 mL) and added to a solution of NaH (31.3 mg, 95%, 1.24 mmol) and imidazole (78.4 mg, 1.15 mmol) in dry THF (3 mL). The mixture was stirred at room temperature overnight. The solvent was evaporated, and the residue was dissolved with water (20 mL) and extracted with ethyl acetate (20 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (97:3 dichloromethane-methanol). The product was dissolved with methanol (4 mL), and then concentrated HCl (1.2 mL) was added. The mixture was stirred at room temperature overnight. The solvent was removed, and the residue was dissolved with water (20 mL), neutralized with NaHCO3 to pH = 7, and extracted with ethyl acetate-THF (1:1, 20 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (25:5:0.3 dichloromethane-methanol-triethylamine) to provide the product 41 as a pale yellow glass (105 mg, 64%). The NMR spectrum showed a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.35 (s, 2 H), 7.20-7.17 (m, 2 H), 7.10-7.03 (m, 12 H), 6.98 (s, 1 H), 6.95-6.93 (m, 3 H), 6.85-6.82 (m, 6 H), 6.75-6.71 (m, 2 H), 6.60-6.56 (m, 2 H), 6.46-6.43 (m, 2 H), 4.94 (s, 2 H), 4.92 (s, 2 H), 4.00 (t, J = 5.2 Hz, 2 H), 3.82 (t, J = 5.2 Hz, 2 H), 2.99 (t, J = 5.2 Hz, 2 H), 2.88 (t, J = 5.2 Hz, 2 H); 13C NMR (75 MHz, methanol-d4) δ 159.8, 159.0, 158.6, 157.7, 145.9, 141.4, 138.4, 136.0, 134.2, 133.6, 133.0, 131.6, 130.9, 129.3, 128.9, 127.9, 120.5, 116.6, 115.7, 115.6, 114.6, 70.3, 70.0, 52.1, 41.8; ESIMS m/z (relative intensity) 412 (MH+, 100); HRESIMS m/z calcd for C26H26N3O2 (MH+) 412.2025, found 412.2023. Anal. Calcd for C26H25N3O2·0.6 MeOH: C, 74.17; H, 6.41; N, 9.76. Found: C, 73.97; H, 6.11; N, 9.75.
Procedure for Preparation of 44 and Z-45 and E-45
A solution of 39 (218 mg, 0.445 mmol) and N-bromosuccinimide (81.3 mg, 0.457 mmol) in dry CCl4 (6 mL) was heated at reflux under argon for 3 h. After cooling down, the solid was removed by filtration and the filtrate was concentrated in vacuo. The residue was dissolved in dry THF (6 mL) and added to a solution of NaH (36 mg, 95%, 1.42 mmol) and 1,2,4-triazole (88 mg, 1.27 mmol) in dry THF (3 mL). The mixture was stirred at room temperature overnight. The solvent was evaporated, and the residue was dissolved with water (20 mL) and extracted with ethyl acetate (20 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (9:1 dichloromethane-acetone) to first provide 42 and then 43. The crude product 42 was dissolved with methanol (3 mL), and then concentrated HCl (0.9 mL) was added. The mixture was stirred at room temperature overnight. The solvent was removed, and the residue was dissolved with water (15 mL), neutralized with NaHCO3 to pH = 7, and extracted with ethyl acetate-THF (1:1, 15 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (25:5:0.3 dichloromethane-methanol-triethylamine) to provide the product 44. The crude product 43 was dissolved with methanol (3 mL), and then concentrated HCl (0.9 mL) was added. The mixture was stirred at room temperature overnight. The solvent was removed, and the residue was dissolved with water (15 mL), neutralized with NaHCO3 to pH = 7, and extracted with ethyl acetate-THF (1:1, 15 mL × 6). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (25:5:0.3 dichloromethane-methanol-triethylamine) to first provide E-45 and then Z-45.
4-(1-(4-(2-Aminoethoxy)phenyl)-2-phenyl-3-(1H-1,2,4-triazol-1-yl)prop-1-en-1-yl)phenol (44)
The product was obtained as a red glass (56.1 mg, 30.5%). The NMR spectrum showed a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 8.00 (m, 2 H), 7.87 (s, 2 H), 7.43-7.40 (m, 2 H), 7.33-7.29 (m, 2 H), 7.07-6.94 (m, 12 H), 6.85-6.79 (m, 4 H), 6.74-6.70 (m, 2 H), 6.61-6.58 (m, 2 H), 6.45-6.42 (m, 2 H), 5.25 (s, 2 H), 5.22 (s, 2 H), 4.02 (t, J = 5.1 Hz, 2 H), 3.85 (t, J = 5.1 Hz, 2 H), 3.01 (t, J = 5.1 Hz, 2 H), 2.91 (t, J = 5.1 Hz, 2 H); 13C NMR (75 MHz, methanol-d4) δ 159.8, 159.0, 158.4, 157.6, 151.9, 145.8, 145.3, 141.0, 136.3, 136.0, 134.5, 134.2, 133.1, 132.7, 132.5, 132.2, 130.9, 129.2, 127.9, 116.3, 115.4, 114.6, 70.2, 69.9, 54.9, 41.8; ESIMS m/z (relative intensity) 413 (MH+, 72), 344 (100); HRESIMS m/z calcd for C25H25N4O2 (MH+) 413.1978, found 413.1972. HPLC purity 95.5% (C-18 reverse phase, MeOH-H2O, 90:10).
(E)-4-(1-(4-(2-Aminoethoxy)phenyl)-2-phenyl-3-(4H-1,2,4-triazol-4-yl)prop-1-en-1-yl)phenol (E-45)
The product was obtained as a red glass (12.8 mg, 7%). 1H NMR (300 MHz, methanol-d4) δ 8.27 (s, 2 H), 7.14-7.08 (m, 7 H), 6.88-6.82 (m, 4 H), 6.65-6.61 (m, 2 H), 5.11 (s, 2 H), 3.89 (t, J = 5.1 Hz, 2 H), 2.94 (t, J = 5.1 Hz, 2 H); 13C NMR (75 MHz, methanol-d4) δ 157.6, 157.1, 145.2, 143.0, 139.0, 134.1, 132.5, 131.4, 131.1, 129.9, 129.4, 128.0, 126.7, 115.1, 113.1, 68.2, 49.2, 40.1; ESIMS m/z (relative intensity) 413 (MH+, 13), 344 (100); HRESIMS m/z calcd for C25H25N4O2 (MH+) 413.1978, found 413.1973. HPLC purity 95.2% (C-18 reverse phase, MeOH-H2O, 90:10).
(Z)-4-(1-(4-(2-aminoethoxy)phenyl)-2-phenyl-3-(4H-1,2,4-triazol-4-yl)prop-1-en-1-yl)phenol (Z-45)
The product was obtained as a yellow glass (13.8 mg, 7.5%). 1H NMR (300 MHz, methanol-d4) δ 8.28 (s, 2 H), 7.24-7.21 (m, 2 H), 7.16-7.11 (m, 5 H), 7.04-7.01 (m, 2 H), 6.77-6.74 (m, 2 H), 6.46-6.43 (m, 2 H), 5.10 (s, 2 H), 4.07 (t, J = 5.2 Hz, 2 H), 3.06 (t, J = 5.2 Hz, 2 H); 13C NMR (75 MHz, methanol-d4) δ 158.3, 156.2, 145.1, 143.0, 139.0, 134.4, 132.4, 131.4, 130.8, 129.9, 129.4, 128.0, 126.7, 114.4, 113.9, 68.3, 49.2, 40.1; ESIMS m/z (relative intensity) 413 (MH+, 60), 344 (100); HRESIMS m/z calcd for C25H25N4O2 (MH+) 413.1978, found 413.1976. HPLC purity 95.1% (C-18 reverse phase, MeOH-H2O, 90:10).
Ethyl 2-(4-(1-(4-Hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)acetate (47)
A suspension of 46 (322 mg, 1.02 mmol) and K2CO3 (498 mg, 3.60 mmol) in acetone (5 mL) was stirred at room temperature for 10 min. A solution of ethyl 2-iodoacetate (208 mg, 0.972 mmol) in acetone (3 mL) was added in small portions over 3 h and the mixture was stirred at room temperature for 1 h. After cooling down, acetone was carefully evaporated and the residue was dissolved in saturated NH4Cl aqueous solution (30 mL) and extracted with ethyl acetate (30 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (4:1 hexanes-ethyl acetate) to provide a 1:1 mixture of E and Z isomers of 47 as a white solid (95.5 mg, 23%): 160-163 °C. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.13-6.99 (m, 14 H), 6.84-6.81 (m, 2 H), 6.76-6.72 (m, 4 H), 6.64-6.61 (m, 2 H), 6.50-6.47 (m, 2 H), 6.42-6.39 (m, 2 H), 4.59 (s, 2 H), 4.44 (s, 2 H), 4.27-4.15 (m, 4 H), 2.47-2.36 (m, 4 H), 1.28-1.18 (m, 6 H), 0.89-0.83 (m, 6 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 170.1, 157.8, 156.8, 155.9, 144.0, 142.5, 142.2, 139.2, 138.9, 138.5, 136.5, 136.1, 133.4, 132.0, 131.0, 129.1, 127.2, 116.2, 115.5, 114.7, 66.8, 66.6, 62.9, 62.8, 30.3, 15.3, 14.8; EIMS m/z (relative intensity) 402 (M+, 100); HRESIMS m/z calcd for C26H26O4Na (MNa+) 425.1729, found 425.1711.
4-(1-(4-(2-Hydroxyethoxy)phenyl)-2-phenylbut-1-en-1-yl)phenol (48)
A suspension of LiAlH4 (160 mg, 3.98 mmol) in dry THF (5 mL) was stirred under argon and cooled to 0 °C. A solution of 47 (93 mg, 0.231 mmol) in dry THF (5 mL) was added. The mixture was warmed to room temperature and stirred under argon overnight. The reaction was quenched with H2O (0.5 mL) and the THF was evaporated. The residue was dissolved in saturated ammonium chloride aqueous solution (20 mL) and extracted with ethyl acetate (15 mL × 5). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (3:2 hexane-ethyl acetate) to provide the product 48 as a white solid (54.2 mg, 65%): mp 186-188 °C. The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.13-7.00 (m, 14 H), 6.89-6.86 (m, 2 H), 6.77-6.72 (m, 4 H), 6.66-6.63 (m, 2 H), 6.54-6.51 (m, 2 H), 6.42-6.39 (m, 2 H), 4.04 (t, J = 4.6 Hz, 2 H), 3.88 (m, 4 H), 3.77 (t, J = 4.6 Hz, 2 H), 2.49-2.41 (m, 4 H), 0.88 (t, J = 7.4 Hz, 6 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 159.0, 158.1, 156.9, 156.0, 144.1, 142.1, 141.9, 139.5, 137.9, 137.5, 136.5, 136.2, 133.3, 133.2, 131.8, 131.0, 129.0, 127.0, 116.0, 115.3, 114.5, 70.5, 70.3, 61.9, 61.8, 30.1, 14.5; ESIMS m/z (relative intensity) 383 (MNa+, 100); HRESIMS m/z calcd for C24H24O3Na (MNa+) 383.1623, found 383.1626. Anal. Calcd for C24H24O3: C, 79.97; H, 6.71. Found: C, 79.92; H, 6.76.
2-(4-(1-(4-Hydroxyphenyl)-2-phenylbut-1-en-1-yl)phenoxy)acetic Acid (49)
A solution of 2 N aq KOH (2.5 mL) was added to a solution of 47 (63.0 mg, 0.157 mmol) in THF (2.5 mL). The mixture was stirred at room temperature overnight. The solvent was evaporated and the residue was diluted with water (10 mL) and acidified with concentrated HCl to pH < 1. The white suspension was extracted with ethyl acetate (10 mL × 3). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (9:1 dichloromethane-methanol) to provide the product 49 as a white solid (57.2 mg, 97%): mp 156-160 °C. The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.16-7.03 (m, 12 H), 7.01-6.98 (m, 2 H), 6.91-6.88 (m, 2 H), 6.77-6.72 (m, 4 H), 6.65-6.62 (m, 2 H), 6.55-6.52 (m, 2 H), 6.41-6.38 (m, 2 H), 4.51 (s, 2 H), 4.34 (s, 2 H), 2.50-2.40 (m, 4 H), 0.91-0.86 (m, 6 H); 13C NMR (75 MHz, methanol-d4) δ 158.5, 157.6, 157.3, 156.4, 144.0, 142.1, 141.8, 139.6, 138.2, 137.8, 136.3, 135.9, 133.1, 133.0, 131.6, 130.9, 128.9, 127.0, 115.9, 115.4, 115.1, 114.6, 67.5, 30.0, 29.9, 14.0; ESIMS m/z (relative intensity) 397 (MNa+, 100); HRESIMS m/z calcd for C24H22O4Na (MNa+) 397.1416, found 397.1431. Anal. Calcd for C24H22O4·0.8 CH2Cl2: C, 67.33; H, 5.38. Found: C, 67.05; H, 5.41.
4-(1-(4-(2-Bromoethoxy)phenyl)-2-phenylbut-1-en-1-yl)phenol (50)47
The benzophenone 30 (125 mg, 0.39 mmol) and propiophenone (199 mg, 1.48 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (4:1 hexanes-ethyl acetate) to afford the product 50 as a white solid (143 mg, 87%): mp 119-122 °C. The NMR spectrum shows a 4:1 mixture of E and Z isomers. The mixture quickly isomerized to be a 1:1 mixtue of E and Z isomers when dissolved in CDCl3 and kept at room temperature overnight. 1H NMR (300 MHz, CDCl3) δ 7.20-7.08 (m, 14 H), 6.92-6.89 (m, 2 H), 6.82-6.78 (m, 4 H), 6.75-6.72 (m, 2 H), 6.58-6.55 (m, 2 H), 6.49-6.46 (m, 2 H), 4.32 (t, J = 6.2 Hz, 2 H), 4.16 (t, J = 6.3 Hz, 2 H), 3.66 (t, J = 6.2 Hz, 2 H), 3.55 (t, J = 6.3 Hz, 2 H), 2.53-2.45 (m, 4 H), 0.95 (t, J = 7.3 Hz, 6 H); ESIMS m/z (relative intensity) 422 (M+, 100). Anal. Calcd for C24H23BrO2: C, 68.09; H, 5.48. Found: C, 68.18; H, 5.56.
4-(4-(2-Chloroethoxy)benzoyl)phenyl Pivalate (53a)
A suspension of 52 (239 mg, 0.801 mmol), 1-bromo-2-chloroethane (910 mg, 6.35 mmol) and K2CO3 (616 mg, 4.46 mmol) in acetone (6 mL) was heated at reflux for 4 h. After cooling down, the solvent was carefully evaporated and the residue was dissolved in water (15 mL) and extracted with ethyl acetate (15 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (80:20 hexanes-ethyl acetate) to provide the product 53a as a white solid (141 mg, 49%): mp 99-101 °C. 1H NMR (300 MHz, CDCl3) δ 7.81-7.77 (m, 4 H), 7.18-7.15 (m, 2 H), 6.98-6.95 (m, 2 H), 4.29 (t, J = 5.8 Hz, 2 H), 3.84 (t, J = 5.8 Hz, 2 H), 1.37 (s, 9 H); 13C NMR (75 MHz, CDCl3) δ 194.3, 176.6, 161.7, 154.1, 135.3, 132.5, 131.3, 130.6, 121.4, 114.1, 68.0, 41.6, 39.2, 27.1; CIMS m/z (relative intensity) 361 (MH+, 100); HRESIMS m/z calcd for C20H22O4Cl (MH+) 361.1207, found 367.1202.
4-(4-(3-Bromopropoxy)benzoyl)phenyl Pivalate (53b)
A suspension of 52 (198 mg, 0.663 mmol) and Cs2CO3 (671 mg, 2.06 mmol) in 1,3-dibromopropane (2 mL) and CH3CN (6 mL) was stirred at room temperature for 4 h. The solvent was carefully evaporated and the residue was dissolved in water (15 mL) and extracted with ethyl acetate (15 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (85:15 hexanes-ethyl acetate) to provide the product 53b as a white solid (219 mg, 79%): mp 87-89 °C. 1H NMR (300 MHz, CDCl3) δ 7.80-7.77 (m, 4 H), 7.18-7.15 (m, 2 H), 6.96-6.92 (m, 2 H), 4.15 (t, J = 5.8 Hz, 2 H), 3.58 (t, J = 5.8 Hz, 2 H), 2.33-2.29 (m, 2 H), 1.36 (s, 9 H); 13C NMR (75 MHz, CDCl3) δ 194.2, 176.6, 162.3, 154.0, 135.4, 132.4, 131.2, 130.2, 121.4, 114.0, 65.5, 39.2, 32.1, 29.8, 27.1; CIMS m/z (relative intensity) 419 (MH+, 100); HRESIMS m/z calcd for C21H23O4BrNa (MNa+) 441.0677, found 441.0672.
4-(1-(4-(3-Bromopropoxy)phenyl)-2-phenylbut-1-en-1-yl)phenyl Pivalate (54b)
The benzophenone 53b (180 mg, 0.429 mmol) and propiophenone (209 mg, 1.56 mmol) were reacted according to the general McMurry cross-coupling reaction procedure, and the product was purified by silica gel column chromatography (9:1 hexanes-ethyl acetate) to provide the product 54b as a colorless oil (215 mg, 96%). The NMR spectrum shows a 3:1 mixture of E and Z isomers. 1H NMR (300 MHz, CDCl3) δ 7.27-7.24 (m, 2 H, isomer 1), 7.20-7.11 (m, 7.3 H), 7.09-7.06 (m, 2 H, isomer 1), 6.92-6.87 (m, 1.4 H, isomer 2), 6.80-6.77 (m, 2 H, isomer 1), 6.74-6.71 (m, 0.7 H, isomer 2), 6.57-6.54 (m, 2 H, isomer 1), 4.14 (t, J = 5.7 Hz, 0.7 H, isomer 2), 3.97 (t, J = 5.7 Hz, 2 H, isomer 1), 3.63 (t, J = 5.7 Hz, 0.7 H, isomer 2), 3.55 (t, J = 5.7 Hz, 2 H, isomer 1), 2.54-2.48 (m, 2.7 H), 2.36-2.32 (m, 0.7 H, isomer 2), 2.27-2.22 (m, 2 H, isomer 1), 1.39 (s, 9 H, isomer 1), 1.30 (s, 3 H, isomer 2), 0.99-0.91 (m, 3.9 H); EIMS m/z (relative intensity) 520 (M+, 13), 57 (100); HRESIMS m/z calcd for C30H33O3BrNa (MNa+) 545.1495, found 545.1492.
4-(1-(4-(2-Chloroethoxy)phenyl)-2-phenylbut-1-en-1-yl)phenol (55a)48
The benzophenone 53a (129 mg, 0.357 mmol) and propiophenone (160 mg, 1.19 mmol) were reacted according to the general McMurry cross-coupling reaction procedure and the crude 54a underwent the general procedure for amide reduction with LiAlH4 sequentially, and the product was purified by silica gel column chromatography (80:20 hexanes-ethyl acetate) to provide the product 55a as a pale yellow oil (79 mg, 58%). The NMR spectrum shows a 1:2 mixture of E and Z isomers. The mixture isomerized to be a 1:1 mixtue of E and Z isomers when dissolved in CDCl3 and kept at room temperature for one week. 1H NMR (300 MHz, CDCl3) δ 7.20-7.09 (m, 14 H), 6.92-6.89 (m, 2 H), 6.82-6.78 (m, 4 H), 6.75-6.72 (m, 2 H), 6.58-6.55 (m, 2 H), 6.49-6.46 (m, 2 H), 4.26 (t, J = 5.8 Hz, 2 H), 4.09 (t, J = 5.8 Hz, 2 H), 3.83 (t, J = 5.8 Hz, 2 H), 3.73 (t, J = 5.8 Hz, 2 H), 2.51-2.46 (m, 4 H), 0.94 (t, J = 7.4 Hz, 6 H). HPLC purity 95.9% (C-18 reverse phase, MeOH-H2O, 85:15).
4-(1-(4-(3-Aminopropoxy)phenyl)-2-phenylbut-1-en-1-yl)phenol (55b)
A mixture of 54b (185 mg, 0.355 mmol) and NaI (273 mg, 1.82 mmol) was dissolved with THF (8 mL), and then an aqueous solution of ammonium hydroxide (30%, 4 mL) was added. The solution was heated to 70 °C and stirred in a sealed tube for 24 h. After cooling down, the solvent was carefully evaporated, and the residue was dissolved in saturated NH4Cl aqueous solution (20 mL) and extracted with ethyl acetate (20 mL × 3). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (85:15 dichloromethane-methanol) to provide the product 55b as a white solid (71.6 mg, 54%): mp 183-186 °C. The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (500 MHz, DMSO-d6) δ 7.15-7.11 (m, 4 H), 7.06-7.02 (m, 8 H), 6.94-6.91 (m, 2 H), 6.88-6.86 (m, 2 H), 6.72-6.70 (m, 2 H), 6.66-6.64 (m, 2 H), 6.56-6.51 (m, 4 H), 6.36-6.34 (m, 2 H), 3.98 (t, J = 6.4 Hz, 2 H), 3.82 (t, J = 6.4 Hz, 2 H), 2.66 (t, J = 6.4 Hz, 2 H), 2.58 (t, J = 6.4 Hz, 2 H), 2.39-2.32 (m, 4 H), 1.76-1.74 (m, 2 H), 1.68-1.63 (m, 2 H), 0.80 (t, J = 7.4 Hz, 6 H); 13C NMR (125 MHz, DMSO-d6) δ 157.7, 156.9, 156.5, 155.6, 142.6, 140.2, 140.0, 138.3, 135.9, 135.6, 134.2, 133.9, 131.7, 130.4, 129.7, 128.3, 128.2, 126.3, 126.2, 115.3, 114.6, 114.3, 113.6, 65.7, 65.5, 38.7, 38.6, 32.9, 32.8, 28.9, 28.8, 13.8; ESIMS m/z (relative intensity) 374 (MH+, 100); HRESIMS m/z calcd for C25H28NO2 (MH+) 374.2120, found 374.2114. Anal. Calcd for C25H27NO2·0.3 MeOH: C, 79.32; H, 7.42; N, 3.66. Found: C, 79.23; H, 7.29; N, 3.81.
(4-Bromophenyl)(4-methoxyphenyl)methanone (57)49
A solution of 56 (2.43 g, 11.1 mmol) and 20 (6 mL, 32.9 mmol) in dry dichloromethane (12 mL) was stirred in an ice bath. Aluminium chloride (1.51 g, 11.2 mmol) was added. The mixture was kept at 0 °C for 1 h and then warmed to room temperature and stirred overnight. The reaction mixture was poured in ice-water (40 mL), stirred for 15 min and extracted with dichloromethane (40 mL × 4). The organic layers were combined and dried over Na2SO4. Solvent was evaporated and the residue was further purified by silica gel column chromatography (1:1 hexanes-dichloromethane) to afford the product 57 as a white solid (2.87 g, 89%): mp 150-153 °C (lit49 151-152 °C). 1H NMR (300 MHz, CDCl3) δ 7.81-7.78 (m, 2 H), 7.65-7.62 (m, 4 H), 6.98-6.95 (m, 2 H), 3.89 (s, 3 H).
(4-Bromophenyl)(4-hydroxyphenyl)methanone (58)50
A solution of 57 (1.45 g, 4.98 mmol) in HBr (48% v/v, 15 mL) and glacial acetic acid (15 mL) was heated at reflux for 7 h. After cooling down, the solvent was carefully removed in vacuo. The residue was dissolved in water (40 mL) and extracted with ethyl acetate (40 mL × 3). The organic layers were combined, washed with saturated NaHCO3 (50 mL) and brine (50 mL), and dried over Na2SO4. The solvent was evaporated, and the residue was further purified by silica-gel column chromatography (7:3 hexanes-ethyl acetate) to provide the product 58 as a white solid (1.23 g, 89%): mp 189-191 °C (lit50 191 °C). 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.53-7.49 (m, 2 H), 7.44-7.38 (m, 4 H), 6.72-6.67 (m, 2 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 195.3, 162.0, 136.8, 132.7, 131.2, 131.0, 128.0, 126.6, 115.0.
4-(1-(4-bromophenyl)-1-(4-hydroxyphenyl)but-1-en-2-yl)phenyl Pivalate (59b)
Compounds 58 (365 mg, 1.32 mmol) and 8 (867 mg, 3.70 mmol) were reacted according to the general McMurry cross-coupling reaction procedure. The product was purified by silica gel column chromatography (4:1 hexanes-ethyl acetate) and then suspended in ethanol-hexanes (1:10, 11 mL) and filtered. The product was dissolved in dichloromethane and allowed to stand at room temperature overnight. Removal of the solvent in vacuo provided the product 59b as a pale yellow oil (386 mg, 61%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, CDCl3) δ 7.48-7.46 (m, 2 H), 7.14-7.08 (m, 8 H), 7.06-7.04 (m, 2 H), 6.90-6.86 (m, 4 H), 6.80-6.78 (m, 2 H), 6.75-6.73 (m, 2 H), 6.69-6.67 (m, 2 H), 6.48-6.46 (m, 2 H), 2.48-2.42 (m, 4 H), 1.35 (s, 9 H), 1.34 (s, 9 H), 0.94-0.90 (m, 6 H); 13C NMR (75 MHz, CDCl3) δ 177.5, 177.4, 154.6, 153.8, 149.2, 149.1, 142.4, 142.0, 141.6, 140.9, 139.5, 139.3, 137.5, 137.4, 135.2, 134.6, 132.4, 132.0, 131.2, 131.1, 130.6, 130.5, 130.4, 121.0, 120.9, 120.5, 119.8, 115.0, 114.5, 39.0, 29.0, 28.8, 27.0, 13.5, 13.4; EIMS m/z (relative intensity) 478 (M+, 86), 257 (100); negative ion HRESIMS m/z calcd for C27H26O3Br (M - H+)− 477.1065, found 477.1079.
4-(1-(4-(3-Aminopropyl)phenyl)-2-phenylbut-1-en-1-yl)phenol (61a)
A suspension of 59a (157 mg, 0.41 mmol), acrylamide (360 mg, 5.1 mmol), and Pd(PPh3)4 (107 mg, 0.093 mmol) in triethylamine (2 mL) and DMF (6 mL) was stirred under argon and heated to 120 °C for 24 h. After cooling down, 1 N HCl (10 mL) was added and the solution was extracted with ethyl acetate (10 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (95:5 dichloromethane-methanol) to provide the product 60a as yellow solid. The NMR spectrum shows the product is not pure, and still contains a certain amount of acrylamide. The crude product 60a was combined with Rh(PPh3)3Cl (14.0 mg, 0.015 mmol), dissolved with methanol (5 mL), and the solution was vigorously stirred at 40 °C under a hydrogen atmosphere overnight. The solid was removed by filtration and the filtrate was concentrated in vacuo. The residue was dissolved with dry THF (5 mL) and added to a suspension of AlCl3 (46 mg, 0.344 mmol) and LiAlH4 (165 mg, 4.40 mmol) in dry THF (3 mL) at 0 °C. The mixture was warmed to room temperature and stirred under argon overnight. The reaction was quenched with H2O (0.5 mL) and the THF was evaporated. The residue was dissolved in saturated NH4Cl aqueous solution (15 mL) and extracted with ethyl acetate (15 mL × 5). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (15:85 methanol-dichloromethane) to provide the product 61a as a colorless glass (45.6 mg, 34%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4 and CDCl3) δ 7.13-7.00 (m, 16 H), 6.77-6.74 (m, 6 H), 6.66-6.63 (m, 2 H), 6.43-6.40 (m, 2 H), 2.73-2.57 (m, 6 H), 2.49-2.36 (m, 6 H), 1.83-1.78 (m, 2 H), 1.68-1.63 (m, 2 H), 0.90-0.84 (m, 6 H); 13C NMR (75 MHz, methanol-d4 and CDCl3) δ 156.9, 156.0, 144.0, 142.9, 142.7, 142.5, 142.1, 141.1, 140.1, 139.8, 139.7, 136.4, 136.0, 133.3, 132.1, 131.9, 131.0, 130.7, 129.3, 129.0, 128.9, 128.5, 127.1, 116.1, 115.4, 42.3, 42.1, 35.4, 35.1, 34.2, 34.0, 30.2, 14.6; ESIMS m/z (relative intensity) 358 (MH+, 100); HRESIMS m/z calcd for C25H28NO (MH+) 358.2171, found 358.2169. Anal. Calcd for C25H27NO·0.1 MeOH: C, 83.58; H, 7.66; N, 3.88. Found: C, 83.43; H, 7.71; N, 3.89.
4,4'-(1-(4-(3-Aminopropyl)phenyl)but-1-ene-1,2-diyl)diphenol (61b)
A suspension of 59b (375 mg, 0.782 mmol), acrylamide (193 mg, 2.72 mmol) and Pd(PPh3)4 (155 mg, 0.134 mmol) in triethylamine (1 mL) and DMF (3 mL) was stirred under argon and heated to 120 °C for 24 h. After cooling down, 1 N HCl (20 mL) was added and the solution was extracted with ethyl acetate (20 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (97:3 dichloromethane-methanol) to provide the product 60b as a black oil. The NMR spectrum showed the product 60b was not pure, and still contained a certain amount of acrylamide. The crude product 60b was combined with Rh(PPh3)3Cl (32 mg, 0.034 mmol) and dissolved with methanol (8 mL), and the mixture was vigorously stirred at 40 °C under a hydrogen atmosphere overnight. The solid was removed by filtration and the filtrate was concentrated in vacuo. The residue was combined with AlCl3 (88 mg, 0.66 mmol) and LiAlH4 (316 mg, 8.33 mmol), dissolved in dry THF (10 mL) and stirred at room temperature overnight. The reaction was quenched with H2O (1 mL) and THF was evaporated. The residue was dissolved in saturated NH4Cl aqueous solution (25 mL) and extracted with ethyl acetate (25 mL × 6). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (first eluting with 15:85 methanol-dichloromethane, and then 25:5:0.3 dichloromethane-methanol-TEA) to provide the product 61b as a yellow glass (53.2 mg, 18%). The NMR spectrum shows a 1:1 mixture of E and Z isomers. 1H NMR (500 MHz, methanol-d4) δ 7.17-7.15 (m, 2 H), 7.10-7.08 (m, 2 H), 6.98-6.96 (m, 2 H), 6.90-6.82 (m, 6 H), 6.78-6.76 (m, 2 H), 6.74-6.72 (m, 2 H), 6.64-6.61 (m, 2 H), 6.58-6.56 (m, 2 H), 6.54-6.52 (m, 2 H), 6.41-6.39 (m, 2 H), 2.84 (t, J = 7.6 Hz, 2 H), 2.73 (t, J = 7.6 Hz, 2 H), 2.67 (t, J = 7.6 Hz, 2 H), 2.51 (t, J = 7.6 Hz, 2 H), 2.44-2.34 (m, 4 H), 1.92-1.87 (m, 2 H), 1.80-1.77 (m, 2 H), 0.90-0.85 (m, 6 H); 13C NMR (125 MHz, methanol-d4) δ 155.7, 155.2, 154.8, 142.0, 141.7, 140.9, 140.3, 138.8, 137.6, 137.5, 134.9, 134.6, 133.3, 131.5, 130.6, 130.4, 130.1, 129.2, 129.0, 127.6, 126.8, 114.4, 114.2, 114.1, 113.6, 39.4, 39.2, 31.9, 31.7, 30.4, 30.0, 28.3, 28.2, 13.2, 12.6; ESIMS m/z (relative intensity) 374 (MH+, 100); HRESIMS m/z calcd for C25H28NO2 (MH+) 374.2120, found 374.2119. Anal. Calcd for C25H27NO2·0.5 MeOH: C, 78.63; H, 7.50; N, 3.60. Found: C, 78.56; H, 7.39; N, 3.63.
(E)-(1-(4-Hydroxyphenyl)but-1-ene-1,2-diyl)bis(4,1-phenylene) Bis(2,2-dimethylpropanoate) (62)
The substituted propiophenone 8 (1.20 g, 5.12 mmol) andbenzophenone 52 (611 mg, 2.05 mmol) were reacted according to the general McMurry cross-coupling reaction procedure. The product was purified by silica gel column chromatography (4:1 hexanes-ethyl acetate) and further tutirated with methanol (15 mL) to afford the product 62 as a white solid (802 mg, 78%): mp 218-219 °C. 1H NMR (500 MHz, methanol-d4 and CDCl3) δ 7.20-7.17 (m, 2 H), 7.09-7.06 (m, 2 H), 6.99-6.97 (m, 2 H), 6.82-6.80 (m, 2 H), 6.65-6.62 (m, 2 H), 6.45-6.42 (m, 2 H), 2.40 (q, J = 7.4 Hz, 2 H), 1.31 (s, 9 H), 1.28 (s, 9 H), 0.87 (t, J = 7.4 Hz, 3 H); 13C NMR (125 MHz, methanol-d4 and CDCl3) δ 177.7, 177.6, 154.8, 149.4, 148.9, 141.2, 140.2, 139.8, 138.0, 133.9, 131.8, 130.4, 130.2, 120.8, 120.6, 114.1, 38.9, 38.8, 28.6, 26.7, 26.6, 13.1; MALDIMS m/z (relative intensity) 500 (M+, 100); HRESIMS m/z calcd for C32H37O5 (MH+) 501.2641, found 501.2643.
(E)-(1-(4-(2-Amino-2-oxoethoxy)phenyl)but-1-ene-1,2-diyl)bis(4,1-phenylene) Bis(2,2-dimethylpropanoate) (63)
A suspension of 62 (730 mg, 1.46 mmol), 2-iodoacetamide (1.10 g, 5.95 mmol), and K2CO3 (1.0 g, 7.24 mmol) in acetone-H2O (19:1, 15 mL) was heated to reflux for 3 h. After cooling down, the solvent was evaporated and the residue was dissolved in saturated aqueous NH4Cl solution (30 mL) and extracted with ethyl acetate (30 mL × 6). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (55:45 hexanes-ethyl acetate) to provide the product 63 as a white solid (455 mg, 56 %): mp 167-169 °C. 1H NMR (300 MHz, CDCl3) δ 7.23-7.20 (m, 2 H), 7.11-7.04 (m, 4 H), 6.89-6.86 (m, 2 H), 6.82-6.79 (m, 2 H), 6.59-6.56 (m, 2 H), 4.35 (s, 2 H), 2.46 (q, J = 7.3 Hz, 2 H), 1.36 (s, 9 H), 1.33 (s, 9 H), 0.92 (t, J = 7.3 Hz, 3 H); 13C NMR (75 MHz, CDCl3) δ 177.0, 171.2, 155.2, 149.8, 149.3, 141.4, 140.7, 139.4, 137.4, 136.5, 132.1, 130.5, 130.3, 121.2, 121.0, 113.7, 66.9, 39.1, 39.0, 29.0, 27.1, 13.5; MALDIMS m/z (relative intensity) 557 (M+, 100); HRESIMS m/z calcd for C34H40NO6 (MH+) 558.2856, found 558.2855.
4,4'-(1-(4-(2-Aminoethoxy)phenyl)but-1-ene-1,2-diyl)diphenol (64)
A suspension of AlCl3 (45 mg, 0.34 mmol) and LiAlH4 (267 mg, 7.03 mmol) in dry THF (5 mL) was stirred under argon and cooled to 0 °C. A solution of 63 (151 mg, 0.269 mmol) in dry THF (6 mL) was added. The mixture was warmed to room temperature and stirred under argon for 2 h. The reaction was quenched with H2O (2 mL) and the solvent was evaporated. The residue was dissolved in saturated NH4Cl aqueous solution (20 mL) and extracted with ethyl acetate (20 mL × 4). The organic layers were combined, dried over Na2SO4, concentrated in vacuo and further purified by silica gel column chromatography (9:1 dichloromethane-methanol) to provide the product 64 as a pale yellow oil (53.7 mg, 53%). The NMR spectrum showed a nearly 1:3 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.09-7.07 (m, 0.7 H, isomer E), 7.00-6.97 (m, 2 H, isomer Z), 6.92-6.87 (m, 3.5 H), 6.76-6.72 (m, 4 H, isomer Z), 6.66-6.63 (m, 0.7 H, isomer E), 6.59-6.54 (m, 4.8 H), 6.43-6.40 (m, 0.7 H, isomer E), 3.99 (t, J = 5.2 Hz, 0.7 H, isomer E), 3.85 (t, J = 5.2 Hz, 2 H, isomer Z), 2.99 (t, J = 5.2 Hz, 0.7 H, isomer E), 2.90 (t, J = 5.2 Hz, 2 H, isomer Z), 2.44-2.39 (m, 2.7 H), 0.92-0.87 (m, 4.1 H).
4,4'-(1-(4-(2-Aminoethoxy)phenyl)but-1-ene-1,2-diyl)diphenol (65)
The amide 9 (84.0 mg, 0.177 mmol, E/Z = 4.5:1) was reduced following the general LiAlH4 reduction procedure and the product was further purified by silica gel column chromatography (9:1 dichloromethane-methanol) to provide the product 65 as a pale yellow oil (46.6 mg, 70%). NMR shows a nearly 3:2 mixture of E and Z isomers. 1H NMR (300 MHz, methanol-d4) δ 7.10-7.07 (m, 2 H, isomer E), 7.00-6.97 (m, 1.2 H, isomer Z), 6.92-6.88 (m, 5.2 H), 6.76-6.72 (m, 2.4 H, isomer Z), 6.66-6.62 (m, 2 H, isomer E), 6.59-6.55 (m, 4.4 H), 6.43-6.40 (m, 2 H, isomer E), 4.00 (t, J = 5.2 Hz, 2 H, isomer E), 3.86 (t, J = 5.2 Hz, 1.2 H, isomer Z), 2.99 (t, J = 5.2 Hz, 2 H, isomer E), 2.90 (t, J = 5.2 Hz, 1.2 H, isomer Z), 2.44-2.37 (m, 3.2 H), 0.92-0.87 (m, 5 H).
Inhibition of Recombinant Human Aromatase (CYP19) by Microsomal Incubations
The activity of recombinant aromatase (CYP19) was determined by measuring the conversion rate of the fluorometric substrate 7-methoxy-4-trifluoromethylcoumarin (MFC) to its fluorescent metabolite 7-hydroxy-4-trifluoromethylcoumarin (HFC). Experimental procedures were consistent with the published methodology.51 All of the incubations were performed using incubation times and protein concentrations that were within the linear range for reaction velocity. The fluorometric substrate, MFC, was dissolved in acetonitrile with the final concentration of 25 mM. All tested samples were dissolved in either methanol or DMSO. The sample solutions (2 μL) were mixed well with 98 μL of NADPH-Cofactor Mix (16.25 μM NADP+, 825.14 μM MgCl2, 825.14 μM glucose-6-phosphate and 0.4 Units/mL glucose-6-phosphate dehydrogenase), and were pre-warmed for 10 min at 37 °C. Enzyme/Substrate Mix was prepared with fluorometric substrate, recombinant human aromatase (CYP19) and 0.1 M potassium phosphate buffer (pH 7.4). Reactions were initiated by adding 100 μL Enzyme/Substrate Mix to bring the incubation volume to 200 μL, and incubated for 30 min. All the reactions were stopped by adding 75 μL of 0.1 M tris base dissolved in acetonitrile. The amount of fluorescent product was determined immediately by measuring fluorescent response using a BioTek (Winooski, VT) Synergy 2 fluorometric plate reader. Excitation-emission wavelengths for MFC metabolite were 409 and 530 nm. The standard curve for MFC metabolite was constructed using the appropriate fluorescent metabolite standards. Quantification of samples was performed by applying the linear regression equation of the standard curve to the fluorescence response. The limit of quantification for the metabolites of MFC was 24.7 pmol with intra- and inter-assay coefficients of variation less than 10%.
Kinetic Analysis of Recombinant Human Aromatase (CYP19)
The rates of metabolite formation in the presence of the test inhibitors were compared with those in the control, in which the inhibitor was replaced with vehicle. The extent of enzyme inhibition was expressed as the percentage of remaining enzyme activity compared to the control. IC50 values were determined as the inhibitor concentrations that brought about half reduction in enzyme activity by fitting all the data to a one-site competition equation using Graphpad Prism 5.0 (GraphPad Software Inc., San Diego, CA). To characterize the inhibitory mechanism of norendoxifen against aromatase (CYP19), all inhibitory data by norendoxifen at different substrate concentrations were plotted as Lineweaver-Burk plots. The inhibitory constant Ki values were determined by nonlinear least square regression analysis using Graphpad Prism 5.0 (GraphPad Software Inc., San Diego, CA). Before modeling the data using nonlinear models, initial information about the inhibitory mechanism was obtained by visual inspection of Lineweaver-Burk plots. The final decision on the mechanism of inhibition was made on model-derived parameters, such as R2 (or R Square) and absolute sum of squares.
Binding Affinities for Recombinant Human ER-α and ER-β
The binding affinities to ER-α and ER-β were determined by measuring the change of polarization value when the fluorescent estrogen ligand, ES2, was displaced by the tested compounds. Experimental procedures were consistent with the protocol provided by Invitrogen. The fluorescent estrogen ligand, ES2, was provided in methanol/water (4:1, v/v) with the concentration of 1800 nM. Recombinant human ER-α and ER-β were provided in buffer (50 mM bis-tris propane, 400 mM KCl, 2 mM DTT, 1 mM EDTA and 10% glycerol), with concentrations of 734 nM and 3800 nM, respectively. All tested samples were dissolved in either methanol or DMSO. The sample solutions (1 μL) were mixed well with 49 μL of ES2 screening buffer (100 mM potassium phosphate, 100 μg/mL BGG and 0.02% NaN3). The ER-α/ES2 complex was prepared with the fluorescent estrogen ligand ES2, human recombinant ER and ES2 screening buffer with the concentration of 9 nM ES2 and 30 nM ER-α. The ER-β/ES2 complex was prepared with the fluorescent estrogen ligand ES2, human recombinant ER-β and ES2 screening buffer with the concentration of 9 nM ES2 and 20 nM ER-β. Reactions were initiated by adding 50μL ER/ES2 complex to bring the incubation volume to 100 μL, and incubated for 2 h avoiding light. The polarization value was determined by measuring fluorescent response using a BioTek (Winooski, VT) Synergy 2 fluorometric plate reader. Excitation-emission wavelengths for fluorescence polarization were 485 and 530 nM. The polarization values in the presence of the test competitors were compared with those in the control, in which the competitor was replaced with vehicle. The extent of competition was expressed as the percentage of remaining polarization compared to the control. EC50 values were determined as the competitor concentrations that brought about half reduction in polarization value by fitting all the data to a one-site competition equation using Graphpad Prism 5.0 (GraphPad Software Inc., San Diego, CA).
Construction of the Z-Norendoxifen-ER-α Complex
The crystal structure of the 4-hydroxytamoxifen and ER-α complex was obtained from the Protein Data Bank (PDB ID: 3ert). The two N-methyl groups of 4-hydroxy tamoxifen were removed and all crystal water molecules were removed (except the water that forms bifurcated hydrogen bonds with Glu353 and Arg394). The resulting ligand-protein complex was thoroughly energy minimized by use of the Amber 10 molecular dynamics package. The Amber parm99 force field was used for the protein during energy minimization. For the ligand, the force field parameters were taken from the general amber force field (GAFF), whereas the atomic partial charges were derived from the AM1-BCC method implemented by antechamber.
Molecular Dynamics Simulations of the Z-Norendoxifen-ER-α Complex
The minimized Z-norendoxifen-ER-α complex was solvated with a truncated octahedron periodic box explicit solvent model filled with TIP3P water. The box was extended 8 Å from the solute atoms, and sodium ions were added as counterions to neutralize the system. The whole system was slowly heated to 300 K and equilibrated for about 100 ps at constant-volume. Then, 2 ns of molecular dynamics simulation at constant-pressure were performed. The lengths of bonds involved in hydrogen atoms were fixed with the SHAKE algorithm, and the time step of simulation was 2.0 fs. Periodic boundary conditions were used and the cutoff for nonbonded interaction was 10 Å. All molecular dynamics simulations were performed with the AMBER 14 package.
Cell Culture and Test Compound Treatment
The estrogen receptor-positive human breast carcinoma cell line MCF-7 was seeded at a density of 105 cells/well in 6-well plates and maintained at 37 °C under a humidified atmosphere of 5% CO2 and 95% air in minimum essential media (MEM) supplemented with 10% fetal bovine serum (FBS). Before the test compound treatments, the cells were preconditioned in charcoal-stripped FBS for 72 h to remove the estrogens from the growth medium containing 10% FBS. The cells were treated with vehicle (0.1% methanol) alone, 1 μM test compound, or 1 μM endoxifen (positive control) for 24 h in the presence of 10 nM β-estradiol (E2) dissolved in MEM supplemented with 10% charcoal-stripped FBS.
Ribonucleic Acid (RNA) Extraction and Concentration Measurement
The MCF-7 cells treated for 24 h with test compounds or experimental controls were harvested for progesterone receptor (PGR) messenger ribonucleic acid (mRNA) extraction. Before ribonucleic acid (RNA) extraction, genomic DNA was eliminated. RNA was extracted from approximately 3×105 cells by RNeasy Plus Mini Kit (Qiagen Inc., Valencia, California, USA). The RNA concentration was measured using the Qubit RNA BR assay (Life Technologies Corp., Carlsbad, CA) for the Qubit 2.0 fluorometer (Life Technologies Corp., Carlsbad, CA). The RNA was stored at −80 °C before further use.
Complementary Deoxyribonucleic Acid (cDNA) Synthesis
Complementary deoxyribonucleic acid (cDNA) for the real-time quantitative polymerase chain reaction (PCR) assay was synthesized from DNase-treated total RNA using the QuantiTect reverse transcription kit (Qiagen Inc., Valencia, California, USA).
Real-Time Quantitative Polymerase Chain Reaction (PCR) for cDNA
The cDNA was amplified with TaqMan Universal PCR Master Mix (Applied Biosystems Inc., Carlsbad, CA), and then PCR was performed in the QuantStudio 12K Flex Real-Time PCR System (Life Technologies Corp., Carlsbad, CA). Progesterone receptor gene (PGR, FAM, Hs01556702, Life Technologies Corp., Carlsbad, CA) was the target gene, while glyceraldehyde-3-phosphate dehydrogenase (GAPDH, VIC, Hs02758991, Life Technologies Corp., Carlsbad, CA) gene expression was quantified to normalize each sample. A total of 40 amplification cycles were performed. Quantitative values of amplification were obtained from the threshold cycle (Ct) defined as the cycle number at which the fluorescent signal is first recorded above the background as determined during the exponential phase of PCR rather than at the endpoint. The 2−ΔΔCt method was used to determine the relative mRNA expression, and the results were expressed as percentages of antagonism effects compared to E2-stimulated PGR mRNA expression (considered as 100%). If amplification was not seen by 40 cycles, the measured RNA was considered to be undetectable.
Figure 6.

Abilities of norendoxifen analogues (1000 nM) to antagonize progesterone receptor mRNA expression stimulated by β-estradiol (10 nM) in MCF-7 cells.
ACKNOWLEDGMENT
This research was supported by the Purdue University Center for Cancer Research and the Indiana University Cancer Center Joint Funding Award 206330, by the Purdue Center for Cancer Research grant P30 CA023168, by a Purdue Research Foundation Research Grant, and by an award from the Floss Endowment, provided to the Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University. W. Lv thanks Dr. Charles Paget for providing the graduate travel award.
ABBREVIATIONS USED
- AIs
aromatase inhibitors
- ATAC
arimidex, tamoxifen, alone or in combination
- ER
estrogen receptor
- MOMCl
methyl chloromethyl ether
- NBS
N-bromosuccinimide
- PDB
protein data bank
- SERM
selective estrogen receptor modulator
Footnotes
ASSOCIATED CONTENT
SMILES molecular formula strings. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Breast Cancer Facts & Figures 2013-2014. American Cancer Society, Inc.; Atlanta: 2013. [Google Scholar]
- 2.Cancer Facts & Figures 2013. American Cancer Society, Inc.; Atlanta: 2013. [Google Scholar]
- 3.Clarke M, Collins R, Davies C, Godwin J, Gray R, Peto R. Early Breast Cancer Trialists Collaborative, G. Tamoxifen for Early Breast Cancer: An Overview of the Randomised Trials. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 4.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N. Tamoxifen for Prevention of Breast Cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J. Natl. Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 5.Ali S, Coombes RC. Endocrine-Responsive Breast Cancer and Strategies for Combating Resistance. Nat. Rev. Cancer. 2002;2:101–112. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 6.Santen RJ. Long Term Tamoxifen Therapy: Can an Antagonist Become an Agonist? J. Clin. Endocrinol. Metab. 1996;81:2027–2029. doi: 10.1210/jcem.81.6.8964823. [DOI] [PubMed] [Google Scholar]
- 7.Bonneterre J, Buzdar A, Nabholtz JMA, Robertson JFR, Thurlimann B, von Euler M, Sahmoud T, Webster A, Steinberg M, Arimidex Writing C, Investigators Comm M. Anastrozole is Superior to Tamoxifen as First-line Therapy in Hormone Receptor Positive Advanced Breast Carcinoma - Results of Two Randomized Trials Designed for Combined Analysis. Cancer. 2001;92:2247–2258. doi: 10.1002/1097-0142(20011101)92:9<2247::aid-cncr1570>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 8.Mouridsen H, Gershanovick M, Sun Y, Perez-Carrion R, Boni C, Monnier A, Apffelstaedt J, Smith R, Sleeboom HP, Jaenicke F, Pluzanska A, Dank M, Becquart D, Bapsy PP, Salminen E, Snyder R, Chaudri-Ross H, Lang R, Wyld P, Bhatnagar A. Phase III Study of Letrozole Versus Tamoxifen as First-line Therapy of Advanced Breast Cancer in Postmenopausal Women: Analysis of Survival and Update of Efficacy from the International Letrozole Breast Cancer Group. J. Clin. Oncol. 2003;21:2101–2109. doi: 10.1200/JCO.2003.04.194. [DOI] [PubMed] [Google Scholar]
- 9.Howell A, Cuzick J, Baum M, Buzdar A, Dowsett M, Forbes JF, Hoctin-Boes G, Houghton I, Locker GY, Tobias JS, Grp AT. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) Trial after Completion of 5 Years' Adjuvant Treatment for Breast Cancer. Lancet. 2005;365:60–62. doi: 10.1016/S0140-6736(04)17666-6. [DOI] [PubMed] [Google Scholar]
- 10.Thurlimann B, Keshaviah A, Coates AS, Mouridsen H, Mauriac L, Forbes JF, Paridaens R, Castiglione-Gertsch M, Gelber RD, Rabaglio M, Smith I, Wardly A, Price KN, Goldhirsch A, Grp BIGC. A Comparison of Letrozole and Tamoxifen in Postmenopausal Women with Early Breast Cancer. N. Engl. J. Med. 2005;353:2747–2757. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 11.Williams N. Effect of Anastrozole and Tamoxifen as Adjuvant Treatment for Early-stage Breast Cancer: 100-Month Analysis of the ATAC Trial. Lancet Oncol. 2008;9:45–53. doi: 10.1016/S1470-2045(07)70385-6. [DOI] [PubMed] [Google Scholar]
- 12.Heshmati HM, Khosla S, Robins SP, O'Fallon WM, Melton LJ, Riggs BL. Role of Low Levels of Endogenous Estrogen in Regulation of Bone Resorption in Late Postmenopausal Women. J. Bone Miner. Res. 2002;17:172–178. doi: 10.1359/jbmr.2002.17.1.172. [DOI] [PubMed] [Google Scholar]
- 13.Ewer MS, Gluck S. A Woman's Heart. The Impact of Adjuvant Endocrine Therapy on Cardiovascular Health. Cancer. 2009;115:1813–1826. doi: 10.1002/cncr.24219. [DOI] [PubMed] [Google Scholar]
- 14.Bird BRJH, Swain SM. Cardiac Toxicity in Breast Cancer Survivors: Review of Potential Cardiac Problems. Clin. Cancer Res. 2008;14:14–24. doi: 10.1158/1078-0432.CCR-07-1033. [DOI] [PubMed] [Google Scholar]
- 15.Bundred NJ. The Effects of Aromatase Inhibitors on Lipids and Thrombosis. Br. J. Cancer. 2005;93:S23–S27. doi: 10.1038/sj.bjc.6602692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan QJ, O'Dea AP, Sharma P. Musculoskeletal Adverse Events Asociated with Adjuvant Aromatase Inhibitors. J. Oncol. 2010:8. doi: 10.1155/2010/654348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jelovac D, Macedo L, Goloubeva OG, Handratta V, Brodie AMH. Additive Antitumor Effect of Aromatase Inhibitor Letrozole and Antiestrogen Fulvestrant in a Postmenopausal Breast Cancer Model. Cancer Res. 2005;65:5439–5444. doi: 10.1158/0008-5472.CAN-04-2782. [DOI] [PubMed] [Google Scholar]
- 18.Baum M, Buzdar AU, Cuzick J, Forbes J, Houghton J, Klijn JGM, Sahmoud T, Grp AT. Anastrozole Alone or in Combination with Tamoxifen Versus Tamoxifen Alone for Adjuvant Treatment of Postmenopausal Women with Early Breast Cancer: First Results of the ATAC Randomised Trial. Lancet. 2002;359:2131–2139. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 19.Lu WJ, Xu C, Pei ZF, Mayhoub AS, Cushman M, Flockhart DA. The Tamoxifen Metabolite Norendoxifen Is a Potent and Selective Inhibitor of Aromatase (CYP19) and a Potential Lead Compound for Novel Therapeutic Agents. Breast Cancer Res. Treat. 2012;133:99–109. doi: 10.1007/s10549-011-1699-4. [DOI] [PubMed] [Google Scholar]
- 20.Lv W, Liu J, Lu D, Flockhart DA, Cushman M. Synthesis of Mixed (E,Z)-, (E)-, and (Z)-Norendoxifen with Dual Aromatase Inhibitory and Estrogen Receptor Modulatory Activities. J. Med. Chem. 2013;56:4611–4618. doi: 10.1021/jm400364h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh D, Lo J, Morton D, Valette D, Xi J, Griswold J, Hubbell S, Egbuta C, Jiang W, An J, Davies HML. Novel Aromatase Inhibitors by Structure-Guided Design. J. Med. Chem. 2012;55:8464–8476. doi: 10.1021/jm300930n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 23.Favia AD, Nicolotti O, Stefanachi A, Leonetti F, Carotti A. Computational Methods for the Design of Potent Aromatase Inhibitors. Expert Opin. Drug Discov. 2013;8:395–409. doi: 10.1517/17460441.2013.768983. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen A, Top S, Pigeon P, Vessieres A, Hillard EA, Plamont MA, Huche M, Rigamonti C, Jaouen G. Synthesis and Structure-Activity Relationships of Ferrocenyl Tamoxifen Derivatives with Modified Side Chains. Chem. Eur. J. 2009;15:684–696. doi: 10.1002/chem.200801108. [DOI] [PubMed] [Google Scholar]
- 25.Lu WJ, Desta Z, Flockhart DA. Tamoxifen Metabolites as Active Inhibitors of Aromatase in the tTeatment of Breast Cancer. Breast Cancer Res. Treat. 2012;131:473–481. doi: 10.1007/s10549-011-1428-z. [DOI] [PubMed] [Google Scholar]
- 26.Davies HML, Nagashima T, Klino JL. Stereoselectivity of Methyl Aryldiazoacetate Cyclopropanations of 1,1-Diarylethylene. Asymmetric Aynthesis of a Cyclopropyl Analogue of Tamoxifen. Organic Lett. 2000;2:823–826. doi: 10.1021/ol005563u. [DOI] [PubMed] [Google Scholar]
- 27.Canne LE, Winston RL, Kent SBH. Synthesis of a Versatile Purification Handle for Use with Boc Chemistry Aolid Phase Peptide Synthesis. Tetrahedron Lett. 1997;38:3361–3364. [Google Scholar]
- 28.Ruenitz PC, Bourne CS, Sullivan KJ, Moore SA. Estrogenic Triarylethylene Acetic Acids: Effect of Structural Variation on Estrogen Receptor Affinity and Estrogenic Potency and Efficacy in MCF-7 Cells. J. Med. Chem. 1996;39:4853–4859. doi: 10.1021/jm960517e. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Lu D, Lu J, Lv W, Cushman M, Desta Z, Flockhart DA. Hydroxynorendoxifen, an Active Tamoxifen Metabolite, Possesses Dual Aromatase Inhibitory and Estrogen Receptor Modulatory Activities. Clin. Pharmacol. Ther. 2014;95:S21–S21. [Google Scholar]
- 30.Liu JZ, Flockhart PJ, Lu DS, Lv W, Lu WJJ, Han X, Cushman M, Flockhart DA. Inhibition of Cytochrome P450 Enzymes by the E- and Z-Isomers of Norendoxifen. Drug Metab. and Dispos. 2013;41:1715–1720. doi: 10.1124/dmd.113.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gauthier S, Mailhot J, Labrie F. New Highly Stereoselective Synthesis of (Z)-4-Hydroxytamoxifen and (Z)-4-Hydroxytoremifene via McMurry Reaction. J. Org. Chem. 1996;61:3890–3893. doi: 10.1021/jo952279l. [DOI] [PubMed] [Google Scholar]
- 32.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular Basis of Agonism and Antagonism in the Oestrogen Receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 33.Jordan VC. Antiestrogens and Selective Estrogen Receptor Modulators as Multifunctional Medicines. 1. Receptor Interactions. J. Med. Chem. 2003;46:883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 34.Jordan VC, Lieberman ME, Cormier E, Koch R, Bagley JR, Ruenitz PC. Structural Requirements for the Pharmacological Activity of Nonsteroidal Antiestrogens In Vitro. Molecular Pharmacology. 1984;26:272–278. [PubMed] [Google Scholar]
- 35.Levenson AS, Jordan VC. The Key to the Antiestrogenic Mechanism of Raloxifene Is Amino Acid 351 (Aspartate) in the Estrogen Receptor. Cancer Res. 1998;58:1872–1875. [PubMed] [Google Scholar]
- 36.Schafer JM, Liu H, Bentrem DJ, Zapf JW, Jordan VC. Allosteric Silencing of Activating Function 1 in the 4-Hydroxytamoxifen Estrogen Receptor Complex Is Induced by Substituting Glycine for Aspartate at Amino Acid 351. Cancer Res. 2000;60:5097–5105. [PubMed] [Google Scholar]
- 37.Lim YC, Desta Z, Flockhart DA, Skaar TC. Endoxifen (4-Hydroxy-N-desmethyltamoxifen) Has Anti-estrogenic Effects in Breast Cancer Cells with Potency Similar to 4-Hydroxy-tamoxifen. Cancer Chemother. Pharmacol. 2005;55:471–478. doi: 10.1007/s00280-004-0926-7. [DOI] [PubMed] [Google Scholar]
- 38.Miquel JF. 1,1-Bis(p-hydroxyphenyl)-2,2-diethylethylene, a New Isomer of Stilbestrol. Acta Chem. Scand. 1958;12:274–280. [Google Scholar]
- 39.Shiina I, Sano Y, Nakata K, Suzuki M, Yokoyama T, Sasaki A, Orikasa T, Miyamoto T, Ikekita M, Nagahara Y, Hasome Y. An Expeditious Synthesis of Tamoxifen, a Representative SERM (Selective Estrogen Receptor Modulator), via the Three-Component Coupling Reaction Among Aromatic Aldehyde, Cinnamyltrimethylsilane, and β-Chlorophenetole. Bioorg. Med. Chem. 2007;15:7599–7617. doi: 10.1016/j.bmc.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 40.Stanciuc O, Balaban AT. Reaction of Pyrylium Salts with Nucleophiles. 23: Triarylethene Derivatives Containing an Oxyalkyleneamino or Oxyalkylene-N-pyridinium Side Chain. J. Pharm. Sci. 1993;82:927–933. doi: 10.1002/jps.2600820911. [DOI] [PubMed] [Google Scholar]
- 41.Bao XF, Lu SY, Liow JS, Zoghbi SS, Jenko KJ, Clark DT, Gladding RL, Innis RB, Pike VW. Radiosynthesis and Evaluation of an F-18-Labeled Positron Emission Tomography (PET) Radioligand for Brain Histamine Subtype-3 Receptors Based on a Nonimidazole 2-Aminoethylbenzofuran Chemotype. J. Med. Chem. 2012;55:2406–2415. doi: 10.1021/jm201690h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lubczyk V, Bachmann H, Gust R. Investigations on Estrogen Receptor Binding. The Estrogenic, Antiestrogenic, and Cytotoxic Properties of C2-Alkyl-Substituted 1,1-Bis(4-hydroxyphenyl)-2-phenylethenes. J. Med. Chem. 2002;45:5358–5364. doi: 10.1021/jm0209230. [DOI] [PubMed] [Google Scholar]
- 43.Forman B, Yu D. Compounds and Methods for Treating Breast Cancer and Other Diseases. U.S. Pat. Appl. Publ. 2005 US 2005/0096384 A1.
- 44.Duan XF, Zeng J, Lu JW, Zhang ZB. Insights into the General and Efficient Cross McMurry Reactions between Ketones. J. Org. Chem. 2006;71:9873–9876. doi: 10.1021/jo061644d. [DOI] [PubMed] [Google Scholar]
- 45.Katzenellenbogen JA, Tatee T, Robertson DW. Preparation of Tritium-Labeled 4-Hydroxy-α-[p-(2-(N-pyrrolidinyl)ethoxy)phenyl]-α'-nitrostilbene (CN-928), a Biologically Important Metabolite of the Antiestrogen CI-628. J. Labelled Compd. Radiopharm. 1981;18:865–879. [Google Scholar]
- 46.Kim SH, Katzenellenbogen JA. Triarylethylene Bisphenols with a Novel Cycle Are Ligands for the Estrogen Receptor. Bioorg. Med. Chem. 2000;8:785–793. doi: 10.1016/s0968-0896(00)00016-x. [DOI] [PubMed] [Google Scholar]
- 47.Burke PJ, Koch TH. Design, Synthesis, and Biological Evaluation of Doxorubicin - Formaldehyde Conjugates Targeted to Breast Cancer Cells. J. Med. Chem. 2004;47:1193–1206. doi: 10.1021/jm030352r. [DOI] [PubMed] [Google Scholar]
- 48.Ali SM, Ahmad A, Shahabuddin S, Ahmad MU, Sheikh S, Ahmad I. Endoxifen is a New Potent Inhibitor of PKC: A Potential Therapeutic Agent for Bipolar Disorder. Bioorg. Med. Chem. Lett. 2010;20:2665–2667. doi: 10.1016/j.bmcl.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 49.Fausett BW, Liebeskind LS. Palladium-Catalyzed Coupling of Thiol Esters with Aryl and Primary and Secondary Alkyl Organoindium Reagents. J. Org. Chem. 2005;70:4851–4853. doi: 10.1021/jo050110u. [DOI] [PubMed] [Google Scholar]
- 50.Prakash GKS, Panja C, Mathew T, Olah GA. BF3-H2O catalyzed Fries Rearrangement of Phenolic Esters. Catal. Lett. 2007;114:24–29. [Google Scholar]
- 51.Lu WJ, Ferlito V, Xu C, Flockhart DA, Caccamese S. Enantiomers of Naringenin as Pleiotropic, Stereoselective Inhibitors of Cytochrome P450 Isoforms. Chirality. 2011;23:891–896. doi: 10.1002/chir.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]