

Abstract
Background
Gibberellin (GA), a classical phytohormone, plays significant roles in plant growth and development. It shares the important intermediate diphosphate precursor, GGPP, with the main lipophilic bioactive components, diterpenoid tanshinones in Salvia miltiorrhiza Bunge, one of the most important Traditional Chinese Medicine materials and an emerging model medicinal plant. Analysis of GA metabolism and regulation may help to demonstrate the biological functions of GAs and the crosstalk between GA metabolism and tanshinone biosynthesis in S. miltiorrhiza. However, genes involved in the conversion of ent-kaurene to GAs have not been systematically studied.
Results
Through genome-wide prediction and molecular cloning, twenty two candidate gibberellin metabolism pathway genes were systematically identified for the first time. It includes a SmKO, two SmKAOs, six SmGA20oxs, two SmGA3oxs and eleven SmGA2oxs, of which twenty genes are novel. The deduced proteins showed sequence conservation and divergence. Gibberellin metabolism pathway genes exhibited tissue-specific expression patterns and responded differentially to exogenous GA3 treatment, indicating differential regulation of gibberellin metabolism in different tissue types in S. miltiorrhiza. SmKAO1, SmKAO2, SmGA2ox2, and SmGA2ox4–SmGA2ox7 were significantly up-regulated; SmGA20ox2, SmGA3ox1, SmGA2ox1, SmGA2ox8, SmGA2ox10 and SmGA2ox11 were significantly down-regulated; while the responses of many other genes varied among different tissue-types and time-points of GA3 treatment, suggesting the complexity of feedback regulation. Tanshinone biosynthesis-related genes, such as SmCPS1 and SmKSL1, were up-regulated in response to GA3 treatment. Among the 22 identified genes, nine responded to yeast extract and Ag+-treatment in S. miltiorrhiza hairy roots. Moreover, tissue-specifically expressed splice variants were identified for SmKO, SmGA20ox3, SmGA2ox3 and SmGA2ox11, of which SmKOv1, SmGA20ox3v and SmGA2ox11v1 were GA3-responsive, suggesting the importance of alternative splicing in regulating GA metabolism.
Conclusions
The results show tissue-specifically expressed, feedback-regulated, stress-responsive and alternatively spliced novel genes and reveal multiple layer regulation of GA metabolism and crosstalk between gibberellin metabolism and tanshinone biosynthesis in S. miltiorrhiza.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2315-5) contains supplementary material, which is available to authorized users.
Keywords: Alternative splicing, Gibberellin, Salvia miltiorrhiza, Tanshinone, Terpenoid, Traditional Chinese Medicine
Background
Gibberellin (GA), discovered by Dr. E. Kurosawa in 1926 [1], is a classical phytohormone. It is a large group of tetracylic diterpenoids with more than 130 members identified in plants, fungi and bacteria [2, 3]. Based on the number of carbon atoms, GAs can be classified into C19-GAs and C20-GAs, which consist of 19 and 20 carbon atoms, respectively. C19-GAs, such as GA1, GA3, GA4, GA5, GA7, GA9 and GA20, are converted from C20-GAs, including GA12, GA15, GA24, GA19, GA44, GA53 and so on. GA1, GA3, GA4 and GA7 of C19-GAs are biologically active GAs in higher plants. GA1 and GA4 are saturated and are the main bioactive GAs with relative abundance varying in different species and tissues, while GA3 and GA7 are double bond-containing GAs with less abundance compared with GA1 and GA4 [4]. GAs play vital roles in many diverse aspects of plant growth and development, such as seed germination [5, 6], shoot elongation [7], leaf expansion [8], flower development [9], and fruit-setting [10, 11].
Similarly with other diterpenoids, GAs are synthesized from trans-geranylgeranyl diphosphate (GGPP), an important intermediate diphosphate precursor produced mainly from pyruvate and glyceraldehyde 3-phosphate via the 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway in the plastid. The formation of GGPP during diterpenoid biosynthesis also depends on the crosstalk between the MEP pathway and the mevalonate (MVA) pathway operated in the cytoplasm, peroxisome, endoplasmic reticulum and mitochondrion [12]. GA metabolism pathway from GGPP to end-products can be generally divided into three stages [2, 13, 14]. In the first stage, ent-kaurene is formed from GGPP in two steps via ent-copalyl diphosphate (CPP) under the catalysis of ent-copalyl diphosphate synthase (CPS) and ent-kaurene synthase (KS). ent-Kaurene is further oxidized to GA12, in the second stage, by two multifunctional cytochrome P450 monooxygenases (P450s), known as ent-kaurene oxidase (KO) and ent-kaurenoic acid oxidase (KAO). KO converts ent-kaurene to ent-kaurenoic acid via ent-kaurenol and ent-kaurenal, while KAO converts ent-kaurenoic acid to GA12 via ent-7α-hydroxykaurenoic acid and GA12-aldehyde. In the last stage, bioactive GAs are produced under the catalysis of GA 20-oxidase (GA20ox) and GA 3-oxidase (GA3ox) and can be deactivation by GA 2-oxidase (GA2ox) in plants [2, 14–17].
Genes involved in GA metabolism have been identified in various plant species, such as Arabdopsis [16–23], pumpkin [24], rice [25], pea [6, 26, 27], and maize [28]. CPS, KS, KO and KAO enzymes involved in the early steps of the GA metabolism pathway are usually encoded by single or few genes [4]. For instance, the rice OsCPS and OsKSL gene families consist of three and eleven members, respectively; however, only OsCPS1 and OsKS1 are responsible for ent-kaurene biosynthesis. Similarly, in the Arabidopsis genome, only a CPS and a KS exist. KO and KAO are members of the large P450 gene family containing 246 and 356 genes in Arabidopsis and rice, respectively [29]. In Arabidopsis, there are only one AtKO and two AtKAO genes. The number of rice OsKO and OsKAO is two and one, respectively. Unlike the enzymes involved in stages one and two, GA20ox, GA3ox and GA2ox, which play catalytic roles in the third stage, are encoded by multiple differentially expressed genes [4]. Arabidopsis has seven AtGA2ox, four AtGA3ox and five AtGA20ox genes (http://www.arabidopsis.org), each of which exhibits a unique expression pattern and plays distinct developmental roles [19–21, 30, 31]. For instance, AtGA3ox1 and AtGA3ox2 are responsible for bioactive GA biosynthesis during vegetative growth, while AtGA3ox1, AtGA3ox3 and AtGA3ox4 are important for the development of reproductive organs [19, 21]. Among the five AtGA20ox genes, AtGA20ox1, AtGA20ox2 and AtGA20ox3 are the dominant paralogs [20]. AtGA20ox3 is functionally redundant with AtGA20ox1 and AtGA20ox2, while AtGA20ox4 and AtGA20ox5 play very minor roles in most developmental stages [20]. Differential expression and distinct developmental roles were also observed for rice 2-oxoglutarate-dependent dioxygenase (2-ODD) genes, which include eight OsGA20ox, two OsGA3ox and eleven OsGA2ox genes (http://www.ricedata.cn/gene) [23, 25, 31, 32]. In addition to differential expression in organs, tissues and developmental stages of plants, the expression of GA metabolism pathway genes is also regulated by environmental cues, such as light, temperature and other stresses [4, 33]. Moreover, transcript levels of some, but not all, GA metabolism pathway genes are under feedback control [18, 34–36]. It includes inhibition of some GA20ox and GA3ox gene expression and activation of some GA2ox gene expression [4, 15, 19].
Salvia miltiorrhiza Bunge is an important Traditional Chinese Medicine (TCM) material widely used in Chinese medicines. It mainly produces two groups of bioactive components, including the water-soluble phenolic acids and the lipid-soluble tanshinones. The latters are a group of diterpenoids sharing the universal precursor, GGPP, with other diterpenoids, including GAs [12, 37]. The biosynthesis of tanshinones from GGPP involves SmCPS1, SmKSL1, CYP76AH1 and other unknown genes [12, 37, 38]. SmCPS1 of the S. miltiorrhiza CPS gene family encodes enzymes responsible for the conversion of GGPP to CPP, which is subsequently cycled and rearranged to miltiradiene under the catalysis of enzymes encoded by SmKSL1, a member of the SmKSL gene family. Recently, a total of 40 genes, members of 19 gene families involved in terpenoid biosynthesis, have been identified and characterized in S. miltiorrhiza through a genome-wide analysis [12]. Of the 40 genes, 33 are involved in the formation of intermediate diphosphate precursors via the MEP and MVA pathways, five are members of the SmCPS gene families, while the other two encode SmKSLs. Analysis of GA metabolism and regulation may greatly help to demonstrate the role of GAs in S. miltiorrhiza growth and development and the crosstalk between GA metabolism and tanshinone biosynthesis, which are very important for genetic improvement of S. miltiorrhiza. However, genes involved in the conversion of ent-kaurene to GAs have not been systematically studied. In this study, genome-wide identification, molecular cloning and expression analysis of the SmKO, SmKAO, SmGA20ox, SmGA3ox and SmGA2ox gene families in S. miltiorrhiza were carried out. The results identified tissue-specifically expressed, feedback-regulated, stress-responsive and alternatively spliced novel genes, revealed multiple layer regulation of GA metabolism, and provided evidence for crosstalk between GA metabolism and tanshinone biosynthesis in S. miltiorrhiza.
Results
Prediction and molecular cloning of GA metabolism pathway genes in S. miltiorrhiza
In order to predict GA metabolism pathway genes in S. miltiorrhiza, we downloaded the deduced amino acid sequences of 19 Arabidopsis and 24 rice KO, KAO, GA2ox, GA3ox and GA20ox proteins from the Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org) and the China Rice Data Center (http://www.ricedata.cn/gene), respectively. BLAST analysis of the downloaded Arabidopsis and rice sequences against the current assembly of the S. miltiorrhiza genome was then performed using the tBLASTn algorithm [39, 40]. The retrieved S. miltiorrhiza genomic DNA sequences putatively encoding proteins with more than 50 % identity to Arabidopsis or rice homologs were predicted for gene models on the Genscan web server (http://genes.mit.edu/GENSCAN.html) [41] and the NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) [40]. As a result, 22 gene models, including one for SmKO, two for SmKAOs, six for SmGA20oxs, two for SmGA3oxs and eleven for SmGA2oxs, were predicted. Among them, sixteen putatively encode full-length proteins, while the other six are partial.
In order to verify the predicted gene models and to obtain full-length coding sequences, molecular cloning was carried out using PCR techniques. The results showed that all of the predicted gene models were experimentally validated and full-length coding sequences of the 22 predicted genes were obtained (Table 1). Based on sequence similarities between the cloned cDNAs and the known genes in other plant species, the identified genes were named SmKO, SmKAO1, SmKAO2, SmGA20ox1–SmGA20ox6, SmGA3ox1, SmGA3ox2, and SmGA2ox1–SmGA2ox11, respectively. The cloned nucleotide sequences and their deduced amino acid sequences may be found in GenBank under the accession numbers shown in Table 1.
Table 1.
Sequence features of GA metabolism pathway genes in S. miltiorrhiza
| Gene name | Accession number | ORFa (bp) | Lenb | MWc (kDa) | pI | Locd |
|---|---|---|---|---|---|---|
| SmKO | KT853074 | 1557 | 519 | 58.73 | 7.63 | S |
| SmKAO1 | KT853075 | 1302 | 434 | 50.00 | 9.33 | S |
| SmKAO2 | KT853076 | 1431 | 477 | 54.82 | 9.43 | S |
| SmGA3ox1 | KT853077 | 1038 | 346 | 37.86 | 6.70 | - |
| SmGA3ox2 | KT853078 | 1092 | 364 | 39.92 | 7.74 | - |
| SmGA20ox1 | KT853079 | 1134 | 378 | 43.01 | 6.38 | - |
| SmGA20ox2 | KT853080 | 1173 | 391 | 43.83 | 6.56 | - |
| SmGA20ox3 | KT853081 | 1155 | 385 | 43.01 | 7.69 | - |
| SmGA20ox4 | KT853082 | 1176 | 392 | 44.09 | 6.11 | C |
| SmGA20ox5 | KT853083 | 1044 | 348 | 38.50 | 5.99 | - |
| SmGA20ox6 | KT853084 | 1050 | 350 | 39.99 | 5.84 | - |
| SmGA2ox1 | KT853085 | 1122 | 374 | 42.12 | 7.54 | C |
| SmGA2ox2 | KT853086 | 966 | 322 | 35.70 | 5.60 | - |
| SmGA2ox3 | KT853087 | 936 | 312 | 34.66 | 6.12 | - |
| SmGA2ox4 | KT853088 | 1005 | 335 | 37.67 | 6.22 | - |
| SmGA2ox5 | KT853089 | 1002 | 334 | 38.15 | 7.29 | C |
| SmGA2ox6 | KT853090 | 987 | 329 | 37.39 | 5.56 | - |
| SmGA2ox7 | KT853091 | 939 | 313 | 35.16 | 5.38 | - |
| SmGA2ox8 | KT853092 | 1005 | 335 | 37.19 | 7.08 | - |
| SmGA2ox9 | KT853093 | 933 | 311 | 34.36 | 5.85 | - |
| SmGA2ox10 | KT853094 | 1137 | 379 | 41.83 | 5.69 | - |
| SmGA2ox11 | KT853095 | 972 | 324 | 36.01 | 6.67 | - |
aORF represents open reading frame
bLen represents the number of amino acid residues
cMW represents molecular weight
dLoc represents the protein localization predicted by TargetP1.1. ‘S’ stands for secretory pathway, showing that the sequence cotains a signal peptide. ‘C’ stands for chloroplast, suggesting that the sequence contains a chloroplast transit peptide. ‘-’ indicates any locations other than the plastid, mitochondrion and secretory pathway
BLAST analysis of the Arabidopsis, rice and S. miltiorrhiza sequences against the S. miltiorrhiza transcriptomic unigenes assembled from ESTs and RNA-seq reads [42] showed that unigenes could be identified for nine of the 22 cloned S. miltiorrhiza GA metabolism pathway genes. It includes SmKO, SmKAO1, SmGA3ox2, SmGA20ox2–SmGA20ox4, SmGA2ox3, SmGA2ox5 and SmGA2ox11. No additional GA metabolism pathway genes were identified. BLAST analysis of the cloned cDNAs against the non-redundant protein sequence (nr) database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) using the BLASTn algorithm with default parameters [40] showed that the coding regions of SmKO and SmKAO1 had just been identified through the analysis of high-throughput RNA-Seq data and reported as CYP701A40 (accession no. KP337739) and CYP88A52 (KP337715), respectively [43]. Additionally, SmKO (KJ606394) was also cloned from the hairy roots of S. miltiorrhiza (line Shanxishangluo) [44]. It further verifies our results from computational prediction and experimental cloning.
Characterization and expression analysis of SmKO
KO catalyzes the conversion of ent-kaurene to ent-kaurenoic acid via ent-kaurenol and ent-kaurenal. The cloned SmKO cDNA encodes a protein with the amino acid number of 519, the theoretical isoelectric point (pI) of 7.63, and the predicted molecular weight (MW) of 58.73 kDa (Table 1). It shares over 60 % identities with KOs in various other plants, such as Sesamum indicum (XP_011083784) and Erythranthe guttata (EYU45074). Phylogenetic analysis of 27 KOs from 24 plant species showed that plant KOs could be divided into three clades (Fig. 1). SmKO clusters with EgKO, SiKO and other nine KOs from seven species in clade 1, AtKO clusters with PtKO and other ten KOs in clade 2, while OsKO1 and OsKO2 clusters with TaKO and AsKO in clade 3 (Fig. 1a). The results indicate that the cloned SmKO encodes a bona fide ent-kaurene oxidase in S. miltiorrhiza.
Fig. 1.
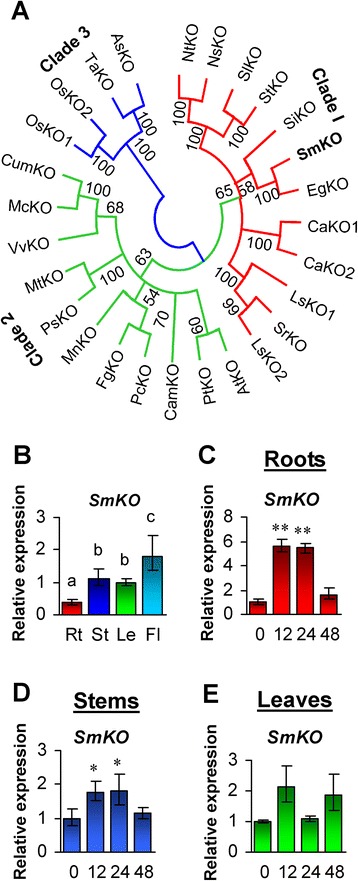
Phylogenetic analysis of KOs and expression profiling of SmKO. a: Phylogenetic relationship of KOs in Aegilops speltoides (As), Arabidopsis thaliana (At), Castanea mollissima (Cam), Coffea arabica (Ca), Cucurbita maxima (Cum), Erythranthe guttata (Eg), Fragaria grandiflora (Fg), Lactuca sativa (Ls), Medicago truncatula (Mt), Momordica charantia (Mc), Morus notabilis (Mn), Nicotiana sylvestris (Ns), Nicotiana tomentosiformis (Nt), Oryza sativa (Os), Pisum sativum (Ps), Populus trichocarpa (Pt), Pyrus communis (Pc), S. miltiorrhiza (Sm), Sesamum indicum (Si), Solanum lycopersicum (Sl), Solanum tuberosum (St), Stevia rebaudiana (Sr), Triticum aestivum (Ta), and Vitis vinifera(Vv). The unrooted neighbor-joining tree was constructed using the MEGA 6.0 [64]. SmKO from S. miltiorrhiza are in bold. Clades 1–3 indicate the three clades identified. b: Fold changes of SmKO genes in roots (Rt), stems (St), leaves (Le) and flowers (Fl) of S. miltiorrhiza plants. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in leaves was arbitrarily set to 1 and the levels in other tissues were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 was considered statistically significant. c–e: Responses of SmKO to exogenous GA3 treatment. Fold changes of SmKO transcripts in roots (c), stems (d) and leaves (e) of S. miltiorrhiza plantlets treated with 100 μM GA3 for 0, 12, 24 and 48 h are shown. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in tissues without treatment (0 h) was arbitrarily set to 1 and the levels in tissues from GA3-treated plantlets were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 (*) and P < 0.01 (**) was considered statistically significant and extremely significant, respectively
Like other P450 proteins, SmKO contains the P450 conserved domain (Additional file 1: Figure S1). Protein subcellular localization prediction indicates that SmKO contains a 15 bp secretory pathway signal peptide at the N-terminus, suggesting this protein is most possibly present in the endoplasmic reticulum (Table 1). Similar results were also obtained for Arabidopsis and rice KOs, including AtKO, OsKO1 and OsKO2 (Table 1, Additional files 2 and 3: Tables S1 and S2), indicating that the conversion of ent-kaurene to ent-kaurenoic acid is associated with the endoplasmic reticulum.
In consistence with the role of GA in plant growth and development, SmKO showed ubiquitous expression in all the tissues analyzed, with the highest in flowers, followed by stems and leaves, and the lowest in roots (Fig. 1b). The expression pattern of SmKO is similar to that of AtKO showing the highest level in inflorescence, less in elongating stems, and relatively low in both rosette and cauline leaves [45]. In order to know the response of GA metabolism pathway genes to active GAs in S. miltiorrhiza, we analyzed the expression of SmKO in roots, stems and leaves of S. miltiorrhiza treated with exogenous 100 μM GA3 for 12, 24 and 48 h, respectively. Relative expression was analyzed using the qRT-PCR method as described previously [12]. The results showed that SmKO was up-regulated in roots and stems of S. miltiorrhiza plants treated with exogenous GA3 for 12 and 24 h (Fig. 1c and 1d). No significant changes were found in leaves at all three time-points of GA3 treatment and in roots and stems at the time-point of 48-h-treatment (Fig. 1c–1e), suggesting tissue-specificity of SmKO response to GA3 treatment.
Characterization and expression analysis of SmKAO1 and SmKAO2
KAO catalyzes the conversion of ent-kaurenoic acid to GA12 via ent-7α-hydroxykaurenoic acid and GA12-aldehyde. One OsKAO and two AtKAO genes were identified in rice and Arabidopsis, respectively [16, 25]. From S. miltiorrhiza, we identified two KAO genes, SmKAO1 and SmKAO2. SmKAO1 encodes a protein with 434 amino acid residues, while SmKAO encodes a protein consisting of 477 amino acid residues (Table 1). Both of them contain the P450 domain. It is consistent with previous results showing that KAOs are members of the P450 family [29]. SmKAO1 and SmKAO2 share about 58 % identity at the amino acid level and are clustered together in the phylogenetic tree constructed with 15 KAOs from 11 plant species (Fig. 2a). Consistently, two KAOs from other plant species, such as Arabidopsis, Helianthus annuus and Pisum sativum, are also clustered together in the phylogenetic tree (Fig. 2a). It suggests that two KAOs from a plant species are usually paralogous proteins. SmKAO1 and SmKAO2 were expressed in all tissues examined (Fig. 2b and 2c). The expression of SmKAO1 was higher in stems, roots and leaves than flowers, while the expression of SmKAO2 was higher in roots, flowers and leaves and less in stems (Fig. 2b and 2c). Analysis of SmKAOs to exogenous GA3 showed that SmKAO1 was up-regulated in roots and leaves at all three time-points of GA3 treatment and in stems at the time-points of 12- and 48-h-treatement (Fig. 2d–2f). SmKAO2 was up-regulated in roots at all three time-points of GA3 treatment, in stems at the time-points of 12- and 48-h-treatement, and in leaves treated with GA3 for 12 and 24 h (Fig. 2g–2i). It suggests differential responses of SmKAO1 and SmKAO2 to GA3 treatment.
Fig. 2.
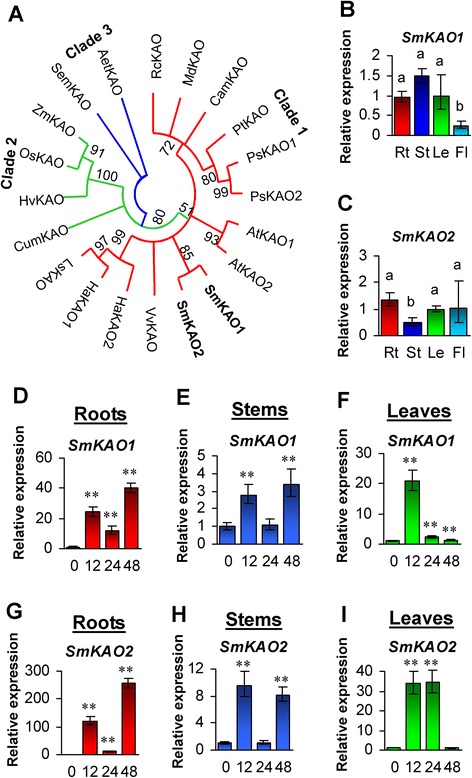
Phylogenetic analysis of KAOs and expression profiling of SmKAO1 and SmKAO2. a: Phylogenetic relationship of KAOs in Aegilops tauschii (Aet), Arabidopsis thaliana (At), Castanea mollissima (Cam), Cucurbita maxima (Cum), Helianthus annuus (Ha), Hordeum vulgare (Hv), Lactuca sativa (Ls), Malus domestica (Md), Oryza sativa (Os), Pisum sativum (Ps), Populus trichocarpa (Pt), Ricinus communis (Rc), S. miltiorrhiza (Sm), Selaginella moellendorffii (Sem), Vitis vinifera(Vv), and Zea mays (Zm). The unrooted neighbor-joining tree was constructed using the MEGA 6.0 [64]. SmKAOs from S. miltiorrhiza are in bold. Clades 1–3 indicate the three clades identified. b and c: Fold changes of SmKAO1 (b) and SmKAO2 (c) genes in roots (Rt), stems (St), leaves (Le) and flowers (Fl) of S. miltiorrhiza plants. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in leaves was arbitrarily set to 1 and the levels in other tissues were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 was considered statistically significant. d–i: Responses of SmKAOs to exogenous GA3 treatment. Fold changes of SmKAO1 (d–f) and SmKAO2 (g–i) transcripts in roots, stems and leaves of S. miltiorrhiza plantlets treated with 100 μM GA3 for 0, 12, 24 and 48 h are shown. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in tissues without treatment (0 h) was arbitrarily set to 1 and the levels in tissues from GA3-treated plantlets were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 (*) and P < 0.01 (**) was considered statistically significant and extremely significant, respectively
Characterization and expression analysis of the SmGA20ox gene family
GA20ox represents an important regulatory node in GA metabolism and plays significant roles in maintaining the endogenous GA level in plants [20, 46, 47]. It catalyzes the conversion of GA12 and GA53 (C20-GAs) to GA9 and GA20 (C19-GAs), respectively. The reaction requires successive oxidation of C-20 from the methyl group of C20-GAs through the alcohol and then aldehyde, from which the C-20 is lost as carbon dioxide [13]. Usually, a single enzyme with full GA20ox catalytic activity can catalyze this reaction sequence [13]. However, few GA20ox enzymes catalyze only partial of the reaction sequence. For instance, there are a total of five Arabidopsis AtGA20ox enzymes, of which, four, including AtGA20ox1–AtGA20ox4, possess full GA20ox activity in vitro, while the other one, named AtGA20ox5, can only catalyze the first two steps of the reaction sequence [18, 20].
From S. miltiorrhiza, a total of six SmGA20ox genes were identified. All of them contain the DIOX_N and 2OG-FeII_Oxy domains found in 2-oxoglutarate/Fe(II)-dependent dioxygenases (Additional file 1: Figure S1). Sequence comparison showed that the six SmGA20ox proteins could be roughly divided into two groups. SmGA20ox1, SmGA20ox2, SmGA20ox4 and SmGA20ox5 share over 60 % sequence identity in a group, while SmGA20ox3 and SmGA20ox6 share about 50 % identity in the other group. Phylogenetic analysis of 27 GA20ox proteins from Arabidopsis, rice, and other eight plant species showed that plant GA20ox proteins could be divided into four clades (Fig. 3a). SmGA20ox1, SmGA20ox2, SmGA20ox4 and SmGA20ox5 group with AtGA20ox1–AtGA20ox4 and other seven GA20ox proteins from various plant species in clade 1. SmGA20ox3 and SmGA20ox6 cluster with AtGA20ox5, OsGA20ox2 and OsGA20ox4 in clade 3. Clade 2 and clade 4 mainly include GA20ox proteins from rice. No SmGA20ox and AtGA20ox proteins belong to the two clades. It indicates the conservation and divergence of GA20ox proteins in plants.
Fig. 3.

Phylogenetic analysis of GA20ox proteins and expression profiling of SmGA20ox genes. a: Phylogentic relationship of GA20ox proteins in Arabidopsis thaliana (At), Lactuca sativa (Ls), Lolium perenne (Lp), Nicotiana tabacum (Nt), Oryza sativa (Os), Phaseolus vulgaris (Pv), Pisum sativum (Ps), Salvia miltiorrhiza (Sm), Solanum dulcamara (Sd), Solanum lycopersicum (Sl), and Spinacia oleracea (So). The unrooted neighbor-joining tree was constructed using the MEGA 6.0 [64]. SmGA20ox proteins from S. miltiorrhiza are in bold. Clades 1–4 indicate the four clades identified. b–g: Fold changes of SmGA20ox1 (b), SmGA20ox2 (c), SmGA20ox3 (d), SmGA20ox4 (e), SmGA20ox5 (f) and SmGA20ox6 (g) in roots (Rt), stems (St), leaves (Le) and flowers (Fl) of S. miltiorrhiza plants. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in leaves was arbitrarily set to 1 and the levels in other tissues were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 was considered statistically significant
Since the majority of GA20ox proteins showed full catalytic activity in converting C20 substrates to C19 products, the expression patterns, rather than enzymatic activities, determine their physiological roles in plants [20]. Using the qRT-PCR method, we analyzed the expression patterns of 6 SmGA20ox genes in roots, stems, leaves and flowers of S. miltiorrhiza plants. Obvious differential expression of SmGA20ox genes was observed. SmGA20ox1 were predominantly expressed in roots, followed by leaves, and the lowest in stems and flowers (Fig. 3b). SmGA20ox2 showed high expression in roots, leaves and flowers, while its expression in stems was significantly low (Fig. 3c). SmGA20ox3 exhibited the highest expression in leaves, less in stems, flowers and roots (Fig. 3d). SmGA20ox4 showed high expression in leaves and stems, less in roots and flowers (Fig. 3e). SmGA20ox5 exhibited the highest expression in stems, followed by roots and leaves, and the lowest in flowers (Fig. 3f). SmGA20ox6 had the highest expression in roots, followed by leaves and flowers, and the lowest in stems (Fig. 3g). Differential expression of GA20ox genes was also observed in other plant species, such as Arabidopsis [20], rice [25] and maize [28], indicating that different members of the GA20ox gene family might play distinct physiological roles. Since the transcripts of multiple SmGA20ox genes exist in a tissue, it is possible that some of them act redundantly in plant growth and development.
Consistently, among the six SmGA20ox genes, SmGA20ox2 was significantly down-regulated in roots, stems and leaves treated with GA3 for 12, 24 and 48 h (Fig. 4d–4f). Down-regulated expression was also found for SmGA20ox1 in roots at the time-point of 12-h-treatment, in stems at the time-points of 12- and 24-h-treatment, and leaves at all three time-points (Fig. 4a–4c). Similarly, SmGA20ox3 was down-regulated in roots and stems at the time-point of 24-h-treatment (Fig. 4g and 4h). SmGA20ox5 was down-regulated in roots at the time-points of 24- and 48-h-treatment (Fig. 4m and 4n). SmGA20ox6 was down-regulated in roots at the time-point of 12- and 48-h-treatment, and in stems and leaves at the time-point of 12-h-treatment (Fig. 4p–4r). It suggests the existence of negative feedback mechanism to regulate the expression of some SmGA20ox genes in S. miltiorrhiza. No significant changes were observed for the expression of SmGA20ox4 in roots, stems and leaves (Fig. 4j–4l), SmGA20ox3 and SmGA20ox5 in leaves (Fig. 4i and 4o), and SmGA20ox1 (Fig. 4a and 4b), SmGA20ox3 (Fig. 4g and 4h), SmGA20ox5 (Fig. 4m and 4n), SmGA20ox6 (Fig. 4p and 4q) in roots and leaves at one or two time-points of GA3 treatment, suggesting that the responses of SmGA20 genes to GA3 treatment depend on tissue types and time-points of GA3 treatment in S. miltiorrhiza.
Fig. 4.
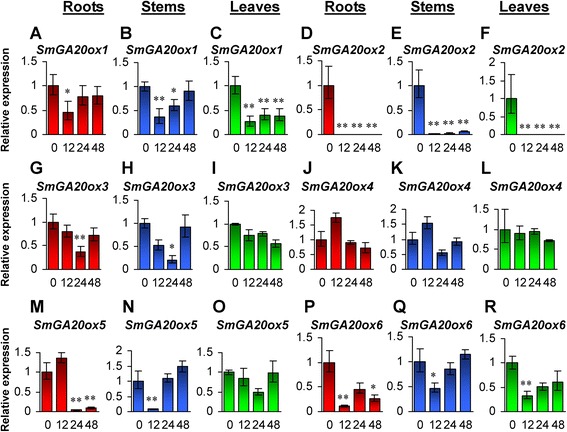
Responses of SmGA20ox genes to exogenous GA3 treatment. Fold changes of SmGA20ox1 (a–c), SmGA20ox2 (d–f), SmGA20ox3 (g–i), SmGA20ox4 (j–l), SmGA20ox5 (m–o) and SmGA20ox6 (p–r) in roots, stems and leaves of S. miltiorrhiza plantlets treated with 100 μM GA3 for 0, 12, 24 and 48 h are shown. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in tissues without treatment (0 h) was arbitrarily set to 1 and the levels in tissues from GA3-treated plantlets were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 (*) and P < 0.01 (**) was considered statistically significant and extremely significant, respectively
Characterization and expression analysis of SmGA3ox1 and SmGA3ox2
GA3ox plays a direct role in conversion of precursor GAs to their biologically active forms and determines the level of bioactive GAs in plants [19]. It catalyzes the formation of final bioactive GA4 and GA1 from GA9 and GA20, respectively, in a single step in plants. It also catalyzes the production of small amounts of GA3, a C-1,2-unsaturated bioactive GA, in a two-step reaction via GA5 in several monocotyledons [4]. From the S. miltiorrhiza genome, we identified and cloned two SmGA3ox genes, named SmGA3ox1 and SmGA3ox2, respectively (Table 1). SmGA3ox1 contains 1038 bp of ORF encoding a protein with 346 amino acids, while SmGA3ox2 contains 1092 bp of ORF encoding a protein with 364 amino acids.
Phylogenetic analysis of 22 GA3ox proteins from 13 plant species showed that plant GA3ox could be divided into 3 clades (Fig. 5a). SmGA3ox1 and SmGA3ox2 were separated into two clades (Fig. 5a), suggesting the divergence of SmGA3ox1 and SmGA3ox2. Consistently, differential expression was observed between SmGA3ox1 and SmGA3ox2 (Fig. 5b and 5c). SmGA3ox1 expressed broadly in the tissues analyzed, while SmGA3ox2 exhibited more tissue-specific expression with the highest level in stems and roots, followed by leaves, and the lowest in flowers. Differential expression was also observed for Arabidopsis and rice GA3ox genes [19, 21, 25]. Of the two rice OsGA3ox genes, OsGA3ox1 was preferentially expressed in the panicles, while OsGA3ox2 was broadly expressed in all organs tested [25]. Among the four AtGA3ox genes, AtGA3ox1 was expressed throughout development; AtGA3ox2 was mainly expressed during seed germination and vegetative growth; while AtGA3ox3 and AtGA3ox4 were predominantly expressed in reproductive organs [19, 21]. Thus, SmGA3ox1 appears to be important for the development of both vegetative and reproductive organs, while SmGA3ox2 is more specific to bioactive GA biosynthesis during vegetative growth of S. miltiorrhiza. Analysis of SmGA3oxs to exogenous GA3 showed that SmGA3ox1 was down-regulated in roots, stems and leaves treated with GA3 for 12, 24 and 48 h (Fig. 5d–5f). SmGA3ox2 was down-regulated in stems and leaves of S. miltiorrhiza treated with GA3 for 12 and 48 h (Fig. 5h and 5i). No changes were found for SmGA3ox2 in roots at all three time-points and in stems and leaves at the time-point of 24-h-treatment (Fig. 5g–5i). It suggests that each member of the GA3ox gene family from a plant species may play different physiological roles.
Fig. 5.
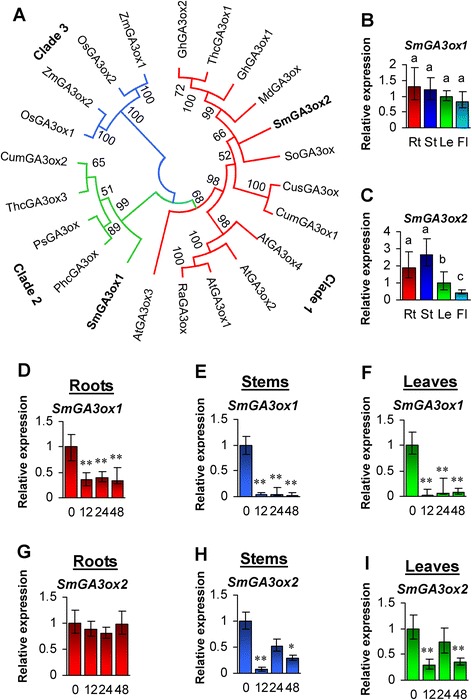
Phylogenetic analysis of GA3ox proteins and expression profiling of SmGA3ox genes. a: Phylogentic relationship of GA3ox proteins in Arabidopsis thaliana (At), Cucumis sativus (Cus), Cucurbita maxima (Cum), Gossypium hirsutum (Gh), Malus domestica (Md), Oryza sativa (Os), Physcomitrella patens (Pp), Pisum sativum (Ps), Rorippa aquatica (Ra), Salvia miltiorrhiza (Sm), Spinacia oleracea (So), Theobroma cacao (Thc), and Zea mays (Zm). The unrooted neighbor-joining tree was constructed using the MEGA 6.0 [64]. SmGA3ox1 and SmGA3ox2 from S. miltiorrhiza are in bold. Clades 1–3 indicate the three clades identified. b and c: Fold changes of SmGA3ox1 (b) and SmGA3ox2 (c) in roots (Rt), stems (St), leaves (Le) and flowers (Fl) of S. miltiorrhiza plants. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in leaves was arbitrarily set to 1 and the levels in other tissues were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 was considered statistically significant. d–i: Responses of SmGA3ox genes to exogenous GA3 treatment. Fold changes of SmGA3ox1 (d–f) and SmGA3ox2 (g–i) transcripts in roots, stems and leaves of S. miltiorrhiza plantlets treated with 100 μM GA3 for 0, 12, 24 and 48 h are shown. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in tissues without treatment (0 h) was arbitrarily set to 1 and the levels in tissues from GA3-treated plantlets were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 (*) and P < 0.01 (**) was considered statistically significant and extremely significant, respectively
Characterization and expression analysis of the SmGA2ox gene family
Contrary to GA3ox involved in bioactive GA formation, GA2ox catalyzes the deactivation of bioactive GAs, such as GA1 and GA4, by introducing a 2β-hydroxy group to the GAs [15, 22, 23]. In addition to the bioactive C19-GAs, GA2ox can also use C19- (GA9 and GA20) and C20-GA precursors (GA12 and GA53) of bioactive GAs as substrates. Therefore, GA2ox is important for turnover of the physiologically active GAs, allowing precise regulation of GA concentration in plant tissues [4, 13]. GA2ox is usually encoded by a small gene family in plants. Based on the substrates, GA2ox proteins can be divided into two groups, one group, known as C19-GA2ox proteins, using bioactive C19-GAs and C19-GA precursors as substrates, while the other group, known as C20-GA2ox, acting on C20-GA precursors [15, 22, 23].
From the current assembly of the S. miltiorrhiza genome, we predicted 11 SmGA2ox genes (Table 1). The prediction was verified by PCR amplification and subsequent sequencing. All of the deduced SmGA2ox proteins contain the conserved domains, DIOX_N and 2OG-FeII_Oxy (Additional file 1: Figure S1). It is consistent with their 2-oxoglutarate/Fe(II)-dependent dioxygenase activity. Moreover, SmGA2ox1, SmGA2ox4 and SmGA2ox6 contain the three unique and conserved motifs (Fig. 6) identified in AtGA2ox7, AtGA2ox8, OsGA2ox5, OsGA2ox6, OsGA2ox9 and SoGA2ox3, which use C20-GA precursors as substrates [22, 23]. It indicates that SmGA2ox1, SmGA2ox4 and SmGA2ox6 are C20-GA2ox proteins. Interestingly, SmGA2ox5 also contain the three motifs; however, the sequences of these motifs are less conserved compared with those in SmGA2ox1, SmGA2ox4 and SmGA2ox6 (Fig. 6). Further analysis of the motifs in AtGA2ox and OsGA2ox proteins showed that OsGA2ox11 also contained the three less conserved motifs (Fig. 6). It implies that SmGA2ox5 and OsGA2ox11 may be C20-GA2ox proteins with specific functions. The three motifs are absent from other seven SmGA2ox proteins, such as SmGA2ox2, SmGA2ox3, SmGA2ox7–SmGA2ox11, indicating they are C19-GA2ox proteins.
Fig. 6.
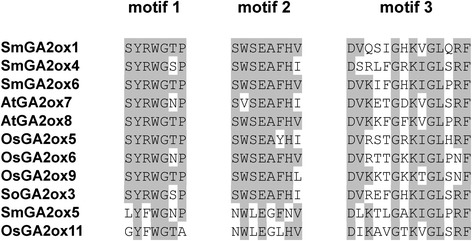
Sequence alignment of the three unique and conserved motifs identified in various GA2ox proteins. A. thaliana (At), O. sativa (Os), Spinacia oleracea (So) and S. miltiorrhiza (Sm)
Phylogenetic relationship among 42 GA2ox proteins from 10 plant species was analyzed. Based on the phylogenetic tree constructed, plant GA2ox proteins could be divided into four clades (Fig. 7). SmGA2ox2, SmGA2ox7, SmGA2ox10, SmGA2ox11, AtGA2ox1–AtGA2ox3 and other ten plant GA2ox proteins group in clade 1. Five rice OsGA2ox proteins, including OsGA2ox3, OsGA2ox4, OsGA2ox7, OsGA2ox8 and OsGA2ox10, cluster in clade 2. SmGA2ox3, SmGA2ox8, SmGA2ox9, AtGA2ox4, AtGA2ox6, OsGA2ox1, OsGA2ox2, SoGA2ox2 and PsGA2oxL group in clade 3. The other eleven, including SmGA2ox1, SmGA2ox4, SmGA2ox5, SmGA2ox6 and OsGA2ox11 and the known C20-GA2ox proteins (AtGA2ox7, AtGA2ox8, OsGA2ox5, OsGA2ox6, OsGA2ox9 and SoGA2ox3), cluster in clade 4. It is similar with previous results from plant GA2ox analysis [22, 23], suggesting members of clades 1, 2 and 3 are C19-GA2ox proteins, while members of clade 4 are C20-GA2ox proteins.
Fig. 7.
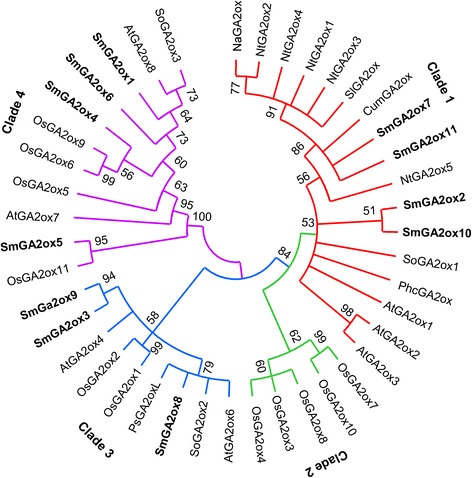
Phylogenetic analysis of GA2ox proteins in S. miltiorrhiza and various other plant species. It includes Arabidopsis thaliana (At), Cucurbita maxima (Cum), Nicotiana attenuata (Na), Nicotiana tabacum (Nt), Oryza sativa (Os), Phaseolus coccineus (Phc), Pisum sativum (Ps), Salvia miltiorrhiza (Sm), Solanum lycopersicum (Sl), and Spinacia oleracea (So). The unrooted neighbor-joining tree was constructed using the MEGA 6.0 [64]. SmGA2ox proteins from S. miltiorrhiza are in bold. Clades 1–4 indicate the four clades identified
To investigate the expression pattern of each SmGA2ox gene, we analyzed their transcript levels in roots, stems, leaves and flowers of S. miltiorrhiza using the qRT-PCR method. Similarly with the results from SmGA20ox and SmGA3ox genes, SmGA2ox genes showed obvious differential expression (Fig. 8). SmGA2ox4, SmGA2ox8 and SmGA2ox9 were mainly expressed in roots (Fig. 8d, 8h and 8i). SmGA2ox2, SmGA2ox6 and SmGA2ox7 showed the highest expression in leaves, followed by roots, flowers and stems (Fig. 8b, 8f and 8g). SmGA2ox3 and SmGA2ox5 were predominantly expressed in flowers (Fig. 8c and 8e). SmGA2ox1 showed the highest expression in leaves and flowers, followed by roots and stems (Fig. 8a). SmGA2ox10 exhibited the highest expression in stems, followed by flowers, leaves and roots (Fig. 8j). SmGA2ox11 was predominantly expressed in flowers and stems (Fig. 8k). Distinct expression patterns were also observed for members of the GA2ox gene family in Arabidopsis [15] and rice [23, 25]. These results suggest the redundant and diversified physiological roles of GA2ox genes played in plants.
Fig. 8.
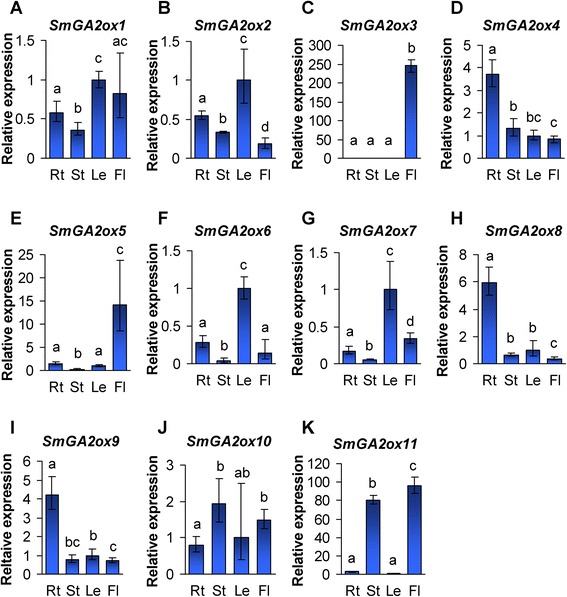
Expression patterns of SmGA2ox genes in S. miltiorrhiza plants. a–k: Fold changes of SmGA2ox1 (a), SmGA2ox2 (b), SmGA2ox3 (c), SmGA2ox4 (d), SmGA2ox5 (e), SmGA2ox6 (f), SmGA2ox7 (g), SmGA2ox8 (h), SmGA2ox9 (i), SmGA2ox10 (j) and SmGA2ox11 (k) genes in roots (Rt), stems (St), leaves (Le) and flowers (Fl) of S. miltiorrhiza plants. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in leaves was arbitrarily set to 1 and the levels in other tissues were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 was considered statistically significant
Among the eleven SmGA2ox genes, five, including SmGA2ox2 and SmGA2ox4–SmGA2ox7, were significantly up-regulated in roots, stems and leaves treated with GA3 for 12, 24 and 48 h (Fig. 9). Up-regulation of GA2ox genes has been previously found for AtGA2ox1 and AtGA2ox2 in Arabidopsis treated with exogenous GA3 [15] and is considered as a result of feedback control [4, 15]. Additionally, SmGA2ox1, SmGA2ox8, SmGA2ox10 and SmGA2ox11 were significantly down-regulated in all or the majority of the GA3-treated tissues analyzed (Fig. 9). SmGA2ox9 was significantly down-regulated in roots and stems at the time-point of 48 h and significantly up-regulated in roots at the time-point of 12-h-treatment, in stems at the time-points of 12- and 24-h-treatment, and in leaves at the time-points of 24- and 48-h-treatment (Fig. 9v–9x). The expression of SmGA2ox3 was too low to be detected in roots, stems and leaves (Fig. 8c) and in the tissues of S. miltiorrhiza treated with GA3. These results suggest that individual members of the SmGA2ox gene family exhibit differential responses to GA3 treatment and the responses depend on tissue types.
Fig. 9.
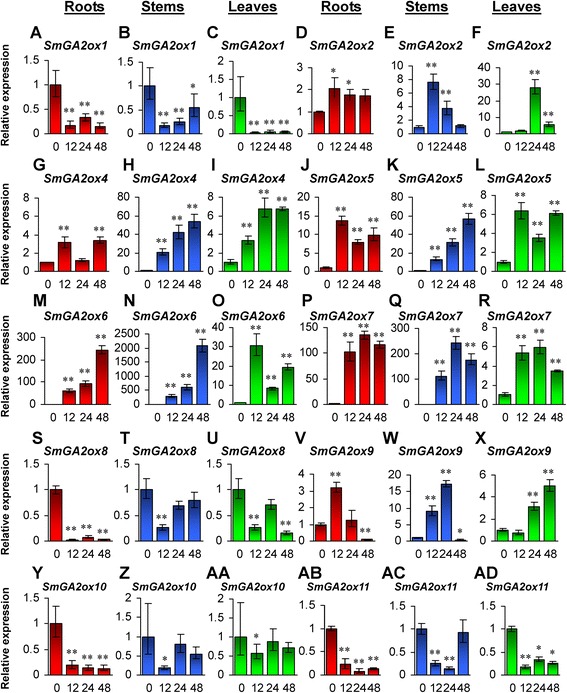
Responses of SmGA2ox genes to exogenous GA3 treatment. a–ad: Fold changes of SmGA2ox1 (a–c), SmGA2ox2 (d–f), SmGA2ox4 (g–i), SmGA20ox5 (j–l), SmGA20ox6 (m–o), SmGA20ox7 (p–r), SmGA2ox8 (s–u), SmGA2ox9 (v–x), SmGA2ox10 (y–aa) and SmGA2ox11 (ab–ad) in roots, stems and leaves of S. miltiorrhiza plantlets treated with 100 μM GA3 for 0, 12, 24 and 48 h. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in tissues without treatment (0 h) was arbitrarily set to 1 and the levels in tissues from GA3-treated plantlets were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 (*) and P < 0.01 (**) was considered statistically significant and extremely significant, respectively
Responses of SmCPSs and SmKSLs to exogenous GA3
CPS and KS are two enzymes catalyze the formation of ent-kaurene from GGPP in two steps via ent-copalyl diphosphate (CPP) in the first stage of GA biosynthesis. In our previous studies, five SmCPS genes in S. miltiorrhiza were identified [12]. Although the function of SmCPS genes has not been fully elucidated, SmCPS1, which was predominantly expressed in root cortices, is involved in tanshinone biosynthesis [12, 37]. SmCPS1 were significantly up-regulated in roots, stems and leaves of S. miltiorrhiza plants treated with GA3 (Fig. 10a–10c). It suggests that tanshinone biosynthesis-related genes may be responsive to exogenous GA3 treatment. Similarly, SmCPS3 and SmCPS5 were significantly up-regulated in GA3-treated plants (Fig. 10d–10i). The expression levels of SmCPS2 and SmCPS4 were too low to be detected in the tissues analyzed.
Fig. 10.
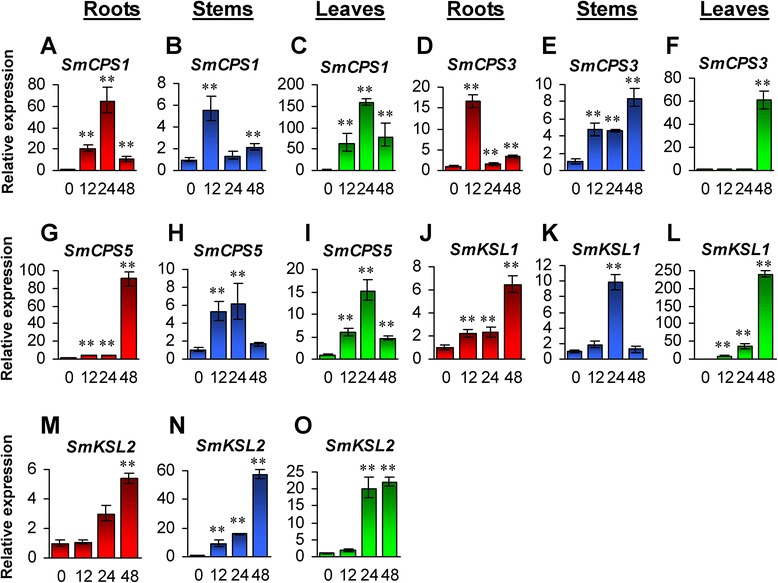
Responses of SmCPS and SmKSL genes to exogenous GA3 treatment. Fold changes of SmCPS1 (a–c), SmCPS3 (d–f), SmCPS5 (g–i), SmKSL1 (j–l) and SmKSL2 (m–o) in roots, stems and leaves of S. miltiorrhiza plantlets treated with 100 μM GA3 for 0, 12, 24 and 48 h are shown. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in tissues without treatment (0 h) was arbitrarily set to 1 and the levels in tissues from GA3-treated plantlets were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 (*) and P < 0.01 (**) was considered statistically significant and extremely significant, respectively
So far, two SmKSLs, known as SmKSL1 and SmKSL2, have been identified from S. miltiorrhiza [12, 48]. SmKSL1 showed the highest expression level in stems, followed by leaves, roots and flowers, while the expression levels of SmKSL2 were similar in all tissues [12, 48]. Previous studies have shown that SmKSL1 is involved in tanshinone biosynthesis [48]. Analyzing the responses of SmKSL1 and SmKSL2 to GA3 treatment showed that both SmKSL1 and SmKSL2 were significantly up-regulated in roots, stems and leaves of S. miltiorrhiza plants treated with exogenous GA3 (Fig. 10j–10o), confirming that tanshinone biosynthesis-related genes may be responsive to exogenous GA3 treatment.
Responses of GA metabolism pathway genes to yeast extract and Ag+ treatment
In addition to exogenous bioactive GAs, GA metabolism pathway genes may also response to environmental cues, such as light, temperature and various stresses [4]. Yeast extract and Ag+ are effective elicitors for the production of terpenoids. Many genes involved in the upstream of GA biosynthesis pathway were significantly up-regulated in S. miltiorrhiza after yeast extract and Ag+ treatment [48]. However, it was unknown whether the GA metabolism pathway genes were responsive to yeast extract and Ag+ treatment. In this study, transcriptome-wide analysis of GA metabolism pathway genes was carried out using RNA-seq data of S. miltiorrhiza hairy roots treated with or without yeast extract (100 μg/ml) and Ag+ (30 μM) [49]. RNA-seq reads from S. miltiorrhiza hairy roots non-treated (0 hpi) and treated for 12 (12 hpi), 24 (24 hpi) and 36 h (36 hpi) were mapped to the cloned ORFs of 22 GA metabolism pathway genes using SOAP2 [50]. A total of nine genes had the RPKM value >1.0 and was considered to be expressed in S. miltiorrhiza hairy roots. It includes SmKO, SmKAO1, SmKAO2, SmGA3ox1, SmGA20ox2, SmGA2ox5, SmGA2ox7, SmGA2ox8 and SmGA2ox11, of which SmGA2ox5 was significantly down-regulated at the time-point of 12 hpi, SmKO and SmGA20ox2 were significantly down-regulated at the time-points of 12 and 24 hpi, SmKAO2, SmGA2ox7, SmGA2ox8 and SmGA2ox11 were significantly down-regulated at all time-points of treatment, while SmGA3ox1 was up-regulated at all time-points of treatment (Fig. 11). SmKAO1 was down-regulated at the time-points of 12 and 24 hpi and up-regulated at the time-point of 36 hpi (Fig. 11). It suggests that the majority of GA metabolism pathway genes are yeast extract and Ag+-responsive.
Fig. 11.
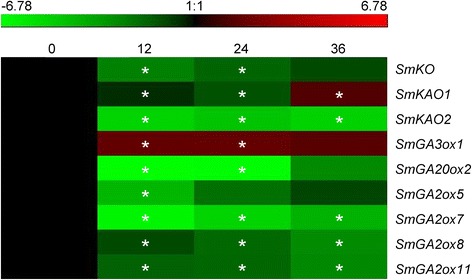
Responses of GA metabolism pathway genes to yeast extract and Ag+ treatment. S. miltiorrhiza hairy roots were treated for 0, 12, 24 and 36 h, respectively. RNA-seq reads were mapped to the cloned ORFs of GA metabolism pathway genes. Genes with RPKM value greater than 1 were analyzed for differential expression using Fisher’s exact test. P < 0.05 was considered as differentially expressed. *indicates significant differential expression compared with the level in hairy roots without treatment
Alternative splicing (AS) of GA metabolism pathway genes
Alternative splicing (AS) is a post-transcriptional mechanism of precursor-mRNA (pre-mRNA) [51, 52]. Through AS, multiple distinct mRNA isoforms are produced from a single gene locus. The process of AS plays significant roles in the diversity of transcriptome and proteome and the abundance of transcripts and proteins and is involved in many aspects of plant growth and development, such as photosynthesis, flowering, cereal grain quality, circadian clock, and response to biotic and abiotic stresses [51, 52]. In a previous study, two differently spliced AtKO (GA3) mRNAs had been cloned, sequenced and detected using Northern hybridization [45]. In order to examine whether genes associated with GA metabolism underwent AS events, we analyzed the assembled S. miltiorrhiza transcriptome [42] for the 22 cloned GA metabolism pathway genes. Sequence comparison among unigenes, the cloned cDNAs and the genomic DNA showed that SmKO, SmGA20ox3, SmGA2ox3 and SmGA2ox11 produced splice variants (Fig. 12a–12d). The number of unigenes corresponding to splice variants is two for SmKO, one for SmGA20ox3, one for SmGA2ox3, and three for SmGA2ox11, respectively (Fig. 12a–12d).
Fig. 12.
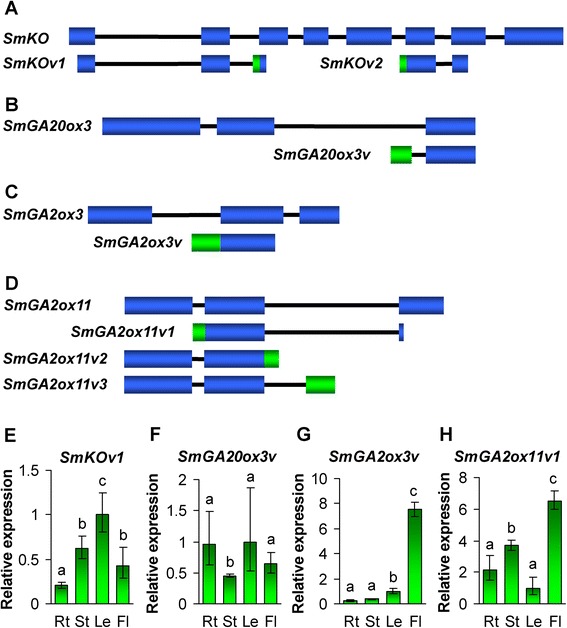
Alternative splicing and tissue-specific expression of various GA metabolism pathway genes in S. miltiorrhiza. a–d: Gene structures of SmKO (a), SmGA20ox3 (b), SmGA2ox3 (c), SmGA2ox11 (d) and their splice variants. Filled boxes represent exons. Lines represent introns. e–h: Fold changes of SmKOv1 (e), SmGA20ox3v (f), SmGA2ox3v (g) and SmGA2ox11v1 (h) in roots (Rt), stems (St), leaves (Le) and flowers (Fl) of S. miltiorrhiza plants. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in leaves was arbitrarily set to 1 and the levels in other tissues were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 was considered statistically significant
qRT-PCR analysis using splice variant-specific primers showed that specific amplicons could be obtained for SmKOv1, SmGA20ox3v, SmGA2ox3v and SmGA2ox11v1 (Fig. 12e–12h). SmKOv1 exhibited the highest expression in leaves, followed by stems and flowers, and the lowest in roots (Fig. 12e). SmGA20ox3v showed higher expression in roots, leaves and flowers than stems (Fig. 12f). SmGA2ox3v was predominantly expressed in flowers (Fig. 12g). SmGA2ox11v1 showed the highest expression in flowers, followed by stems, and the lowest in roots and leaves (Fig. 12h). The expression patterns of splice variants in roots, stems, leaves and flowers were similar to normal transcripts (Figs. 1b, 3d, 8c and 8k), although the overall expression levels of splice variants were significantly lower. SmKOv1, SmGA20ox3v and SmGA2ox11v1 showed differential response to exogenous GA3 treatment in roots, stems and leaves of S. miltiorrhiza (Fig. 13). SmKOv1 was significantly up-regulated at the time-point of 12-h-treatment, while down-regulated at the time-points of 24- and 48-h-treatment in S. miltiorrhiza roots (Fig. 13a). It was significantly down-regulated in stems at the time-points of 12- and 48-h-treatment and in roots at all three time-points of GA3 treatment (Fig. 13b and 13c). The responses of SmKOv1 to GA3 treatment were different from SmKO (Fig. 1c–1e). SmGA20ox3v showed significant down-regulation in roots at the time-points of 24- and 48-h-treatment and in stems at the time-points of 12- and 24-h-treatment, while no significant change was observed in roots at the time-point of 12-h-treatment, in stems at the time-point of 48-h-treatment, and in leaves at all three time-points of GA3 treatment (Fig. 13d–13f). SmGA2ox11v1 exhibited significant down-regulated in roots and leaves at all three time-points and in stems at the time-points of 12- and 24-h-treatment (Fig. 13g–13i). The responsive patterns of SmGA20ox3v and SmGA2ox11v1 were similar to SmGA20ox3 (Fig. 4g–4i) and SmGA2ox11 (Fig. 9ab–9ad), respectively. Taken together, these results suggest the importance of AS in regulating GA metabolism in S. miltiorrhiza.
Fig. 13.
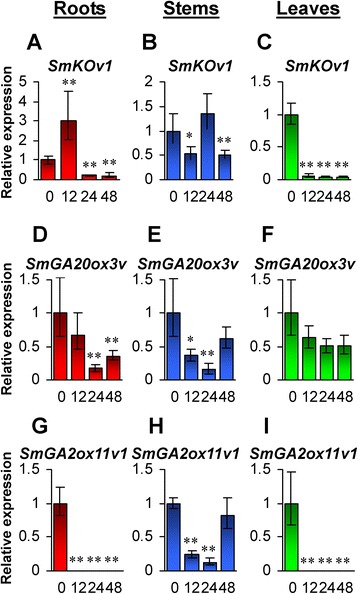
Responses of splice variants to exogenous GA3 treatment. a–i: Fold changes of SmKOv1 (a–c), SmGA20ox3v (d–f) and SmGA2ox11v1 (g–i) in roots, stems and leaves of S. miltiorrhiza plantlets treated with 100 μM GA3 for 0, 12, 24 and 48 h. The expression levels were analyzed using the quantitative RT-PCR method. Expression level in tissues without treatment (0 h) was arbitrarily set to 1 and the levels in tissues from GA3-treated plantlets were given relative to this. Error bars represent standard deviations of mean value from three biological and four technical replicates. ANOVA (analysis of variance) was calculated using SPSS. P < 0.05 (*) and P < 0.01 (**) was considered statistically significant and extremely significant, respectively
Discussion
Characterization of the GA metabolism pathway genes in S. miltiorrhiza
S. miltiorrhiza Bunge is one of the most important Traditional Chinese Medicine (TCM) materials. It is also an emerging model medicinal plant. Identification of GA metabolism pathway genes in S. miltiorrhiza may help to elucidate the biological function of these genes, the regulatory mechanism of GA metabolism, and the interplay between the GA metabolism pathway and other terpenoid metabolism pathways. Genome-wide prediction, molecular cloning and expression analysis of the 22 GA metabolism pathway genes encoding SmKO, SmKAO, SmGA20ox, SmGA3ox and SmGA2ox proteins in S. miltiorrhiza provide a foundation for further demonstrating the GA metabolism pathway and for commercial application. However, due to the incompleteness of the S. miltiorrhiza whole genome assembly, it could be not a complete set of GA metabolism pathway genes.
KO and KAO are two multifunctional cytochrome P450 monooxygenases catalyzing the conversion of ent-kaurene to GA12 in the second stage of GA metabolism pathway [2, 13, 14]. They are encoded by one or two genes in a plant species. The Arabidopsis genome contains an AtKO and two AtKAOs, while the rice genome has two OsKOs and an OsKAO. Similarly, from S. miltiorrhiza, we identified a SmKO and two SmKAOs. It is consistent with previous conclusion that enzymes involved in the early steps of the GA metabolism pathway are usually encoded by single or few genes [4]. Analysis of phylogenetic relationship showed that KO and KAO proteins found in a plant species, such as rice OsKO1 and OsKO2, Coffea arabica CaKO1 and CaKO2, Lactuca sativa LsKO1 and LsKO2, Arabidopsis AtKAO1 and AtKAO2, Pisum sativum PsKAO1 and PsKAO2 and S. miltiorrhiza SmKAO1 and SmKAO2, clustered together in the unrooted neighbor-joining trees (Figs. 1a and 2a). It indicates that KOs and KAOs from a plant species are usually paralogous originating from gene duplication.
Using ChloroP, AtKO had been predicted to contain a plastid transit peptide [17]. Transient expression studies of AtKO-green fluorescent protein (GFP) fusion in tobacco leaves showed that AtKO-GFP was targeted to the chloroplast [17]. In vitro protein import assays with isolated pea chloroplasts showed that AtKO was located on the outer surface of the chloroplast envelope [17]. However, it has been suggested that the subcellular localization of AtKO requires further confirmation, since the strong fluorescence from the chlorophylls may result in false positive results [14]. In this study, subcellular localization analysis of the deduced SmKO, SmKAO1 and SmKAO2 proteins using TargetP 1.1 [53] showed that each of them contained a secretory pathway signal peptide at the N-terminus (Table 1). It indicates that these proteins are most possibly located in the endoplasmic reticulum of S. miltiorrhiza cells. The secretory pathway signal peptides were also found at the N-terminal of KOs and KAOs from Arabidopsis and rice (Additional files 2 and 3: Tables S1 and S2). It implies that the conversion of ent-kaurene to GA12 is also located in the endoplasmic reticulum in Arabidopsis and rice. The results are consistent with those from Arabidopsis AtKAO1 and AtKAO2 analyses [17] and from early biochemistry studies showing that the conversion of ent-kaurenol, ent-7α-hydroxyk aurenoic acid and GA12 are located in the endoplasmic reticulum [54].
Bioactive gibberellins are converted from GA12 through the non-13-hydroxylation pathway and the early 13-hydroxylation pathway under the catalysis of GA20ox and GA3ox [4]. Recently, rice CYP714 members, CYP714B1 and CYP714B2, were reported to encode GA 13-oxidase (GA13ox) catalyzing the conversion of GA12 to GA53, which is the first step of the early GA 13-hydroxylation pathway [55]. In Arabidopsis, a member of the CYP714 member, termed CYP714A2, was found to convert GA12 to 12α-hydroxyl GA12 (GA111) as a major product and 13-hydroxy GA12 (GA53) as a minor product, while the other member, termed CYP714A1, catalyzed the conversion of GA12 to 16-carboxylated GA12 [56]. From S. miltiorrhiza, a total of nineteen 2-ODDs were identified. It includes six SmGA20oxs and two SmGA3oxs catalyzing the biosynthesis of bioactive GAs and eleven SmGA2oxs involved in the deactivation of bioactive GAs. However, no genomic DNA sequence was retrieved when BLAST analysis of Arabidopsis CYP714A1 and CYP714A2 and rice CYP714B1 and CYP714B2 against the current assembly of the S. miltiorrhiza genome was carried out using the tBLASTn algorithm [39, 40]. It could be due to the incompleteness of the S. miltiorrhiza whole genome assembly or low sequence homology among GA13ox proteins from S. miltiorrhiza, Arabidopsis and rice.
Among the nineteen S. miltiorrhiza 2-ODDs, sixteen were predicted to be located in the cytoplasm. Similarly, 14 of the 16 Arabidopsis 2-ODDs and 18 of the 21 rice 2-ODDs were predicted to be located in the cytoplasm. It suggests that the final stage of GA metabolism is conservatively occurred in the cytoplasm. Unexpectedly, various 2-ODDs from S. miltiorrhiza, Arabidopsis and rice, such as SmGA20ox4, SmGA2ox1, SmGA2ox5, AtGA20ox4, AtGA2ox7, OsGA2ox2, OsGA3ox1 and OsGA2ox11, contain chloroplast transit peptides at the N-terminal. Although the function of these plastid-located 2-ODDs is not clear, some of them may play limited and specific roles in plants. For instance, AtGA20ox5 catalyzes only the first two steps of the successive oxidation reaction sequence [20]. No significant phenotype changes were observed when AtGA20ox5 was disrupted in Arabidopsis plants [20]. Thus, cytoplasm- and plastid-located 2-ODDs probably play distinct physiological roles, which need to be investigated further.
Multiple layer regulation of GA metabolism in S. miltiorrhiza
As one of the five classical phytohormones, GA plays significant roles in many aspects of plant growth and development. Precise regulation of GA content appears to be critical in plant normal development and response to various environmental cues, such as light, temperature and other stresses [4, 33]. Multiple layer regulation may greatly help keeping GA content at the appropriate level and satisfying the needs of cells and organs at different developmental stages and under different environmental conditions. In this study, we showed that GA metabolism in S. miltiorrhiza was controlled by multiple mechanisms, such as alternative splicing, tissue-/organ-specific metabolism pathway, positive and negative feedback regulation, and yeast extract and Ag+-responsive gene expression.
Alternative splicing represents an important post-transcriptional mechanism in regulation of gene expression during plant growth and development [51, 52]. Transcriptome-wide analysis of splice variants showed the occurrence of AS in more than 40 % of the nine candidate GA metabolism pathway genes with assembled unigenes in S. miltiorrhiza. Splice variants showed tissue-specific expression patterns and were responsive to exogenous GA3 treatement. These results suggest the importance of AS in regulating GA metabolism in S. miltiorrhiza.
Gene expression profiling revealed that the identified GA metabolism pathway genes exhibit tissue-/organ-specific expression patterns. Additionally, exogenous GA3 treatment showed that individuals of GA metabolism pathway genes responded differentially and the responses depended on tissue types and organs. Differential expression of GA metabolism pathway genes suggests that each member of a gene family may play different physiological roles. Tissue-/organ-specific expression patterns and response to GA3 treatment provide evidence for the hypothesis that an additional layer of regulation may reside in the separation of the GA metabolism pathway into distinct cell types in tissues and organs requiring GA for development [19]. The results indicate the existence of tissue-/organ-specific metabolism pathway involving different enzyme isoforms.
It has been shown that the expression of some GA oxidase genes is under feedback control by bioactive GAs [4, 15, 18, 19, 34–36]. It includes down-regulation of some GA20ox and GA3ox genes (also known as negative feedback regulation) and up-regulation of some GA2ox genes (also known as positive forward regulation). For instance, GA3 treatment of Arabidopsis ga1-2 plants substantially reduced the abundance of AtGA20ox1–AtGA20ox3 transcripts in floral shoots [18]. Consistently, Arabidopsis AtGA20ox1 (GA5) and AtGA3ox1 (GA4) transcription was repressed in wild-type plants treated with exogenous GA3 [34, 36] Compared with wild-type plants, expression levels of three potato StGA20ox genes, named StGA20ox1–StGA20ox3, were very much increased in the ga1 dwarf mutant of potato, while treatment of potato ga1 plants with GA3 strongly reduced transcript abundance of all three StGA20ox genes [35]. In contrast to AtGA20ox and AtGA3ox genes, expression levels of Arabidopsis AtGA2ox1 and AtGA2ox2 were increased by application of GA3 [15]. Bioactive GA content reduction caused by GA20ox and GA3ox gene down-regulation and GA inactivation resulting from GA2ox gene up-regulation appear to be important for maintaining active GA concentrations within certain limits [15]. On the other hand, it has been shown that feedback regulation of GA levels does not occur in all organs [14] and the expression of some GA metabolism pathway genes, such as Arabidopsis AtKO1 [45] and AtGA3ox2 [57], wheat TaCPS, TaKS, TaKO and TaKAO [58], are not subject to feedback regulation by GAs. Down-regulation of SmGA20ox and SmGA3ox genes and up-regulation of SmGA2ox genes suggest the existence of feedback regulation mechanism of GA metabolism in S. miltiorrhiza. The underlying mechanism for up-regulation of SmKO, SmKAO1 and SmKAO2 (positive feedback regulation) and down-regulation of various SmGA2ox genes (negative forward regulation) in tissues treated with exogenous GA3 remain to be elucidated.
The responses of GA metabolism to environmental cues have been previously analyzed for light and temperature [4, 33]. In this study, we found that nine GA metabolism pathway genes were expressed in hairy roots of S. miltiorrhiza. Among them, seven, including SmKO, SmKAO2, SmGA20ox2, SmGA2ox5, SmGA2ox7, SmGA2ox8 and SmGA2ox11, were significantly down-regulated at different time-points of yeast extract and Ag+-treatment. SmGA2ox1 was significantly up-regulated at all time-points of treatment, while SmKAO1 was down-regulated at the time-points of 12 and 24 hpi and up-regulated at the time-point of 36 hpi. The identification of yeast extract and Ag+-responsive GA metabolism pathway genes provide additional evidence for the regulation of GA metabolism by stresses.
Crosstalk between GA metabolism and tanshinone biosynthesis in S. miltiorrhiza
Both GAs and tanshinones are diterpenoids sharing GGPP as the common precursor [12, 37]. Previously, SmCPS1 and SmKSL1 were identified to be key enzyme genes involved in tanshinone biosynthesis in S. miltiorrhiza [12, 37, 38]. In this study, both SmCPS1 and SmKSL1 were significantly up-regulated in S. miltiorrhiza plants treated with GA3. Significant change of SmCPS1 and SmKSL1 gene expression levels in GA3-treated S. miltiorrhiza plants suggests tanshinone biosynthesis-related genes were responsive to exogenous GA3 treatment. It indicates the existence of crosstalk between GA metabolism and tanshinone biosynthesis. The expression changes of these genes could be important for redirecting GGPP into other branch of the diterpenoid biosynthesis pathway.
Conclusions
Through systematical identification and characterization, including genome-wide identification, molecular cloning and expression analysis, we showed in this study for the first time a total of 22 candidate GA metabolism pathway genes. It represents five gene families, encoding a SmKO, two SmKAOs, six SmGA20oxs, two SmGA3oxs and eleven SmGA2oxs. Gene feature and phylogenetic analysis showed the conservation and divergence of GA metabolism pathway genes in S. miltiorrhiza. Many genes identified in this study were tissue-specifically expressed, feedback-regulated, stress-responsive and alternatively spliced. We found that nine of the twenty two GA metabolism pathway genes were responsive to yeast extract and Ag+-treatment in S. miltiorrhiza hairy roots and tissue-specifically expressed splice variants existed for SmKO, SmGA20ox3, SmGA2ox3 and SmGA2ox11. Among the splice variants identified, SmKOv1, SmGA20ox3v and SmGA2ox11v1 were GA3-responsive. Moreover, we showed that SmCPS1 and SmKSL1, two tanshinone biosynthesis-related genes, were up-regulated in response to GA3 treatment. Taken together, our results reveal multiple layer regulation of GA metabolism and crosstalk between GA metabolism and tanshinone biosynthesis in S. miltiorrhiza.
Methods
Plant materials and GA treatment
Salvia miltiorrhiza Bunge (line 993) plants were grown under natural growth conditions in a field nursery at the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China. Roots, stems, leaves and flowers were collected from two-year-old plants and stored immediately in liquid nitrogen until use. Plantlets used for GA treatment were prepared as described previously [12, 42]. Plantlets were sub-cultivated on 1/2 MS agar medium [59] for 6 weeks under a 16/8 h light/dark photoperiod at 25 °C and then transferred to 1/2 MS liquid medium for 2 days. GA3 stock solution in 1/2 MS was added to the medium to obtain a final concentration of 100 μM. Plantlets were treated for 12, 24 and 48 h, respectively. Plantlets without GA treatment were used as controls. Leaves, stems and roots from GA-treated plantlets and controls were collected at the same time and stored in liquid nitrogen until use. Three independent biological replicates were performed for each experiment.
Database search and gene prediction
The deduced amino acid sequences of 19 Arabidopsis and 24 rice KO, KAO, GA20ox, GA3ox and GA2ox proteins (Additional files 2 and 3: Tables S1 and S2) were obtained from the Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org) and the China Rice Data Center (http://www.ricedata.cn/gene), respectively. S. miltiorrhiza SmKO, SmKAO, SmGA20ox, SmGA3ox and SmGA2ox genes were predicted as described previously [12, 60, 61]. Briefly, BLAST analysis of Arabidopsis and rice KO, KAO, GA20ox, GA3ox and GA2ox amino acid sequences against the S. miltiorrhiza genome assembly, which consists of 611,208 contigs representing about 92 % of the entire genome and 96 % of the protein-coding genes [12, 39], were carried out using the tBLASTn algorithm. An e-value cut-off of 1e–5 was applied to the homologue recognition. All retrieved nucleotide sequences encoding proteins with more than 50 % identity to Arabidopsis or rice homologs were used for gene prediction on the Genscan web server (http://genes.mit.edu/GENSCAN.html) [41]. The predicted gene models were further examined and corrected manually through comparison with related genes in other plant species identified by BLAST analysis of the retrieved S. miltiorrhiza nucleotide sequences against the non-redundant protein sequence (nr) database (http://blast.ncbi.nlm.nih.gov/Blast.cgi) using the BLASTx algorithm with default parameters [40].
RNA extraction and cDNA cloning
Total RNA was extracted from S. miltiorrhiza tissues using the Trizol reagent (Sigma, USA) and the RNA Extract kit (Huayueyang, China). The integrity of total RNA was analyzed on a 1.2 % agarose gel. RNA quantity was determined using a NanoDrop 2000C spectrophotometer (Thermo Scientific, USA). Reverse transcription was performed on total RNA using the SMART RACE cDNA amplification kit (Clontech, USA). cDNA was PCR-amplified under the following conditions: predenaturation at 94 °C for 2 min, 30 cycles of amplification at 94 °C for 30 s, 56 °C for 30 s and 72 °C for 2 min, followed by a final extension at 72 °C for 15 min. Gene specific primers used for full-length cDNA amplifications are listed in Additional file 4: Table S3. Amplicons with expected size were gel-purified, cloned and sequenced.
Bioinformatic analysis and phylogenetic tree construction
The theoretical isoelectric point (pI) and molecular weight (MW) were predicted using the Compute pI/MW tool on the ExPASy server (http://web.expasy.org/compute_pi/) [62]. Conserved domains were analyzed by searching the deduced amino acid sequences against the NCBI Conserved Domain Database (CDD, http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) with the expected E-value threshold of 0.01 and the maximum size of hits to be 1000 amino acids [63]. The localizations of deduced proteins were predicted on the TargetP1.1 server (http://www.cbs.dtu.dk/services/TargetP/) [53]. Phylogenetic trees were constructed for full-length protein sequences using MEGA version 6.0 by the neighbor-joining method with 1,000 bootstrap replicates [64]. The Poisson correction parameter and pairwise deletion of gaps were applied as described previously [12]. For each analysis, only nodes supported by bootstrap values greater than 50 % are shown.
Quantitative real-time reverse transcription-PCR (qRT-PCR)
DNA contamination removal and reverse transcription was carried out on 1.0 μg total RNA using the RT-PCR kit (Clontech, USA) in a 20 μl volume. The resulting cDNA was diluted to 400 μl with sterile water. PCR was carried out in a 20 μl volume containing 1.0 μl diluted cDNA, 0.8 μl of 10.0 μM forward primer, 0.8 μl of 10.0 μM reverse primer and 10.0 μl 1 × SYBR Premix Ex Taq I (TaKaRa, Japan) using the Bio-Rad CFX96 system (Bio-Rad, USA) under the following conditions: predenaturation at 95 °C for 30 s, 40 cycles of amplification at 95 °C for 5 s, 60 °C for 18 s and 72 °C for 15 s. qPCR was performed in three biological and four technical replicates. Gene-specific primers listed in Additional file 5: Table S4 were designed using Primer-Premier 5.1 (Palo Alto, Canada). The length of amplicons is between 100 and 350 bp. SmUBQ10 was used as a reference as described previously [12]. The specificity of amplification was assessed by dissociation curve analysis. Results from gene-specific amplification were analyzed using the comparative Cq method which uses an arithmetic formula, 2-ΔΔCq, to achieve results for relative quantification [65]. Cq represents the threshold cycle. Standard derivation was calculated from three independent biological replicates and four technical replicates. Analysis of variance (ANOVA) was calculated using SPSS (Version 21.0, IBM, USA). P < 0.05 and P < 0.01 were considered as statistically significant and extremely significant, respectively.
Analysis of yeast extract and Ag+-responsive genes
RNA-seq data from S. miltiorrhiza hairy roots non-treated (0 hpi) and treated with yeast extract (100 μg/ml) and Ag+ (30 μM) for 12 (12 hpi), 24 (24 hpi) and 36 h (36 hpi) were downloaded from GenBank under the accession number SRR924662 [49]. RNA-seq reads were mapped to the cloned ORFs of GA metabolism pathway genes using SOAP2 [50]. The data were analyzed as described previously [42, 61]. Genes with the RPKM (RNA-seq reads mapped to an ORF per total million reads from a treatment per kilobases of the ORF length) value greater than 1 were analyzed for differential expression using Fisher’s exact test as described previously [61]. P < 0.05 was considered as differentially expressed.
Availability of supporting data
S. miltiorrhiza GA metabolisms pathway gene sequences supporting the results of this article are available in GenBank under the accession numbers KT853074–KT853095. The other supporting data sets are included within the article and its additional files.
Acknowledgements
We thank Prof. Shilin Chen and the S. miltiorrhiza genome sequencing group for kindly providing the S. miltiorrhiza genome sequence. We appreciate Prof. Xian’en Li for providing S. miltiorrhiza plants. This work was supported by grants from the Natural Science Foundation of China (grant no.31370327), the Beijing Natural Science Foundation (grant no. 5152021), the Major Scientific and Technological Special Project for Significant New Drugs Creation (grant no. 2012ZX09301002-001-031), and the Central Public-Interest Scientific Institution Basal Research Fund of Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences.
Abbreviations
- 2-ODD
2-oxoglutarate-dependent dioxygenase
- ANOVA
analysis of variance
- AS
alternative splicing
- CPP
ent-copalyl diphosphate
- CPS
ent-copalyl diphosphate synthase
- GA
gibberellin
- GA20ox
GA 20-oxidase
- GA2ox
GA 2-oxidase
- GA3ox
GA 3-oxidase
- GFP
green fluorescent protein
- GGPP
trans-geranylgeranyl diphosphate
- KAO
ent-kaurenoic acid oxidase
- KO
ent-kaurene oxidase
- KS
ent-kaurene synthase
- MEP
2-C-methyl-D-erythritol 4-phosphate
- MVA
mevalonate
- MW
molecular weight
- ORF
open reading frame
- P450
cytochrome P450 monooxygenase
- pI
isoelectric point
- qRT-PCR
quantitative real-time reverse transcription-PCR
- TCM
Traditional Chinese Medicine
Additional files
Conserved domains of enzymes involved in gibberellin metabolism in S. miltiorrhiza. Conserved domains of enzymes involved in gibberellin metabolism in S. miltiorrhiza are shown. (DOC 183 kb)
Sequence features of gibberellin metabolism genes in A. thaliana. The accession numbers, cDNA length, number of amino acid residues, molecular weight, pI and predicted protein localization are shown. (DOC 59 kb)
Sequence features of gibberellin metabolism genes in O. sativa. The accession numbers, cDNA length, number of amino acid residues, molecular weight, pI and predicted protein localization are shown. (DOC 73 kb)
Primers used for full-length coding region cloning. Complete set of primers used for amplication of full-length coding region. (DOC 62 kb)
Primers used for qRT-PCR. Complete set of primers used for qRT-PCR. (DOC 87 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
QD carried out molecular cloning, bioinformatics analysis, qRT-PCR, and participated in drafting the manuscript. CL analyzed the RNA-seq data. DL performed S. miltiorrhiza database search. SL conceived of the study, guided the experiment, participated in its design and coordination, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Qing Du, Email: bjdfydq@163.com.
Caili Li, Email: licaili390@163.com.
Dongqiao Li, Email: butterfly_qiao@163.com.
Shanfa Lu, Email: sflu@implad.ac.cn.
References
- 1.Kende H, Zeevaart JAD. The five “classical” plant hormones. Plant Cell. 1997;9:1197–210. doi: 10.1105/tpc.9.7.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hedden P, Phillips AL. Gibberellin metabolism: new insights revealed by the genes. Trends Plant Sci. 2000;5:523–30. doi: 10.1016/S1360-1385(00)01790-8. [DOI] [PubMed] [Google Scholar]
- 3.MacMillan J. Occurrence of gibberellins in vascular plants, fungi, and bacteria. J Plant Growth Regul. 2002;20:387–442. doi: 10.1007/s003440010038. [DOI] [PubMed] [Google Scholar]
- 4.Hedden P. eLS. Chichester: John Wiley & Sons, Ltd; 2012. Gibberellin biosynthesis. [Google Scholar]
- 5.Tyler L, Thomas SG, Hu JH, Dill A, Alonso JM, Ecker JR, et al. Della proteins and gibberellin-regulated seed germination and floral development in Arabidopsis. Plant Physiol. 2004;135:1008–19. doi: 10.1104/pp.104.039578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayele BT, Ozga JA, Reinecke DM. Regulation of GA biosynthesis genes during germination and young seedling growth of pea (Pisum sativum L.) J Plant Growth Regul. 2006;25:219–32. doi: 10.1007/s00344-006-0007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Junttila O, Jensen E, Pearce DW, Pharis RP. Stimulation of shoot elongation in Salix pentandra by gibberellin GA3; activity appears to be dependent upon hydroxylation to GA1 via GA3. Plant Physiol. 1992;84:113–20. doi: 10.1111/j.1399-3054.1992.tb08773.x. [DOI] [Google Scholar]
- 8.Hedden P, Proebsting WM. Genetic analysis of gibberellin biosynthesis. Plant Physiol. 1999;119:365–70. doi: 10.1104/pp.119.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blazquez MA, Green R, Nilsson O, Sussman MR, Weigel D. Gibberellins promote flowering of Arabidopsis by activating the LEAFY promoter. Plant Cell. 1998;10:791–800. doi: 10.1105/tpc.10.5.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santes CM, Hedden P, Gaskin P, Garcia-Martinez JL. Gibberellins and related compounds in young fruits of pea and their relationship to fruit-set. Phytochemistry. 1995;40:1347–55. doi: 10.1016/0031-9422(95)00489-T. [DOI] [Google Scholar]
- 11.Serrani JC, Sanjuán R, Ruiz-Rivero O, Fos M, García-Martínez JL. Gibberellin regulation of fruit set and growth in tomato. Plant Physiol. 2007;145:246–57. doi: 10.1104/pp.107.098335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma Y, Yuan L, Wu B, Li X, Chen S, Lu S. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. J Exp Bot. 2012;63:2809–23. doi: 10.1093/jxb/err466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hedden P, Kamiya Y. Gibberellin biosynthesis: enzymes, genes and their regulation. Ann Rev Plant Physiol Plant Mol Biol. 1997;48:431–60. doi: 10.1146/annurev.arplant.48.1.431. [DOI] [PubMed] [Google Scholar]
- 14.Olszewski N, Sun TP, Gubler F. Gibberellin signaling: biosynthesis, catabolism, and response pathways. Plant Cell. 2002;14:S61–80. doi: 10.1105/tpc.010476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas SG, Phillips AL, Hedden P. Molecular cloning and functional expression of gibberellin 2-oxidases, multifunctional enzymes involved in gibberellin deactivation. Proc Natl Acad Sci USA. 1999;96:4698–703. doi: 10.1073/pnas.96.8.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helliwell CA, Chandler PM, Poole A, Dennis ES, Peacock WJ. The CYP88A cytochrome P450, ent-kaurenoic acid oxidase, catalyzes three steps of the gibberellin biosynthesis pathway. Proc Natl Acad Sci USA. 2001;98:2065–70. doi: 10.1073/pnas.98.4.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helliwell CA, Sullivan JA, Mould RM, Gray JC, Peacock WJ, Dennis ES. A plastid envelope location of Arabidopsis ent-kaurene oxidase links the plastid and endoplasmic reticulum steps of the gibberellin biosynthesis pathway. Plant J. 2001;28:201–8. doi: 10.1046/j.1365-313X.2001.01150.x. [DOI] [PubMed] [Google Scholar]
- 18.Phillips AL, Ward DA, Uknes S, Appleford NE, Lange T, Huttly AK, et al. Isolation and expression of three gibberellin 20-oxidase cDNA clones from Arabidopsis. Plant Physiol. 1995;108:1049–57. doi: 10.1104/pp.108.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchum MG, Yamaguchi S, Hanada A, Kuwahara A, Yoshioka Y, Kato T, et al. Distinct and overlapping roles of two gibberellin 3-oxidases in Arabidopsis development. Plant J. 2006;45:804–18. doi: 10.1111/j.1365-313X.2005.02642.x. [DOI] [PubMed] [Google Scholar]
- 20.Plackett AR, Powers SJ, Fernandez-Garcia N, Urbanova T, Takebayashi Y, Seo M, et al. Analysis of the developmental roles of the Arabidopsis gibberellin 20-oxidases demonstrates that GA20ox1, -2, and -3 are the dominant paralogs. Plant Cell. 2012;24:941–60. doi: 10.1105/tpc.111.095109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu J, Mitchum MG, Barnaby N, Ayele BT, Ogawa M, Nam E, et al. Potential sites of bioactive gibberellin production during reproductive growth in Arabidopsis. Plant Cell. 2008;20:320–36. doi: 10.1105/tpc.107.057752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee DJ, Zeevaart JA. Molecular cloning of GA2-oxidase 3 from spinach and its ectopic expression in Nicotiana sylvestris. Plant Physiol. 2005;138:243–54. doi: 10.1104/pp.104.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo SF, Yang SY, Chen KT, Hsing YI, Zeevaart JA, Chen LJ, et al. A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. Plant Cell. 2008;20:2603–18. doi: 10.1105/tpc.108.060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lange T, Kappler J, Fischer A, Frisse A, Padeffke T, Schmidtke S, et al. Gibberellin biosynthesis in developing pumpkin seedlings. Plant Physiol. 2005;139:213–23. doi: 10.1104/pp.105.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakamoto T, Miura K, Itoh H, Tatsumi T, Ueguchi-Tanaka M, Ishiyama K, et al. An overview of gibberellin metabolism enzyme genes and their related mutants in rice. Plant Physiol. 2004;134:1642–53. doi: 10.1104/pp.103.033696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davidson SE, Swain SM, Reid JB. Regulation of the early GA biosynthesis pathway in pea. Planta. 2005;222:1010–19. doi: 10.1007/s00425-005-0045-7. [DOI] [PubMed] [Google Scholar]
- 27.Stavang JA, Lindgård B, Erntsen A, Lid SE, Moe R, Olsen JE. Thermoperiodic stem elongation involves transcriptional regulation of gibberellin deactivation in pea. Plant Physiol. 2005;138:2344–53. doi: 10.1104/pp.105.063149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song J, Guo B, Song F, Peng H, Yao Y, Zhang Y, et al. Genome-wide identification of gibberellins metabolic enzyme genes and expression profiling analysis during seed germination in maize. Gene. 2011;482:34–42. doi: 10.1016/j.gene.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Nelson DR, Schuler MA, Paquette SM, Werck-Reichhart D, Bak S. Comparative genomics of rice and Arabidopsis. Analysis of 727 cytochrome P450 genes and pseudogenes from a monocot and a dicot. Plant Physiol. 2004;135:756–72. doi: 10.1104/pp.104.039826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rieu I, Ruiz-Rivero O, Fernandez-Garcia N, Griffiths J, Powers SJ, Gong F, et al. The gibberellin biosynthetic genes AtGA20ox1 and AtGA20ox2 act, partially redundantly, to promote growth and development throughout the Arabidopsis life cycle. Plant J. 2008;53:488–504. doi: 10.1111/j.1365-313X.2007.03356.x. [DOI] [PubMed] [Google Scholar]
- 31.Han F, Zhu B. Evolutionary analysis of three gibberellin oxidase genes in rice, Arabidopsis, and soybean. Gene. 2011;473:23–35. doi: 10.1016/j.gene.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Plackett ARG, Thomas SG, Wilson ZA, Hedden P. Gibberellin control of stamen development: a fertile field. Trends Plant Sci. 2011;16:568–78. doi: 10.1016/j.tplants.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Kamiya Y, García-Martínez JL. Regulation of gibberellin biosynthesis by light. Curr Opin Plant Biol. 1999;2:398–403. doi: 10.1016/S1369-5266(99)00012-6. [DOI] [PubMed] [Google Scholar]
- 34.Chiang HH, Hwang I, Goodman HM. Isolation of the Arabidopsis GA4 locus. Plant Cell. 1995;7:195–201. doi: 10.1105/tpc.7.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrera E, Jackson SD, Prat S. Feedback control and diurnal regulation of gibberellin 20-oxidase transcript levels in potato. Plant Physiol. 1999;119:765–74. doi: 10.1104/pp.119.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu YL, Li L, Gage DA, Zeevaart JA. Feedback regulation of GA5 expression and metabolic engineering of gibberellin levels in Arabidopsis. Plant Cell. 1999;11:927–36. doi: 10.1105/tpc.11.5.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao W, Hillwig ML, Huang L, Cui G, Wang X, Kong J, et al. A functional genomics approach to tanshinone biosynthesis provides stereochemical insights. Org Lett. 2009;11:5170–73. doi: 10.1021/ol902051v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo J, Zhou YJ, Hillwig ML, Shen Y, Yang L, Wang Y, et al. CYP76AH1 catalyzes turnover of miltiradiene in tanshinones biosynthesis and enables heterologous production of ferruginol in yeasts. Proc Natl Acad Sci U S A. 2013;110:12108–13. doi: 10.1073/pnas.1218061110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song JY, Luo HM, Li CF, Sun C, Xu J, Chen SL. Salvia miltiorrhiza as medicinal model plant. Yao Xue Xue Bao. 2013;48:1099–106. [PubMed] [Google Scholar]
- 40.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burge CB, Karlin S. Finding the genes in genomic DNA. Curr Opin Struct Biol. 1998;8:346–54. doi: 10.1016/S0959-440X(98)80069-9. [DOI] [PubMed] [Google Scholar]
- 42.Li D, Shao F, Lu S. Identification and characterization of mRNA-like noncoding RNAs in Salvia miltiorrhiza. Planta. 2015;241:1131–43. doi: 10.1007/s00425-015-2246-z. [DOI] [PubMed] [Google Scholar]
- 43.Chen H, Wu B, Nelson DR, Wu K, Liu C. Computational identification and systematic classification of novel cytochrome P450 genes in Salvia miltiorrhiza. PLoS One. 2014;9:e115149. doi: 10.1371/journal.pone.0115149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu YT, Gao W, Liu YJ, Cheng QQ, Su P, Liu YZ, et al. Cloning and bioinformatics analysis of ent-kaurene oxidase synthase gene in Salvia miltiorrhiza. Zhongguo Zhong Yao Za Zhi. 2014;39:4174–9. [PubMed] [Google Scholar]
- 45.Helliwell CA, Sheldon CC, Olive MR, Walker AR, Zeevaart JA, Peacock WJ, et al. Cloning of the Arabidopsis ent-kaurene oxidase gene GA3. Proc Natl Acad Sci USA. 1998;95:9019–24. doi: 10.1073/pnas.95.15.9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang SS, Raman AS, Ream JE, Fujiwara H, Cerny RE, Brown SM. Overexpression of 20-oxidase confers a gibberellin overproduction phenotype in Arabidopsis. Plant Physiol. 1998;118:773–81. doi: 10.1104/pp.118.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coles JP, Phillips AL, Croker SJ, García-Lepe R, Lewis MJ, Hedden P. Modification of gibberellin production and plant development in Arabidopsis by sense and antisense expression of gibberellin 20-oxidase genes. Plant J. 1999;17:547–56. doi: 10.1046/j.1365-313X.1999.00410.x. [DOI] [PubMed] [Google Scholar]
- 48.Cui G, Huang L, Tang X, Zhao J. Candidate genes involved in tanshinone biosynthesis in hairy roots of Salvia miltiorrhiza revealed by cDNA microarray. Mol Biol Rep. 2011;38:2471–8. doi: 10.1007/s11033-010-0383-9. [DOI] [PubMed] [Google Scholar]
- 49.Gao W, Sun HX, Xiao H, Cui G, Hillwig ML, Jackson A, et al. Combining metabolomics and transcriptomics to characterize tanshinone biosynthesis in Salvia miltiorrhiza. BMC Genomics. 2014;15:73. doi: 10.1186/1471-2164-15-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–7. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 51.Barbazuk WB, Fu Y, McGinnis KM. Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Res. 2008;18:1381–92. doi: 10.1101/gr.053678.106. [DOI] [PubMed] [Google Scholar]
- 52.Filichkin S, Priest HD, Megraw M, Mockler TC. Alternative splicing in plants: directing traffic at the crossroads of adaptation and environmental stress. Curr Opin Plant Biol. 2015;24:125–35. doi: 10.1016/j.pbi.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 53.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–71. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 54.Graebe JE. Gibberellin biosynthesis and control. Ann Rev Plant Physiol. 1987;38:419–65. doi: 10.1146/annurev.pp.38.060187.002223. [DOI] [Google Scholar]
- 55.Magome H, Nomura T, Hanada A, Takeda-Kamiya N, Ohnishi T, Shinma Y, et al. CYP714B1 and CYP714B2 encode gibberellin 13-oxidases that reduce gibberellin activity in rice. Proc Natl Acad Sci USA. 2013;110:1947–52. doi: 10.1073/pnas.1215788110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nomura T, Magome H, Hanada A, Takeda-Kamiya N, Mander LN, Kamiya Y, et al. Functional analysis of Arabidopsis CYP714A1 and CYP714A2 reveals that they are distinct gibberellin modification enzymes. Plant Cell Physiol. 2013;54:1837–51. doi: 10.1093/pcp/pct125. [DOI] [PubMed] [Google Scholar]
- 57.Yamaguchi S, Smith MW, Brown RGS, Kamiya Y, Sun TP. Phytochrome regulation and differential expression of gibberellin 3ß-hydroxylase genes in germinating Arabidopsis seeds. Plant Cell. 1998;10:2115–26. doi: 10.1105/tpc.10.12.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang Y, Yang W, Pei Z, Guo X, Liu D, Sun J, et al. The genes for gibberellin biosynthesis in wheat. Funct Integr Genomics. 2012;12:199–206. doi: 10.1007/s10142-011-0243-2. [DOI] [PubMed] [Google Scholar]
- 59.Murashige T, Skoog I. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Plant Physiol. 1962;15:473–7. doi: 10.1111/j.1399-3054.1962.tb08052.x. [DOI] [Google Scholar]
- 60.Li C, Lu S. Genome-wide characterization and comparative analysis of R2R3-MYB transcription factors shows the complexity of MYB-associated regulatory networks in Salvia miltiorrhiza. BMC Genomics. 2014;15:277. doi: 10.1186/1471-2164-15-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li C, Li D, Shao F, Lu S. Molecular cloning and expression analysis of WRKY transcription factor genes in Salvia miltiorrhiza. BMC Genomics. 2015;16:200. doi: 10.1186/s12864-015-1411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bjellqvist B, Basse B, Olsen E, Celis JE. Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions. Electrophoresis. 1994;15:529–39. doi: 10.1002/elps.1150150171. [DOI] [PubMed] [Google Scholar]
- 63.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43:D222–6. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]