

Abstract
Activin A, a member of the TGF-β superfamily of cytokines, was originally identified as an inducer of follicle stimulating hormone release, but has since been ascribed roles in normal physiological processes, as an immunoregulatory cytokine and as a driver of fibrosis. In the last 10–15 years, it has also become abundantly clear that activin A plays an important role in the regulation of asthmatic inflammation and airway remodelling. This review provides a brief introduction to the activin A/TGF-β superfamily, focussing on the regulation of receptors and signalling pathways. We examine the contradictory evidence for generalized pro- vs. anti-inflammatory effects of activin A in inflammation, before appraising its role in asthmatic inflammation and airway remodelling specifically by evaluating data from both murine models and clinical studies. We identify key issues to be addressed, paving the way for safe exploitation of modulation of activin A function for treatment of allergic asthma and other inflammatory lung diseases.
Introduction
Asthma is a chronic inflammatory lung disease characterized by variable airflow obstruction resulting in wheeze, chest tightness and shortness of breath. One of the main features of asthma is constriction of the airway smooth muscle [bronchoconstriction or airway hyperresponsivenesss (AHR)] that can be induced or exacerbated by exposure to a wide range of environmental triggers including cold air, allergens, infections (e.g. viruses), airborne particulates and pollutants and certain drugs [1,2]. The disease occurs in genetically susceptible individuals and commonly begins in childhood. The increased prevalence and severity of allergic asthma in developed countries over the last few decades has led to the concept of an asthma ‘epidemic’ [3]. Asthma can be categorized as allergic or non-allergic, the former being experienced by approximately 80% of asthma sufferers and the latter commonly developing later in life [4]. Allergic asthma can be further divided into ‘Th2 high’ and ‘Th2 low’ subtypes, the Th2 high subtype distinguished by increased IL-13 production and induction of related genes [2]. A subset of asthmatics with severe asthma has been identified, characterized by glucocorticoid non-responsiveness, predominantly neutrophilic inflammation and increased Th17 cells and production of the associated cytokine IL-17 [5]. Asthma is characterized by tissue structural changes including sub-basement membrane collagen deposition, angiogenesis, goblet cell metaplasia and increased smooth muscle mass, collectively termed airway remodelling [2,6]. The relationship between airway remodelling and AHR remains contentious, although remodelling is generally thought to lead to increased AHR and decreased lung function. Airway remodelling is present in young children at or before the onset of clinical asthma symptoms [7] and may precede or occur in parallel with inflammation [6].
Evidence suggests that asthmatic airway epithelium is structurally and functionally abnormal, with increased susceptibility to injury and defective repair responses [2]. The airway epithelium can secrete a multitude of cytokines, chemokines and growth factors which regulate and drive inflammation and remodelling, including IL-1α, IL-25, IL-33, GM-CSF and TGF-β [1,8]. TGF-β is strongly implicated in asthma pathogenesis via its ability to inhibit AHR and promote airway remodelling [9–17]. Activin A has structural and functional homology to TGF-β. Studies by our group have shown altered compartmentalization of activin A in asthmatic airway epithelium and bronchoalveolar lavage (BAL) in the mouse [18,19]. Nevertheless, there have been divergent findings regarding the pro- vs. anti-inflammatory effects of activin A, both generally and in allergic asthma specifically. This review critically appraises the evidence for these apparently contradictory findings and discusses the growing body of data implicating activin A as an important regulator of asthmatic inflammation and airway remodelling. Comparison between activin A and TGF-β will be made throughout this review, given their many overlapping roles in physiological and pathological processes.
The TGF-β superfamily
The TGF-β superfamily is divided into the TGF-β/activin/inhibin/nodal subfamily and the BMP (bone morphogenic protein)/GDF (growth and differentiation factor)/MIS (Mullerian inhibiting substance) subfamily, grouped by similarities in sequence and signalling pathways. While the BMP subfamily signals by binding initially to transmembrane serine threonine kinase type I (BMP) receptors, the TGF-β and activin A family ligands signal by binding to type II receptors, but do not bind directly to type I receptors [20]. TGF-β isoforms (TGF-β1, TGF-β2 and TGF-β3), myostatin (GDF8, a regulator of skeletal muscle mass) and GDF11 are secreted as latent complexes (ligand, prodomain and latent binding protein) which are sequestered to the extracellular matrix [21]. In contrast, the other members of the TGF-β superfamily are secreted in an ‘active’ state, although there is evidence that they are still associated with their prodomain and that this interaction facilitates binding to the extracellular matrix [21]. Activin signalling is regulated by various extracellular binding proteins, discussed further below.
Activins
Activins are produced by a wide variety of immune and non-immune cell types at nearly all stages of development including in adult tissue. Activins have a wide range of physiological actions and roles during development and have both overlapping and unique functions. Activin proteins exist as either homo- or hetero-dimers of β-subunits linked by disulphide bonds. Activin βA, βB, βC, βD and βE subunits have been identified [22], although the βD subunit has only been found in Xenopus. βA and βB transcripts are found in most tissues, while expression of βC and βE subunits is largely restricted to the liver [23]. Homodimers of βA and βB subunits (activin A and activin B, respectively) are the most widely studied of the activins. Activin A is highly conserved across species, with 100% conservation of the amino acid sequence between mouse and man [24]. Activin βA knockout mice are neonatal lethal, demonstrating the importance of activin A during development, whereas activin βB knockouts have developmental defects but are viable [25,26]. Activin A localizes to heparan sulphated proteoglycans in the extracellular matrix via its prodomain, and this interaction is important for its localization within tissues [27].
Activin receptors and signalling
Activins signal by binding to one of two type II receptors on the cell surface (ActRIIA or ActRIIB) which leads to exposure of the type I receptor binding site on activin. Subsequently, the type I receptors ActRIB (ALK4) or ActRIC (ALK7) (for activin A and activin B, respectively) are recruited to the ligand/ActRII complex and phosphorylated and activated by the type II receptor kinase [20,23]. The activated type I receptor phosphorylates and activates the receptor-regulated Smad (R-Smad) proteins Smad2 and Smad3 which recruit the co-mediator Smad4, and this complex translocates into the nucleus to activate gene transcription [23,28]. Although TGF-β binds to distinct type I and type II receptors (TβR-II and TβR-I/ALK-5), it signals via a Smad2/3 and Smad4 pathway shared with activin A, suggesting overlap in biological function. In addition to the classical Smad pathway, activin and TGF-β can also signal via alternative pathways involving the MAP kinases (ERK1/2, JNK and p38 MAPK) [23,24]. These Smad-independent signalling pathways highlight the complexity of the activin/TGF-β signalling cascade.
Regulation of activin receptors
TGF-β superfamily receptor activation is tightly regulated by two classes of molecules. One group of molecules act as ligand binding traps to sequester the ligand, examples of which include α2 macroglobulin, which binds TGF-β, and follistatin, which binds activin A. The other group of molecules are membrane-bound co-receptors which promote ligand binding to the receptors. An example is betaglycan, which does not bind activin but binds to inhibin A and facilitates inhibin access to activin receptors, essentially outcompeting activin binding [20]. Other co-receptors include BAMBI (BMP and activin receptor membrane-bound inhibitor), a pseudo-receptor with sequence similarity to type I receptors that cannot induce protein phosphorylation and inhibits BMP, activin and TGF-β signalling [29], and Cripto, which binds to the activin-ActRII complex and inhibits the recruitment of the type I receptor thereby preventing activin signalling [30].
Regulation of activin signalling via Smads
The signalling activity of activins is also modified by regulatory proteins. The Smad anchor for receptor activation protein (SARA) recruits Smad2 to the TGF-β receptors for phosphorylation [31,32]. A recently identified protein ERBIN interacts with SARA, Smad2 and Smad3 and competes for binding with SARA and Smad2/Smad3, differentially regulating TGF-β/activin A signalling [33]. Additional control is exerted by activin receptor interacting proteins (ARIPS) which regulate localization of activin receptors and positively or negatively regulate activin signalling [23]. The inherent DNA-binding activity of Smads is further regulated by various transcriptional activators or repressors which regulate gene transcription [28]. Following activation, TGF-β receptors are subject to negative regulation by the inhibitory Smads (I-Smad) Smad6 and Smad7, which act by competing for binding to the receptor with the activating Smads (e.g. Smad2, Smad3), and promoting receptor ubiquitination and degradation of type I receptors [34]. I-Smads can also inhibit signalling by interaction with various transcriptional repressors or by disrupting formation of functional Smad-DNA complexes [34]. In addition to inhibiting formation of functional type I and type II receptor complexes, as described above, BAMBI cooperates with Smad7 to inhibit TGF-β signalling [35]. Overall, these numerous regulatory mechanisms operating at multiple levels offer fine control of signalling via the TGF-β/activin signalling pathway necessary to maintain good health.
Regulation of activin signalling by extracellular binding factors
Follistatin
Follistatin is a high-affinity activin-binding protein which neutralizes the bioactivity of activin and several other members of the TGF-β superfamily. Two follistatin molecules surround activin, blocking both the type I and type II receptor binding sites, thereby preventing receptor binding and the initiation of the signalling cascade [36,37]. Follistatin also binds with lower affinity to other TGF-β superfamily members including growth and differentiation factor 9 (GDF9), myostatin and BMP 2, 5, 7 and 8 [24]. Follistatin does not bind TGF-β1 or TGF-β2, but binds TGF-β3 and antagonizes its function [38]. Follistatin is highly conserved between all species studied and 97% conserved between humans and mice [39]. The follistatin gene is approximately 6 kb and made up of 6 exons, producing 2 major species through alternative splicing [39], being follistatin 288 and follistatin 315 (FS288 and FS315, respectively). FS288 exists as a COOH-terminal truncated isoform and binds heparin sulphate proteoglycans in the extracellular matrix and is therefore cell surface associated [40]. In contrast, the FS315 isoform is secreted but does not bind heparin [40] and binds activin A with approximately tenfold lower affinity than FS288 [41]. The high affinity interaction between activin and FS288 suggests that the ECM matrix could be a major store of activin A. Indeed, heparin injected during surgical procedures causes the release of activin A and follistatin into the circulation [42]. The binding of activin to FS288 in pituitary cells leads to internalization and degradation, providing an additional mechanism to control activin availability [43].
Follistatin-related gene
Follistatin-related gene (FLRG), also known as follistatin-related protein or follistatin-like 3, is a recently identified member of the follistatin family [23]. FLRG binds to activin with lower affinity than follistatin. Like follistatin, FLRG also binds myostatin and BMP2 but with lower affinity than for activin [44,45]. FLRG only has two follistatin domains instead of the three present in follistatin, and it also lacks a heparin binding domain [44]. This lack of ability to bind heparin sulphated proteoglycans in the ECM may be the reason why FLRG is less effective than follistatin at inhibiting activin A bioactivity in several cell culture systems [24].
Inhibin
The inhibins are structural homologues of activin A, existing as heterodimers constituting an inhibin α-subunit dimerized with an activin βA or βB subunit, forming inhibin A and inhibin B, respectively (α/βA or α/βB, respectively) [46]. Inhibin acts as a negative regulator of activin activity by binding the activin type II receptor (ActRII), sharing the same binding site as activin A, although the affinity of this interaction is quite low. However, in the presence of the inhibin co-receptor betaglycan, inhibin forms a high affinity interaction with the activin type II receptor, inhibiting the ability of activin to bind and blocking activin-induced signalling [46].
Activin A in inflammation
Activin A plays a role in development of the immune system and is produced by and/or modulates the function of numerous immune cells including B cells, T cells, macrophages, mast cells, neutrophils and dendritic cells (DC), and this topic has been comprehensively reviewed elsewhere [24,47]. Activin A is implicated in the regulation of inflammation in a wide variety of diseases including skin injury, inflammatory bowel disease, arteriosclerosis, inflammatory arthropathies and brain injury [24,47]. However, a major unresolved issue is whether activin A promotes or inhibits inflammation [24,48].
Anti-inflammatory effects of activin A
There is considerable evidence for an anti-inflammatory effect of activin A. In vitro activin A inhibits proliferation of thymocytes and peripheral blood lymphocytes [49,50]. Activin A also inhibits B cell generation and survival by antagonizing IL-6-induced survival signals [51] and inhibits IL-6-induced proliferation of B cell hybridomas [52]. Inhibition of the activin A signalling pathway with follistatin enhances CD40L-mediated production of numerous inflammatory mediators by activated monocyte-derived DC (IL-6, IL-8, IL-12p70, CCL2 (MCP-1), CCL5 (RANTES), CXCL10), consistent with an anti-inflammatory action of activin A [53]. In vitro activin A inhibits DC maturation and the T cell stimulatory capacity of DC [54] and increases endocytic activity of lipopolysaccharide (LPS)-matured monocyte-derived DC, implying maintenance of an immature phenotype [55]. Foxp3+ regulatory T cells (Treg) play a central role in maintaining immune tolerance in the steady state and inhibiting asthma exacerbations [56–58]. Activin A promotes TGF-β-dependent development of Foxp3+ Treg in vitro and induces conversion of naïve CD4+ T cells into Foxp3+ iTreg in vivo [59]. Activin A also promotes development of IL-10-producing Foxp3− Treg in vitro [60]. Furthermore, blockade of the TGF-β/activin A Smad signalling pathway selectively in mature T cells via transgene-mediated overexpression of the inhibitory Smad7 exacerbates allergic airway inflammation [14]. Mice which overexpress activin A in the skin have reduced Th2 polarization and decreased serum OVA-specific IgE levels following epicutaneous OVA sensitization [61]. Collectively these findings reveal multi-faceted anti-inflammatory functions of activin A.
Pro-inflammatory effects of activin A
Conversely, numerous studies provide evidence for pro-inflammatory function of activin A. In contrast to the studies describing inhibitory effects of activin A on DC function [54], activin A was shown to induce directional migration of immature mouse and human myeloid DC in vitro and ex vivo [62] and to promote maturation of monocytes into DC in vitro [55]. A recent study evaluating repeated allergen skin challenges in atopic subjects showed that activin A expression was increased in the skin 24 h after the first allergen challenge and correlated with infiltrating ‘conventional’ BDCA-1+ DC (cDC), suggesting that activin A may be responsible for recruitment or retention of DC. Activin A increased the CCR10/CCR4 ratio on human cDC in vitro, providing a possible explanation for the accumulation of DC in the skin [63]. Direct evidence for a pro-inflammatory role of activin A was shown by the finding that activin A overexpression in mouse airways induces severe pulmonary inflammation with features of acute respiratory distress syndrome, and this was reversed by activin A neutralization [64]. Consistent with this, increased serum activin A (or activin B) levels are associated with increased risk of death in patients with acute respiratory failure [65]. In contrast to the findings of an inhibitory effect of activin A via reduced Th2 responses and IgE levels on skin allergic sensitization [61], another study has demonstrated that neutralization of circulating activin A decreases serum allergen-specific IgE and IL-4 levels after allergen sensitization in vivo, indicating a Th2-promoting effect of activin A [66]. Furthermore, it has been shown that mice which overexpress activin A in the skin have increased pro-inflammatory cytokine (Il1b and Tslp) gene expression following epicutaneous OVA sensitization [61]. Numerous studies have used follistatin to block activin A bioactivity and inhibit inflammation. Intraperitoneal follistatin injection inhibits colonic inflammation and increases survival in mouse models of colitis [67] and increases survival after injection of a lethal dose of LPS [68]. Moreover, data from our group show that follistatin treatment of newborn mice increases survival and reduces lung inflammation and mucus production in a mouse model of cystic fibrosis [69]. These findings establish pro-inflammatory functions for activin A in vivo, in various organs including the lung, and suggest that activin A neutralization or blockade would be therapeutically beneficial in a variety of diseases.
Reconciliation of the pro- vs. anti-inflammatory data
Activin A activates genes in a concentration-dependent manner during embryonic development and is therefore a morphogen [70]. Thus, the conflicting anti- vs. pro-inflammatory effects of activin A [24,48] may be partially explained by concentration-dependent effects on immune function, with the activin A concentration in the local tissue microenvironment presumably modulating immune function in a dose-dependent manner via differential gene induction. It should be noted that much (but not all) of the data describing anti-inflammatory effects of activin A comes from in vitro studies, whereas the majority of data showing pro-inflammatory effects of activin A comes from in vivo studies and is arguably more physiologically relevant. Furthermore, differences in the in vitro model system, cell type, dose, route and/or timing of activin A treatment and method of inhibiting/blocking activin A add further complexity. Overall, it is probably an oversimplification to attempt to argue that activin A will always be anti- or pro-inflammatory – rather, a spectrum of responses is possible, and each disease scenario should be investigated carefully taking into consideration the intricacies of activin A regulation and function. In regard to the potential overlap in activin A and TGF-β function, we hypothesize that activin A and TGF-β can exert distinct effects on immune responses due to the fact that they [1] are secreted in different states (latent vs. active), [2] signal via distinct receptors and [3] are regulated by different ligand binding traps, as outlined earlier in this review.
The role of activin A in allergic asthma
A role for activin A in human asthma
A number of reports suggest a role for activin A in the pathogenesis of allergic asthma. Activin A is increased in the serum of severe asthmatics [11], expression of activin βA and activin receptor genes is increased in CD4+ T cells from allergic asthmatics [71], and signalling by activin and TGF-β is rapidly initiated in asthmatics following allergen challenge [12]. Activin A is expressed by mast cells and macrophages in asthmatic human lung [72] and is secreted by human mast cells and lung fibroblasts stimulated with PMA/Ca2+ ionophore or IgE receptor cross-linking [72] and Th2 cytokines [11], respectively. Recent findings show that pre-stored mature activin A is rapidly released from neutrophils in response to TNF, suggesting a possible involvement in severe/neutrophilic asthma [73]. Involvement of activin A in asthma pathogenesis is perhaps not surprising because activin A and TGF-β reciprocally induce the expression of each other [11,75,74].
A role for activin A in experimental allergic asthma
Asthma can be modelled in mice, and despite recognized limitations [76], mouse models have been useful in shedding light on asthma pathogenesis and immunoregulation. The regulation and function of activin A have been extensively studied in experimental models of allergic asthma. Acute allergic airway inflammation induced by short-term (3–4 times) challenge with the model allergen ovalbumin (OVA) induces a rapid increase in BAL fluid activin A concentrations [19,72,77], and activin βA mRNA expression is increased in mouse mast cells and monocytes following IgE receptor cross-linking in vitro [72,78] and in whole lung following allergen challenge [11]. Similarly, we have observed increased BAL fluid activin A levels in a mouse model of chronic allergic asthma [18]. Analogously, BAL fluid TGF-β levels are increased in models of acute and chronic allergic asthma [9,79]. In normal unmanipulated mice, we observe strong uniform activin A and TGF-β immunostaining in the airway epithelium. However, following acute and chronic allergen challenge, activin A and TGF-β immunolocalization in airway epithelium is decreased [18,19].
Regulation of activin A and TGF-β localization in allergic asthma
Analogous to the above changes in the mouse, there is a loss of TGF-β from airway epithelium in asthmatics relative to non-asthmatics [80] and an increase in BAL fluid TGF-β levels in atopic asthmatics following segmental allergen challenge [10,16]. Furthermore, our preliminary data show decreased activin A immunoreactivity in airway epithelium from severe asthmatics compared to normal lung (Fig. 1). These findings suggest that activin A and TGF-β is pre-stored in airway epithelium (and possibly other subepithelial cells) and liberated into the surrounding tissue following induction of allergic airway inflammation. In contrast, another study did not find any significant change in activin A or TGF-β immunoreactivity in airway epithelium of asthmatics at 24 hour post-allergen challenge relative to baseline [12], while a third study reported increased activin A immunostaining in airway epithelium from atopic asthmatics relative to healthy controls [60]. It is possible that the inconsistent observations of airway epithelial activin A and TGF-β immunolocalization could be due to differences in clinical status, asthma subtype/severity between patient groups or kinetic differences in intra-epithelial localization/storage.
Figure 1.

Strong activin A immunolocalization in normal human airway epithelium is decreased in asthma. Lung tissue sections were stained immunohistochemically with antibody specific for the activin βA subunit (E4; IgG2b) (a, c, e, f) or rabbit anti-mouse IgG2b isotype control antibody (b, d). (a–d) Serial sections from normal human lung stained with βA-specific antibody or isotype control antibody. (e, f) Lung from a patient with severe asthma (e) and a patient with asthma and hypersensitivity pneumonitis (f). Immunoperoxidase, haematoxylin counterstain, original magnification 200×.
Expression of activin A signalling components in allergic asthma
Expression of activin receptors is modulated during allergic airway inflammation. In a mouse model of chronic allergic asthma, we showed loss of immunostaining for ALK4 (ActRIB), ActRIIA and ActRIIB in airway epithelium [18], and follistatin treatment partially prevented the decrease in ALK4 immunostaining. Decreased frequency of ActRIIA-immunostained bronchial epithelial cells and numbers of ALK4- and ActRIIA-positive subepithelial inflammatory cells were seen in biopsies from mild atopic asthmatics, although there was no change in the ALK4 staining in bronchial epithelium [60]. In contrast, others have reported increased ALK4 expression in airway epithelium and subepithelial fibroblasts in a model of acute allergic airway inflammation, with a modest increase in ActRIIA [80], and increased numbers of ALK4 and ActRIIA-positive epithelial cells were seen in atopic asthmatics 24 h after allergen challenge [12]. The differences in the activin receptor expression observed may be related to the time-points chosen, with increased expression seen acutely following allergen challenge which decreases upon sustained challenge, as seen in the chronic asthma model and biopsies from asthmatic airways. A recent study detected antibodies to activin A type 1 receptor in the serum of asthmatics compared to controls, correlating with clinical disease. This finding suggests that epithelial damage from airway inflammation during asthma may result in the exposure of cryptic self-antigens or their determinants, resulting in immune responses to self-antigens (activin A included), which may contribute to asthma pathogenesis [82].
There is abundant evidence of altered expression of Smad molecules in clinical and experimental asthma. In a model of acute allergic asthma, Smad2 was moderately induced in alveolar epithelial cells, while Smad3 was strongly up-regulated in bronchial epithelium and alveolar epithelial cells [81]. In a mouse model of chronic allergic asthma, therapeutic treatment with anti-TGF-β antibody decreased phospho Smad2 expression and increased expression of the inhibitory Smad7, together with inhibition of airway remodelling [13]. Analogously, recent findings show that intranasal BMP7 treatment inhibited allergic airway inflammation and collagen production in a model of chronic allergic asthma, concomitant with decreased phosphorylation of Smad2 and Smad3 [83]. Consistent with the mouse data, TGF-β, and to a lesser extent activin A, activates the Smad2 and Smad3 pathway in human fibroblasts in vitro [11]. Similarly, Smad2 and Smad3 are activated in epithelial and subepithelial cells of atopic asthmatics 24 hour after allergen challenge [12,84]. Others have shown that Smad7 expression is decreased in bronchial epithelial cells from asthmatics, with its expression inversely correlated with basement membrane thickness [85]. Overall these findings show that Smad2 and Smad3 signalling pathways are activated in allergic asthma, while Smad7 is decreased. These changes presumably occur as an adaptive response of the asthmatic lung to regulate TGF-β, and by extension activin A signalling.
Evidence for an anti-inflammatory role for activin A in allergic asthma
It has recently been proposed that activin A plays a predominantly anti-inflammatory role in allergic asthma [48]. Evidence supporting this argument comes from experiments where blocking activin A with neutralizing antibody exacerbated airway eosinophilia and Th2 cytokine production in the lung-draining lymph node (LN), with IL-10-producing Treg likely playing an important role [60]. Similarly, systemic (intraperitoneal) administration of recombinant activin A prior to each allergen challenge suppressed allergic airway inflammation [60]. However, other experiments by this group showed that blocking activin A with neutralizing antibody had no effect on inflammation in a HDM allergic asthma model in which Smad2 overexpression in the airways exacerbated airway remodelling and AHR [86]. The different outcomes could be related to the choice of OVA or HDM as allergen due to their differential triggering of Toll-like receptors [87].
Evidence for a pro-inflammatory role for activin A in allergic asthma
In counterpoint, several lines of evidence argue for a pro-allergic/pro-asthmatic role for activin A. Activin A induces differentiation of Th9 cells in vitro and the blockade of activin A and TGF-β with neutralizing antibodies together, but not alone, inhibited Th9 differentiation, IL-9 and IL-13 production from lung-draining LN cells, and recruitment of eosinophils and mast cells to the lung in a HDM asthma model, while IL-25 levels were reduced by blockade of activin A alone or combined activin A/TGF-β blockade. In contrast, Th2 cell numbers and levels of ‘classical’ pro-allergic cytokines (IL-4, IL-5) and chemokines were unaffected [88]. Blocking activin A via intranasal instillation of follistatin immediately prior to allergen challenge inhibited mucus production in the lung and allergen-specific Th2 cytokine production in the lung-draining LN in a model of acute allergic asthma [19]. In a mouse model of chronic allergic asthma, blockade of activin A via intranasal follistatin (5 μg per allergen challenge) significantly inhibited serum allergen-specific IgE and modestly inhibited allergen-specific Th2 cytokine production in the lung-draining LN [18]. Further supporting a pro-inflammatory role for activin A in allergic asthma, intratracheal instillation of activin A (0.02 μg) 45–60 min prior to allergen challenge in an acute asthma model (4 allergen challenges) significantly increased IL-4 production in the lung-draining LN (Fig. 2; unpublished, LeMasurier, Rolland, O'Hehir, Hardy), with a similar trend seen for IL-13. Notably, this is opposite to the finding observed by Semitekolou et al. [60] following intraperitoneal activin A injection, as mentioned above.
Figure 2.
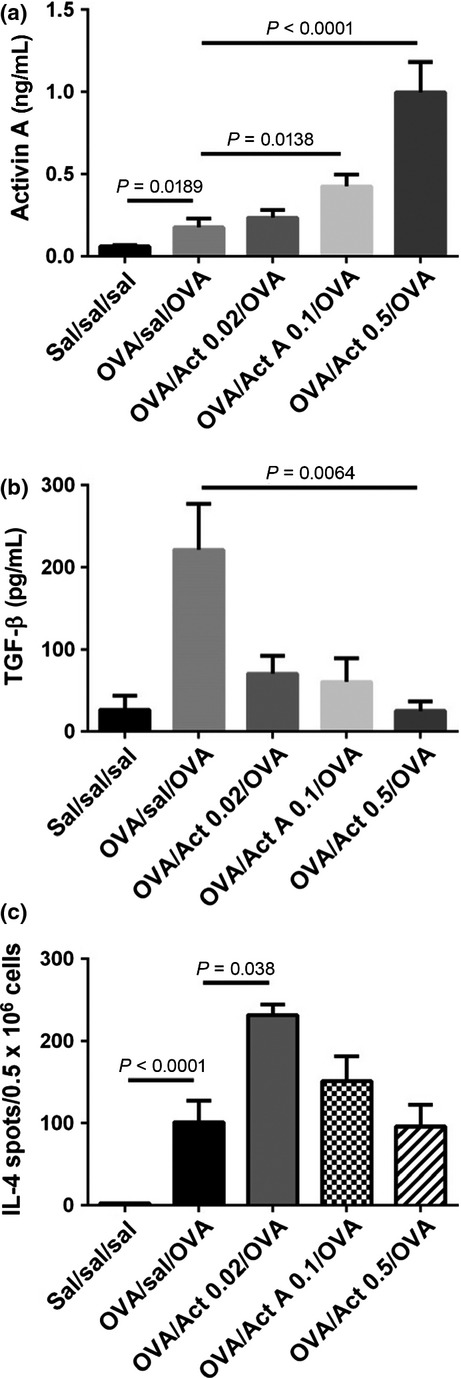
Activin A instilled into the airways prior to allergen challenge decreases BAL fluid TGF-β levels and dose dependently modulates IL-4 production in the lung-draining lymph node in a mouse model of acute allergic asthma. BALB/c mice were sensitized with OVA (50 μg) in aluminium hydroxide on d0 and challenged with OVA (25 μg) in 50 μl saline (d10–12). Recombinant human activin A (0.02, 0.1, 0.5 μg) was instilled intratracheally 45–60 min prior to allergen challenge. Controls received saline instead of OVA or activin A. Mice were killed 24 h after the final challenge for analysis. (a, b) BAL fluid concentrations of activin A and TGF-β as measured by βA-specific ELISA [19] and a TGF-β1 ELISA kit (#DY1679; R&D Systems, Minneapolis, MN, USA). (c) Frequency of IL-4-producing mediastinal lymph node cells as measured by ELISPOT, respectively [19]. Mean ± SEM, n = 7–9 mice/group (pools of 2–3 mice/group for ELISPOT).
Collectively these findings demonstrate the importance of the method of blocking activin A (follistatin vs. neutralizing antibody), and the site in which this occurs (i.e. locally in the lung vs. systemic via intraperitoneal injection). Thus, although the role for activin A in the regulation of inflammation and asthma is not fully resolved, it is clear that activin A is an important modulator of allergic inflammation in general and allergic asthma in particular. However, translation of these findings into the clinic will require careful validation of potential activin inhibitory therapeutics, including dose and route of administration.
The role of activin A in asthmatic airway remodelling
Activin A as a driver of asthmatic airway remodelling – murine asthma models
While there is some debate about the role of activin A in regulation of inflammation generally and allergic asthma in particular, the evidence supporting a role for activin A as a driver of asthmatic airway remodelling is much clearer, at least in murine asthma models. A recent study showed that mice which overexpress Smad2 in the airways have enhanced airway remodelling and increased activin A and TGF-β in lung homogenates following intranasal HDM exposure, and airway remodelling was inhibited by injection of activin A neutralizing antibody prior to HDM challenge [86]. Similarly, the combination of anti-TGF-β and anti-activin A antibodies decreased mucus hypersecretion, mast cell numbers and subepithelial collagen deposition in a HDM model of asthmatic airway remodelling [88]. We found increased BAL fluid activin A levels in a mouse model of chronic asthma together with induction of airway remodelling (airway mucus production, subepithelial collagen and smooth muscle deposition), and these changes were inhibited by follistatin instillation into the lung prior to allergen challenge [18]. The fact that activin A and TGF-β reciprocally induce the expression of one another [11,74,75,89] could be taken to suggest that activin A induces fibrosis via induction of TGF-β. However, receptor blocking experiments have shown that activin A induction of collagen production occurs even if the effect of TGF-β is blocked [75], implying a central role for activin A independent of TGF-β.
Activin A in the regulation of human airway remodelling – role of airway epithelium
The data demonstrating a role for activin A as a driver of asthmatic airway remodelling in humans are more circumstantial, but nevertheless, strongly supported by previous studies. In humans, activin A has been implicated in the development of remodelling in interstitial pulmonary fibrosis [90], as well as liver cirrhosis, wound repair and renal disease [24]. In vitro activin A induces proliferation of human lung fibroblasts [11], human airway smooth muscle cells [72] and normal human bronchial epithelial cells [12]. Moreover, activin A stimulates differentiation of human lung fibroblasts into precursors of muscle cells known as myofibroblasts [91]. Our preliminary data shows that activin A immunolocalization in airway epithelium is decreased in asthmatics (Fig. 1), akin to the situation in the mouse asthma model [18]. Human rhinovirus infection of human airway epithelial cells increases activin A production in vitro, providing a plausible explanation for the link between childhood human rhinovirus infection and increased risk of asthma via early induction of airway remodelling [92]. It was recently shown that the rapid (within 1 h) increase in serum activin A levels following LPS injection into mice is regulated at the post-transcriptional level from newly translated and stored protein [93]. Thus, we propose that stimulation with a variety of inflammatory mediators, including TNF [12] and IL-13 [94], induces secretion of pre-stored or rapidly synthesized activin A from airway epithelium into the surrounding tissue space where it drives fibrotic changes. This is consistent with the concept that the damage to, or stimulation of airway epithelium by various mediators including pollutants, allergens, viruses and even mechanical force stimulates release of inflammatory and pro-fibrotic mediators which drive and sustain asthmatic inflammation and airway remodelling (Fig. 3).
Figure 3.

Proposed model of activin A immunoregulatory effects in the lung. Environmental triggers including infection, pollutants and allergens, as well as certain inflammatory cytokines, cause secretion of activin A from airway epithelial cells via triggering of TLR ligands, protease effects on tight junctions, and binding to cytokine receptors, with or without concomitant cell damage. The secreted activin A exerts immunoregulatory effects on numerous cell types/pathways potentially applicable in asthma in a dose-dependent manner via differential gene induction. Activin A promotes maturation of monocytes (and possibly macrophages) into DC and modulation of DC functions including migration, chemokine secretion, chemokine receptor expression and endocytosis. Additionally, activin A can promote conversion of naïve CD4+ T cells into Foxp3−IL-10+ Treg or Foxp3+ iTreg, the latter being TGF-β dependent, and promotes Th2 responses including IL-4, IL-9, IL-13 and IL-25 production. Activin A is produced by innate immune cells following cross-linking of high-affinity IgE receptors or by exposure to TNF or IL-13. Activin A drives progressive airway remodelling including smooth muscle cell proliferation and collagen deposition.
Conclusions and future directions
The cumulative evidence clearly shows an important role for activin A in the regulation of inflammation in general and allergic asthma in particular. Nevertheless, the above studies highlight a number of major gaps in our understanding of activin A biology. A key area requiring further research is the role of activin A dose on inflammation and immune cell function in vivo. A related question concerns the role of activin A dose on fibrosis, for example smooth muscle proliferation and collagen deposition. Before modulation of the activin A pathway can be utilized clinically, a clearer understanding of the best method of blocking activin A is required. Follistatin has the advantage that it is an endogenous protein; however, given its potential to bind other members of the TGF-β superfamily, albeit at lower affinity, this issue requires further investigation. Such comparisons of different blocking methods (e.g. antibody vs. follistatin) should be performed in parallel in the same animal model. Furthermore, the route via which activin A is blocked and/or tissue site are additional variables requiring detailed exploration. Long-term follistatin administration has thus far proved safe in studies in primates and mice [18,69,95]. Nevertheless, given the wide range of physiological processes that activin A is involved in regulating, careful analysis of potential side-effects will be essential, as for pre-clinical testing of any new pharmaceutical or biological.
Acknowledgments
The authors thank Dr. Sara Prickett (Monash University) for critiquing the manuscript and Jeanne LeMasurier and Jun Yao (Monash University) for assistance with the mouse experiments. The authors thank A/Professor Mark Hedger and Professor David de Kretser, of Monash University, for assistance with the activin A immunohistochemistry (Fig. 1) and the activin A ELISA (Fig. 2). The authors thank Professor John Wilson, of The Alfred Hospital, for provision of human lung tissue specimens.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. 2008;8:193–204. doi: 10.1038/nri2275. [DOI] [PubMed] [Google Scholar]
- 2.Holgate ST. Asthma: a simple concept but in reality a complex disease. Eur J Clin Invest. 2011;41:1339–52. doi: 10.1111/j.1365-2362.2011.02534.x. (12) [DOI] [PubMed] [Google Scholar]
- 3.Umetsu DT, McIntire JJ, Akbari O, Macaubas C, DeKruyff RH. Asthma: an epidemic of dysregulated immunity. Nat Immunol. 2002;3:715–20. doi: 10.1038/ni0802-715. [DOI] [PubMed] [Google Scholar]
- 4.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 5.Trevor JL, Deshane JS. Refractory asthma: mechanisms, targets, and therapy. Allergy. 2014;69:817–27. doi: 10.1111/all.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durrani SR, Viswanathan RK, Busse WW. What effect does asthma treatment have on airway remodeling? Current perspectives. J Allergy Clin Immunol. 2011;128:439–48. doi: 10.1016/j.jaci.2011.06.002. quiz 49–50. [DOI] [PubMed] [Google Scholar]
- 7.Pohunek P, Warner JO, Turzikova J, Kudrmann J, Roche WR. Markers of eosinophilic inflammation and tissue re-modelling in children before clinically diagnosed bronchial asthma. Pediatr Allergy Immunol. 2005;16:43–51. doi: 10.1111/j.1399-3038.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 8.Willart MA, Deswarte K, Pouliot P, et al. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med. 2012;209:1505–17. doi: 10.1084/jem.20112691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alcorn JF, Rinaldi LM, Jaffe EF, et al. Transforming growth factor-beta1 suppresses airway hyperresponsiveness in allergic airway disease. Am J Respir Crit Care Med. 2007;176:974–82. doi: 10.1164/rccm.200702-334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batra V, Musani AI, Hastie AT, et al. Bronchoalveolar lavage fluid concentrations of transforming growth factor (TGF)-beta1, TGF-beta2, interleukin (IL)-4 and IL-13 after segmental allergen challenge and their effects on alpha-smooth muscle actin and collagen III synthesis by primary human lung fibroblasts. Clin Exp Allergy. 2004;34:437–44. doi: 10.1111/j.1365-2222.2004.01885.x. [DOI] [PubMed] [Google Scholar]
- 11.Karagiannidis C, Hense G, Martin C, et al. Activin A is an acute allergen-responsive cytokine and provides a link to TGF-beta-mediated airway remodeling in asthma. J Allergy Clin Immunol. 2006;117:111–8. doi: 10.1016/j.jaci.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 12.Kariyawasam HH, Pegorier S, Barkans J, et al. Activin and transforming growth factor-beta signaling pathways are activated after allergen challenge in mild asthma. J Allergy Clin Immunol. 2009;124:454–62. doi: 10.1016/j.jaci.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J Immunol. 2005;174:5774–80. doi: 10.4049/jimmunol.174.9.5774. [DOI] [PubMed] [Google Scholar]
- 14.Nakao A, Miike S, Hatano M, et al. Blockade of transforming growth factor beta/Smad signaling in T cells by overexpression of Smad7 enhances antigen-induced airway inflammation and airway reactivity. J Exp Med. 2000;192:151–8. doi: 10.1084/jem.192.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohno I, Nitta Y, Yamauchi K, et al. Transforming growth factor beta 1 (TGF beta 1) gene expression by eosinophils in asthmatic airway inflammation. Am J Respir Cell Mol Biol. 1996;15:404–9. doi: 10.1165/ajrcmb.15.3.8810646. [DOI] [PubMed] [Google Scholar]
- 16.Redington AE, Madden J, Frew AJ, et al. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;1:642–7. doi: 10.1164/ajrccm.156.2.9605065. [DOI] [PubMed] [Google Scholar]
- 17.Vignola AM, Chanez P, Chiappara G, et al. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am J Respir Crit Care Med. 1997;156(2 Pt 1):591–9. doi: 10.1164/ajrccm.156.2.9609066. [DOI] [PubMed] [Google Scholar]
- 18.Hardy CL, Nguyen HA, Mohamud R, et al. The activin A antagonist follistatin inhibits asthmatic airway remodelling. Thorax. 2013;68:9–18. doi: 10.1136/thoraxjnl-2011-201128. [DOI] [PubMed] [Google Scholar]
- 19.Hardy CL, O'Connor AE, Yao J, et al. Follistatin is a candidate endogenous negative regulator of activin A in experimental allergic asthma. Clin Exp Allergy. 2006;36:941–50. doi: 10.1111/j.1365-2222.2006.02523.x. [DOI] [PubMed] [Google Scholar]
- 20.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 21.Harrison CA, Al-Musawi SL, Walton KL. Prodomains regulate the synthesis, extracellular localisation and activity of TGF-beta superfamily ligands. Growth Factors. 2011;29:174–86. doi: 10.3109/08977194.2011.608666. [DOI] [PubMed] [Google Scholar]
- 22.Harrison CA, Gray PC, Vale WW, Robertson DM. Antagonists of activin signaling: mechanisms and potential biological applications. Trends Endocrinol Metab. 2005;16:73–8. doi: 10.1016/j.tem.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Tsuchida K, Nakatani M, Hitachi K, et al. Activin signaling as an emerging target for therapeutic interventions. Cell Commun Signal. 2009;7:15. doi: 10.1186/1478-811X-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedger MP, Winnall WR, Phillips DJ, de Kretser DM. The regulation and functions of activin and follistatin in inflammation and immunity. Vitam Horm. 2011;85:255–97. doi: 10.1016/B978-0-12-385961-7.00013-5. [DOI] [PubMed] [Google Scholar]
- 25.Matzuk MM, Kumar TR, Vassalli A, et al. Functional analysis of activins during mammalian development. Nature. 1995;374:354–6. doi: 10.1038/374354a0. [DOI] [PubMed] [Google Scholar]
- 26.Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 1994;8:414–27. doi: 10.1101/gad.8.4.414. [DOI] [PubMed] [Google Scholar]
- 27.Li S, Shimono C, Norioka N, et al. Activin A binds to perlecan through its pro-region that has heparin/heparan sulfate binding activity. J Biol Chem. 2010;285:36645–55. doi: 10.1074/jbc.M110.177865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–73. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Onichtchouk D, Chen YG, Dosch R, et al. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature. 1999;401:480–5. doi: 10.1038/46794. [DOI] [PubMed] [Google Scholar]
- 30.Gray PC, Harrison CA, Vale W. Cripto forms a complex with activin and type II activin receptors and can block activin signaling. Proc Natl Acad Sci USA. 2003;100:5193–8. doi: 10.1073/pnas.0531290100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu G, Chen YG, Ozdamar B, et al. Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science. 2000;287:92–7. doi: 10.1126/science.287.5450.92. [DOI] [PubMed] [Google Scholar]
- 32.Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–91. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 33.Sflomos G, Kostaras E, Panopoulou E, et al. ERBIN is a new SARA-interacting protein: competition between SARA and SMAD2 and SMAD3 for binding to ERBIN. J Cell Sci. 2011;124(Pt 19):3209–22. doi: 10.1242/jcs.062307. [DOI] [PubMed] [Google Scholar]
- 34.Yan X, Liu Z, Chen Y. Regulation of TGF-beta signaling by Smad7. Acta Biochim Biophys Sin. 2009;41:263–72. doi: 10.1093/abbs/gmp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan X, Lin Z, Chen F, et al. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J Biol Chem. 2009;284:30097–104. doi: 10.1074/jbc.M109.049304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrington AE, Morris-Triggs SA, Ruotolo BT, Robinson CV, Ohnuma S, Hyvonen M. Structural basis for the inhibition of activin signalling by follistatin. EMBO J. 2006;25:1035–45. doi: 10.1038/sj.emboj.7601000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson TB, Lerch TF, Cook RW, Woodruff TK, Jardetzky TS. The structure of the follistatin: activin complex reveals antagonism of both type I and type II receptor binding. Dev Cell. 2005;9:535–43. doi: 10.1016/j.devcel.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Nogai H, Rosowski M, Grun J, et al. Follistatin antagonizes transforming growth factor-beta3-induced epithelial-mesenchymal transition in vitro: implications for murine palatal development supported by microarray analysis. Differentiation. 2008;76:404–16. doi: 10.1111/j.1432-0436.2007.00223.x. [DOI] [PubMed] [Google Scholar]
- 39.Lin SY, Morrison JR, Phillips DJ, de Kretser DM. Regulation of ovarian function by the TGF-beta superfamily and follistatin. Reproduction. 2003;126:133–48. doi: 10.1530/rep.0.1260133. [DOI] [PubMed] [Google Scholar]
- 40.Sugino K, Kurosawa N, Nakamura T, et al. Molecular heterogeneity of follistatin, an activin-binding protein. Higher affinity of the carboxyl-terminal truncated forms for heparan sulfate proteoglycans on the ovarian granulosa cell. J Biol Chem. 1993;268:15579–87. [PubMed] [Google Scholar]
- 41.Hashimoto O, Kawasaki N, Tsuchida K, Shimasaki S, Hayakawa T, Sugino H. Difference between follistatin isoforms in the inhibition of activin signalling: activin neutralizing activity of follistatin isoforms is dependent on their affinity for activin. Cell Signal. 2000;12:565–71. doi: 10.1016/s0898-6568(00)00099-1. [DOI] [PubMed] [Google Scholar]
- 42.Phillips DJ, Jones KL, McGaw DJ, et al. Release of activin and follistatin during cardiovascular procedures is largely due to heparin administration. J Clin Endocrinol Metab. 2000;85:2411–5. doi: 10.1210/jcem.85.7.6666. [DOI] [PubMed] [Google Scholar]
- 43.Hashimoto O, Nakamura T, Shoji H, Shimasaki S, Hayashi Y, Sugino H. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J Biol Chem. 1997;272:13835–42. doi: 10.1074/jbc.272.21.13835. [DOI] [PubMed] [Google Scholar]
- 44.Tsuchida K, Arai KY, Kuramoto Y, Yamakawa N, Hasegawa Y, Sugino H. Identification and characterization of a novel follistatin-like protein as a binding protein for the TGF-beta family. J Biol Chem. 2000;275:40788–96. doi: 10.1074/jbc.M006114200. [DOI] [PubMed] [Google Scholar]
- 45.Hill JJ, Davies MV, Pearson AA, et al. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. 2002;277:40735–41. doi: 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- 46.Walton KL, Makanji Y, Harrison CA. New insights into the mechanisms of activin action and inhibition. Mol Cell Endocrinol. 2012;359:2–12. doi: 10.1016/j.mce.2011.06.030. [DOI] [PubMed] [Google Scholar]
- 47.de Kretser DM, O'Hehir RE, Hardy CL, Hedger MP. The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol Cell Endocrinol. 2012;359:101–6. doi: 10.1016/j.mce.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 48.Kariyawasam HH, Semitekolou M, Robinson DS, Xanthou G. Activin-A: a novel critical regulator of allergic asthma. Clin Exp Allergy. 2011;41:1505–14. doi: 10.1111/j.1365-2222.2011.03784.x. [DOI] [PubMed] [Google Scholar]
- 49.Hedger MP, Clarke L. Isolation of rat blood lymphocytes using a two-step Percoll density gradient. Effect of activin (erythroid differentiation factor) on peripheral T lymphocyte proliferation in vitro. J Immunol Methods. 1993;163:133–6. doi: 10.1016/0022-1759(93)90247-5. [DOI] [PubMed] [Google Scholar]
- 50.Hedger MP, Drummond AE, Robertson DM, Risbridger GP, de Kretser DM. Inhibin and activin regulate [3H]thymidine uptake by rat thymocytes and 3T3 cells in vitro. Mol Cell Endocrinol. 1989;61:133–8. doi: 10.1016/0303-7207(89)90198-6. [DOI] [PubMed] [Google Scholar]
- 51.Zipori D, Barda-Saad M. Role of activin A in negative regulation of normal and tumor B lymphocytes. J Leukoc Biol. 2001;69:867–73. [PubMed] [Google Scholar]
- 52.Yu EW, Dolter KE, Shao LE, Yu J. Suppression of IL-6 biological activities by activin A and implications for inflammatory arthropathies. Clin Exp Immunol. 1998;112:126–32. doi: 10.1046/j.1365-2249.1998.00522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robson NC, Phillips DJ, McAlpine T, et al. Activin-A: a novel dendritic cell-derived cytokine that potently attenuates CD40 ligand-specific cytokine and chemokine production. Blood. 2008;111:2733–43. doi: 10.1182/blood-2007-03-080994. [DOI] [PubMed] [Google Scholar]
- 54.Segerer SE, Muller N, Brandt J, et al. The glycoprotein-hormones activin A and inhibin A interfere with dendritic cell maturation. Reprod Biol Endocrinol. 2008;6:17. doi: 10.1186/1477-7827-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scutera S, Riboldi E, Daniele R, et al. Production and function of activin A in human dendritic cells. Eur Cytokine Netw. 2008;19:60–8. doi: 10.1684/ecn.2008.0121. [DOI] [PubMed] [Google Scholar]
- 56.Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol. 2008;8:142–52. doi: 10.1038/nri2236. [DOI] [PubMed] [Google Scholar]
- 57.Khare A, Krishnamoorthy N, Oriss TB, Fei M, Ray P, Ray A. Cutting edge: inhaled antigen upregulates retinaldehyde dehydrogenase in lung CD103+ but not plasmacytoid dendritic cells to induce Foxp3 de novo in CD4+ T cells and promote airway tolerance. J Immunol. 2013;191:25–9. doi: 10.4049/jimmunol.1300193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lloyd CM, Murdoch JR. Tolerizing allergic responses in the lung. Mucosal Immunol. 2010;3:334–44. doi: 10.1038/mi.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber S, Stahl FR, Schrader J, et al. Activin A promotes the TGF-beta-induced conversion of CD4+CD25- T cells into Foxp3+ induced regulatory T cells. J Immunol. 2009;182:4633–40. doi: 10.4049/jimmunol.0803143. [DOI] [PubMed] [Google Scholar]
- 60.Semitekolou M, Alissafi T, Aggelakopoulou M, et al. Activin-A induces regulatory T cells that suppress T helper cell immune responses and protect from allergic airway disease. J Exp Med. 2009;206:1769–85. doi: 10.1084/jem.20082603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kypriotou M, Rivero D, Haller S, et al. Activin a inhibits antigen-induced allergy in murine epicutaneous sensitization. Front Immunol. 2013;4:246. doi: 10.3389/fimmu.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salogni L, Musso T, Bosisio D, et al. Activin A induces dendritic cell migration through the polarized release of CXC chemokine ligands 12 and 14. Blood. 2009;113:5848–56. doi: 10.1182/blood-2008-12-194597. [DOI] [PubMed] [Google Scholar]
- 63.Vittorakis S, Samitas K, Tousa S, et al. Circulating conventional and plasmacytoid dendritic cell subsets display distinct kinetics during in vivo repeated allergen skin challenges in atopic subjects. BioMed Res Int. 2014;2014:231036. doi: 10.1155/2014/231036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Apostolou E, Stavropoulos A, Sountoulidis A, et al. Activin-A overexpression in the murine lung causes pathology that simulates acute respiratory distress syndrome. Am J Respir Crit Care Med. 2012;185:382–91. doi: 10.1164/rccm.201105-0784OC. [DOI] [PubMed] [Google Scholar]
- 65.de Kretser DM, Bensley JG, Pettila V, et al. Serum activin A and B levels predict outcome in patients with acute respiratory failure: a prospective cohort study. Crit Care. 2013;17:R263. doi: 10.1186/cc13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ogawa K, Funaba M, Tsujimoto M. A dual role of activin A in regulating immunoglobulin production of B cells. J Leukoc Biol. 2008;83(6):1451–8. doi: 10.1189/jlb.1007710. [DOI] [PubMed] [Google Scholar]
- 67.Dohi T, Ejima C, Kato R, et al. Therapeutic potential of follistatin for colonic inflammation in mice. Gastroenterology. 2005;128:411–23. doi: 10.1053/j.gastro.2004.11.063. [DOI] [PubMed] [Google Scholar]
- 68.Jones KL, Mansell A, Patella S, et al. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci USA. 2007;104:16239–44. doi: 10.1073/pnas.0705971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hardy CL, King SJ, Mifsud NA, et al. The activin A antagonist follistatin inhibits cystic fibrosis-like lung inflammation and pathology. Immunol Cell Biol. 2015 doi: 10.1038/icb.2015.7. [Epub 3 March 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gurdon JB, Standley H, Dyson S, et al. Single cells can sense their position in a morphogen gradient. Development. 1999;126:5309–17. doi: 10.1242/dev.126.23.5309. [DOI] [PubMed] [Google Scholar]
- 71.Wohlfahrt JG, Kunzmann S, Menz G, et al. T cell phenotype in allergic asthma and atopic dermatitis. Int Arch Allergy Immunol. 2003;131:272–82. doi: 10.1159/000072139. [DOI] [PubMed] [Google Scholar]
- 72.Cho SH, Yao Z, Wang SW, et al. Regulation of activin A expression in mast cells and asthma: its effect on the proliferation of human airway smooth muscle cells. J Immunol. 2003;170:4045–52. doi: 10.4049/jimmunol.170.8.4045. [DOI] [PubMed] [Google Scholar]
- 73.Chen Y, Wu H, Winnall WR, et al. Tumour necrosis factor-alpha stimulates human neutrophils to release preformed activin A. Immunol Cell Biol. 2011;89:889–96. doi: 10.1038/icb.2011.12. [DOI] [PubMed] [Google Scholar]
- 74.Ohnishi N, Miyata T, Ohnishi H, et al. Activin A is an autocrine activator of rat pancreatic stellate cells: potential therapeutic role of follistatin for pancreatic fibrosis. Gut. 2003;52:1487–93. doi: 10.1136/gut.52.10.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wada W, Kuwano H, Hasegawa Y, Kojima I. The dependence of transforming growth factor-beta-induced collagen production on autocrine factor activin A in hepatic stellate cells. Endocrinology. 2004;145:2753–9. doi: 10.1210/en.2003-1663. [DOI] [PubMed] [Google Scholar]
- 76.Kumar RK, Foster PS. Modeling allergic asthma in mice: pitfalls and opportunities. Am J Respir Cell Mol Biol. 2002;27:267–72. doi: 10.1165/rcmb.F248. [DOI] [PubMed] [Google Scholar]
- 77.Le AV, Cho JY, Miller M, McElwain S, Golgotiu K, Broide DH. Inhibition of allergen-induced airway remodeling in Smad 3-deficient mice. J Immunol. 2007;178:7310–6. doi: 10.4049/jimmunol.178.11.7310. [DOI] [PubMed] [Google Scholar]
- 78.Von Bubnoff D, Matz H, Cazenave JP, Hanau D, Bieber T, De La Salle H. Kinetics of gene induction after Fc epsilon RI ligation of atopic monocytes identified by suppression subtractive hybridization. J Immunol. 2002;169:6170–7. doi: 10.4049/jimmunol.169.11.6170. [DOI] [PubMed] [Google Scholar]
- 79.McMillan SJ, Lloyd CM. Prolonged allergen challenge in mice leads to persistent airway remodelling. Clin Exp Allergy. 2004;34:497–507. doi: 10.1111/j.1365-2222.2004.01895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Magnan A, Retornaz F, Tsicopoulos A, et al. Altered compartmentalization of transforming growth factor-beta in asthmatic airways. Clin Exp Allergy. 1997;27:389–95. [PubMed] [Google Scholar]
- 81.Rosendahl A, Checchin D, Fehniger TE, ten Dijke P, Heldin CH, Sideras P. Activation of the TGF-beta/activin-Smad2 pathway during allergic airway inflammation. Am J Respir Cell Mol Biol. 2001;25:60–8. doi: 10.1165/ajrcmb.25.1.4396. [DOI] [PubMed] [Google Scholar]
- 82.Liu M, Subramanian V, Christie C, Castro M, Mohanakumar T. Immune responses to self-antigens in asthma patients: clinical and immunopathological implications. Hum Immunol. 2012;73:511–6. doi: 10.1016/j.humimm.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stumm CL, Halcsik E, Landgraf RG, Camara NO, Sogayar MC, Jancar S. Lung remodeling in a mouse model of asthma involves a balance between TGF-beta1 and BMP-7. PLoS ONE. 2014;9:e95959. doi: 10.1371/journal.pone.0095959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Torrego A, Hew M, Oates T, Sukkar M. Fan Chung K. Expression and activation of TGF-beta isoforms in acute allergen-induced remodelling in asthma. Thorax. 2007;62:307–13. doi: 10.1136/thx.2006.063487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nakao A, Sagara H, Setoguchi Y, et al. Expression of Smad7 in bronchial epithelial cells is inversely correlated to basement membrane thickness and airway hyperresponsiveness in patients with asthma. J Allergy Clin Immunol. 2002;110:873–8. doi: 10.1067/mai.2002.129236. [DOI] [PubMed] [Google Scholar]
- 86.Gregory LG, Mathie SA, Walker SA, Pegorier S, Jones CP, Lloyd CM. Overexpression of Smad2 drives house dust mite-mediated airway remodeling and airway hyperresponsiveness via activin and IL-25. Am J Respir Crit Care Med. 2010;182:143–54. doi: 10.1164/rccm.200905-0725OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boyce JA, Austen KF. No audible wheezing: nuggets and conundrums from mouse asthma models. J Exp Med. 2005;201:1869–73. doi: 10.1084/jem.20050584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jones CP, Gregory LG, Causton B, Campbell GA, Lloyd CM. Activin A and TGF-beta promote T(H)9 cell-mediated pulmonary allergic pathology. J Allergy Clin Immunol. 2012;129:1000–10. doi: 10.1016/j.jaci.2011.12.965. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamashita S, Maeshima A, Kojima I, Nojima Y. Activin A is a potent activator of renal interstitial fibroblasts. J Am Soc Nephrol. 2004;15:91–101. doi: 10.1097/01.asn.0000103225.68136.e6. [DOI] [PubMed] [Google Scholar]
- 90.Matsuse T, Ikegami A, Ohga E, et al. Expression of immunoreactive activin A protein in remodeling lesions associated with interstitial pulmonary fibrosis. Am J Pathol. 1996;148:707–13. [PMC free article] [PubMed] [Google Scholar]
- 91.Ohga E, Matsuse T, Teramoto S, et al. Effects of activin A on proliferation and differentiation of human lung fibroblasts. Biochem Biophys Res Commun. 1996;228:391–6. doi: 10.1006/bbrc.1996.1672. [DOI] [PubMed] [Google Scholar]
- 92.Leigh R, Oyelusi W, Wiehler S, et al. Human rhinovirus infection enhances airway epithelial cell production of growth factors involved in airway remodeling. J Allergy Clin Immunol. 2008;121:1238–45. doi: 10.1016/j.jaci.2008.01.067. e4. [DOI] [PubMed] [Google Scholar]
- 93.Wu H, Chen Y, Winnall WR, Phillips DJ, Hedger MP. Acute regulation of activin A and its binding protein, follistatin, in serum and tissues following lipopolysaccharide treatment of adult male mice. Am J Physiol Regul Integr Comp Physiol. 2012;303:R665–75. doi: 10.1152/ajpregu.00478.2011. [DOI] [PubMed] [Google Scholar]
- 94.Hardy CL, Lemasurier JS, Olsson F, et al. Interleukin-13 regulates secretion of the tumor growth factor-{beta} superfamily cytokine activin A in allergic airway inflammation. Am J Respir Cell Mol Biol. 2010;42:667–75. doi: 10.1165/rcmb.2008-0429OC. [DOI] [PubMed] [Google Scholar]
- 95.Kota J, Handy CR, Haidet AM, et al. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci Transl Med. 2009;1:6ra15. doi: 10.1126/scitranslmed.3000112. [DOI] [PMC free article] [PubMed] [Google Scholar]