

Abstract
The kinase/endonuclease IRE1 is the most conserved signal transducer of the unfolded protein response (UPR), an intracellular signaling network that monitors and regulates the protein folding capacity of the endoplasmic reticulum (ER). Upon sensing protein folding perturbations in the ER, IRE1 initiates the unconventional splicing of XBP1 mRNA culminating in the production of the transcription factor XBP1s, which expands the ER's protein folding capacity. We show that an RNA‐intrinsic conformational change causes the intron of XBP1 mRNA to be ejected and the exons to zipper up into an extended stem, juxtaposing the RNA ends for ligation. These conformational rearrangements are important for XBP1 mRNA splicing in vivo. The features that point to such active participation of XBP1 mRNA in the splicing reaction are highly conserved throughout metazoan evolution, supporting their importance in orchestrating XBP1 mRNA processing with efficiency and fidelity.
Keywords: endoribonuclease, ER stress, RNA conformational change, unfolded protein response, XBP1 splicing
Subject Categories: Protein Biosynthesis & Quality Control, RNA Biology
Introduction
Protein folding deficiencies in the endoplasmic reticulum (ER) result in accumulation of un‐ or mis‐folded polypeptides, a stress condition that triggers the unfolded protein response (UPR). The UPR is a network of signal transduction pathways that regulate the coordinated reduction of client protein load into the ER and the expansion of the ER folding capacity, thereby ensuring that the organelle remains in homeostasis 1. In metazoans, the UPR is orchestrated by three principal ER‐resident sensors/signal transducers of the ER folding status: the membrane tethered transcription factor ATF6 and the transmembrane kinases PERK and IRE1 2. Activation of ATF6 results in its translocation to the Golgi apparatus where it is processed by resident proteases, liberating its cytosolic domain as a soluble transcription factor from the ER membrane 3. Activation of PERK results in global translational attenuation after phosphorylation of the eukaryotic translation initiation factor 2 (eIF2) 4. This reduces the load of proteins entering the ER. eIF2 phosphorylation also leads to the production of the transcription factor ATF4, which augments the gene expression program driven by ATF6. Lastly, activation of IRE1 results in activation of a conserved downstream transcription factor, XBP1 5, 6, 7. The IRE1‐mediated branch is the most evolutionarily conserved: IRE1 is the only existing ER stress sensor/transducer outside of metazoans.
IRE1 is a type I transmembrane protein that consists of a sensor domain residing in the ER lumen and a Ser/Thr kinase domain fused to a ribonuclease (RNase) domain residing in the cytosol. ER stress leads to oligomerization, trans‐autophosphorylation, and allosteric activation of IRE1's RNase domain 8, 9, which transmits the UPR signal to the cell nucleus by a unique splicing mechanism: IRE1 cleaves HAC1 (yeast) or XBP1 (metazoans) mRNAs at non‐conventional splice sites 5, 7, 10, 11. The free exons are joined by the tRNA ligase Trl1 in yeast 12, or the RTCB tRNA ligase complex in metazoans 13, 14, 15. The spliced HAC1 and XBP1 mRNAs encode potent transcription factors that activate several hundred genes that correct ER folding defects 16, 17. Besides splicing, metazoan IRE1 also cleaves ER‐bound mRNAs, preventing their translation and thereby diminishing the protein folding load in the ER. This additional functional output is known as regulated IRE1‐dependent mRNA decay (RIDD) 18, 19.
IRE1 recognizes RNA hairpins in its splicing substrates, cleaving a scissile bond 3′ of a guanosine always found in position 3 of a 7‐mer loop 20. The recognition motif of RIDD substrate RNAs is less conserved, and the determinants that shunt specific mRNAs into the RIDD pathway remain obscure. Biochemical and structural evidence suggest that oligomerization precedes activation and that at least one IRE1 dimer is required for a single cleavage event 8, 21. How the splicing reaction, requiring two cleavage events and the subsequent ligation of the correct exon ends, is orchestrated with fidelity also remains an outstanding question. Here, we provide evidence that XBP1 mRNA is not a passive substrate but an active protagonist in the splicing reaction.
Results
Conserved features in metazoan XBP1 mRNAs are required for recognition and cleavage by IRE1
To investigate the RNA features required for XBP1 mRNA splicing, we performed multiple‐sequence‐alignment analyses of RNA segments adjacent to the splice sites from evolutionarily distant metazoans (Figs 1A and EV1A). These analyses identified the conserved exon–intron boundaries conforming to the C(C/U)G¦CAGC consensus sequence of the splice junctions separated by the non‐conventional intron (Figs 1A and EV1A and see 20). RNA secondary‐fold predictions suggested that these sequences fold into a conserved bifurcated stem‐loop (BSL) structure comprised of three stems (Fig 1B, left structure, and Fig EV1B, top structures): a central stem, S1, and two arm stems, S2 and S3. S1 is formed by exon–exon pairing. Note that Fig 1B only shows the portion of the stem that is proximal to the bifurcation. S1 can invariably be extended to form a long stem interrupted by bulges (Fig EV1C), whereas S2 and S3 are short stems formed by exon–intron pairing. Analyses of recently published datasets obtained using two different genome‐wide methods to determine RNA secondary structures in living cells 22, 23 support our computational predictions of the secondary fold of the XBP1 BSL in vertebrates (Fig EV2).
Figure 1. Conserved features in metazoan XBP1 mRNAs.

- Multiple sequence alignment of XBP1‐BSL. IRE1 cleavage sites are indicated in boldface. The guanosines 3′ of the scissile bond are colored in red. Grey boxes: 7‐mer loops harboring the cleavage sites. Dash‐outlined box: unconventional XBP1 intron. Asterisks: conserved bases. Colored arrows: stems in human XBP1‐BSL or in its corresponding spliced structure.
- Secondary structures of the human XBP1‐BSL (from sequence in A) and its corresponding spliced RNA. Arrowheads: scissile bonds (unspliced structure); exon–exon junction (spliced structure). Closed circles: Watson–Crick base pairs. Open circles: Wobble base pairs. Colored arrows: stems in unspliced and spliced XBP1‐ BSL structures. S1, central stem; S2, S3, arm stems; ES1, extended stem.
- Predicted tertiary structure of the unspliced human XBP1‐BSL. The intron is colored in grey. The guanosines 3′ of the scissile bond are colored in red. Arrowheads: scissile bonds.
Figure EV1. Evolutionary conservation of sequence and structural features of XBP1‐BSL .
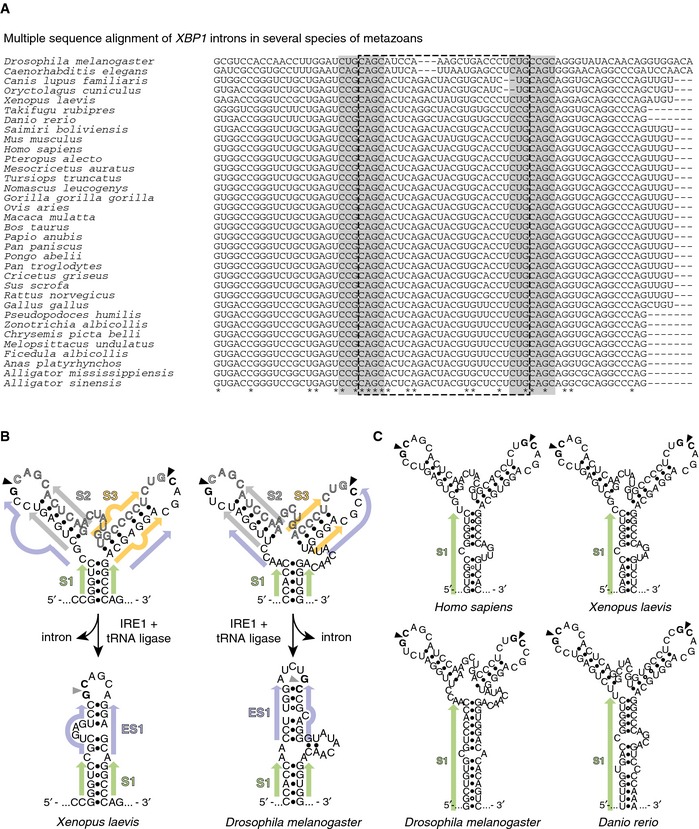
- Multiple sequence alignment of XBP1‐BSL from phylogenetically diverse metazoans. Grey boxes: 7‐mer loops harboring the conserved cleavage sites. Dash‐outlined box: XBP1 intron.
- Secondary structures of select sequences in (A), and their corresponding spliced RNAs. Arrowheads: scissile bonds (unspliced structure); exon–exon junction (spliced structure). Closed circles: Watson–Crick base pairs. Open circles: Wobble base pairs. Colored arrows: stems in unspliced or spliced XBP1‐ BSL structures. S1, central stem; S2, S3, arm stems; ES1 extended stem.
- Secondary structures of select sequences in (A) showing the extension of S1.
Figure EV2. In vivo structure of XBP1‐BSL .
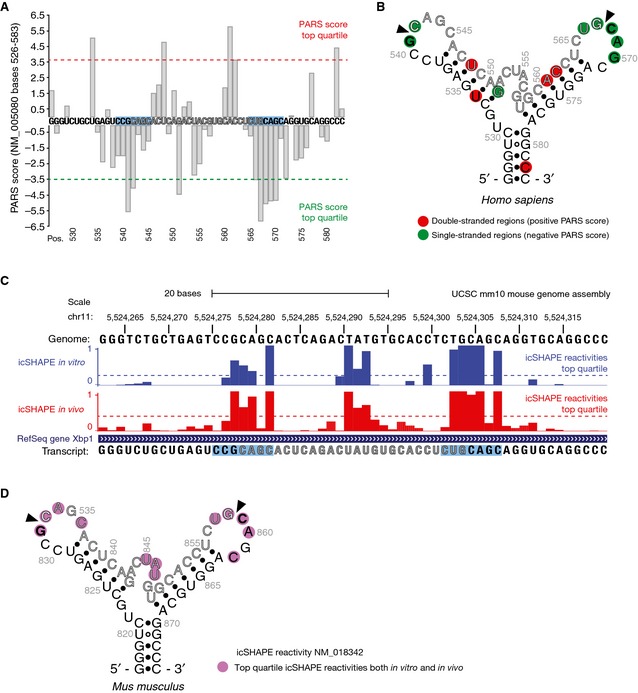
- Parallel analysis of RNA structure (PARS) performed on the transcript encoding unspliced XBP1 mRNA of human origin (NCBI Accession NM_005080). Data were obtained from the Gene Expression Omnibus accession GSE50676. Nuclease digestion profiles for RNase V1 (cleaving double‐stranded RNA) and S1 nuclease (cleaving single‐stranded RNA) after deep‐sequencing are combined to generate a PARS score, defined as the ratio of mapped reads obtained from RNA libraries prepared after digestion with either nuclease 22. PARS scores for nucleotides at positions 526–583 are shown. The cut‐off values for the PARS scores corresponding to the top quartile (most significant) are indicated. The intron is colored in grey. The blue boxes indicate the 7‐mer loop harboring the IRE1 cleavage site.
- Secondary structure of the region corresponding to the nucleotides shown in (A). Colored circles indicate the corresponding PARS score per position.
- UCSC Genome Browser view showing the RNA secondary structure signature of Xbp1 mRNA of mouse origin as determined by in vivo click selective 2′‐hydroxyl acylation and profiling experiments (icSHAPE). The bar graphs indicate the icSHAPE reactivities reported in the Gene Expression Omnibus accession GSE64169 for the corresponding transcript (NCBI Accession NM_018342). Cutoff values for the icSHAPE reactivity values corresponding to the top quartile (most significant) are indicated. The intron is colored in grey. The blue boxes indicate the 7‐mer loop harboring the IRE1 cleavage site.
- Secondary structure of the region corresponding to the nucleotides 817–874 of the transcript shown in (C). Colored circles indicate the corresponding icSHAPE reactivity value above the top quartile threshold per position.
By contrast, similar structure prediction analyses performed on the corresponding spliced mRNA molecules from which we removed the intron and adjoined the exons computationally suggested that the spliced RNA folds into an extended stem loop (Fig 1B, right structure, and Fig EV1B, bottom structures). In this structure, the exon–exon base pairing of S1 is preserved but now is extended by a previously unrecognized new stem ES1 that results from base pairing between the exons. ES1 is a short stem interrupted by embedded bulges. Strikingly, we found that ES1 is conserved even among distant metazoans (Fig EV1B, bottom structures), suggesting that it may be part of an evolutionarily conserved architectural requirement for splicing.
Three‐dimensional structure predictions on human XBP1 mRNA suggested that the loops containing the exon–intron boundaries are placed within ~32 Å of each other (Fig 1C), which renders cleavage of both sites by a single IRE1 dimer sterically improbable. We surmise that IRE1 assemblies that are obligatorily larger than a dimer are necessary to concomitantly engage both splice sites (see Discussion for details).
To determine the functional importance of the features thus identified, we constructed a short RNA transcript harboring the BSL of human XBP1 mRNA (XBP1‐BSL) (Fig EV3A, see Materials and Methods for details). Incubation of XBP1‐BSL with recombinant human IRE1α‐KR43 (containing IRE1α's kinase and RNase domains and a 43‐residue portion of the N‐terminal linker that tethers the kinase domain to IRE1's transmembrane domain) yielded the expected cleavage products on denaturing polyacrylamide gels (Fig 2A). In agreement with previous findings 7, 24, replacement of the guanosine residues 5′ of the scissile bond at either or both splice junctions by adenosines abrogated the cleavage reaction (Fig 2B–D), while not disrupting the predicted secondary structure fold (Fig 2E). Moreover, these results showed that the cleavage of each splice site proceeds independently and with comparable rates (compare Fig 2B and C), suggesting that IRE1 does not cleave the splice junctions in an obligate order.
Figure EV3. Biochemical analysis of the XBP1 splicing product.
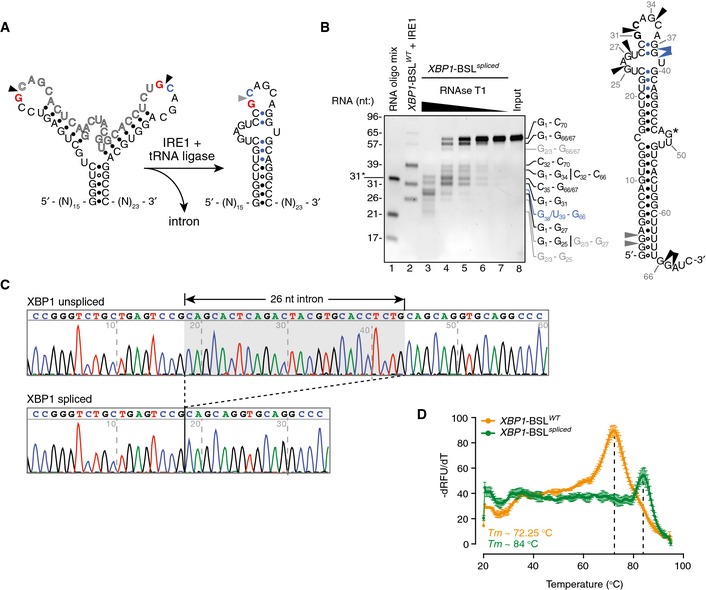
- Secondary structures of the short RNA transcripts harboring the human XBP1‐BSL (left structure) or its corresponding spliced RNA (right structure). Arrowheads: scissile bonds (unspliced structure); exon–exon junction (spliced structure). The exon boundaries are indicated by a red guanosine, a blue cytosine. Blue closed circles: Watson–Crick base pairing between the 5′ and 3′ exons that form ES1.
- Left: TBE–urea gel showing the ribonuclease T1 (RNase T1) mapping of the XBP1‐BSL spliced RNA. RNA ladders: RNA oligo mix (lane 1), and IRE1‐cleaved XBP1‐BSLWT (lane 2). 31* indicates the migration of a 2′‐ACE‐(acid‐labile orthoester)‐protected RNA oligonucleotide. The XBP1‐BSL‐derived RNA ladder was cleaved with 0.5 μM of IRE1α‐KR43. The fragments generated by RNase T1 are indicated on the right side of the gel and color coded according to their origin (see schematic). Right: schematic of the XBP1‐BSL spliced transcript. Blue: closed circles: Watson–Crick base pairs between the 5′ and 3′ exons that form ES1. Black arrowheads: preferred cleavage sites by RNase T1. Blue arrowheads: overdigestion products generated when the distal C‐G pairs of ES1 are melted. Grey arrowheads: possible cleavage products resulting from the melting of G‐U wobble base pairs at the beginning of the main stem S1. G* indicates a single inaccessible single‐stranded guanosine.
- Electropherograms showing the sequencing analysis of in vitro‐generated splice products using XBP1‐BSLWT as a substrate.
- Melting curves of the XBP1‐ BSLWT or XBP1‐ BSL spliced RNA transcripts. The melting curve analysis is consistent with the faster‐than‐expected mobility of the spliced RNA in TBE–urea–PAGE gels (this RNA migrates faster than its predicted molecular size of 70 nt, see B), which suggests it is partially denatured during TBE–urea–PAGE electrophoresis. Mean ± SD.
Figure 2. Cleavage of XBP1 mRNA by IRE1.
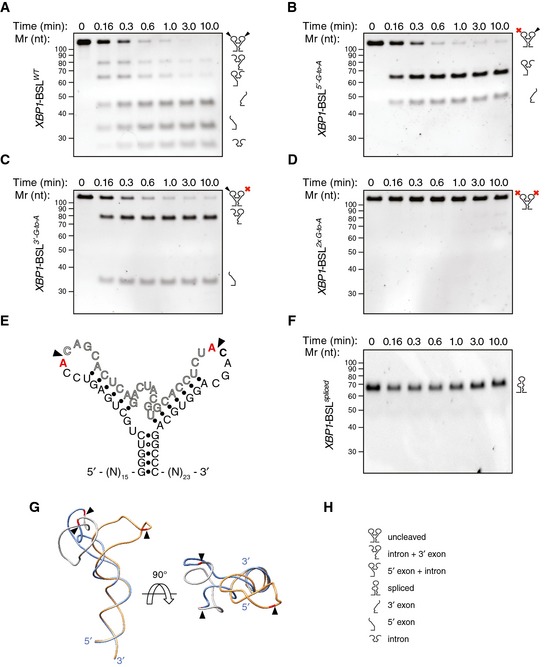
-
ATBE–urea–PAGE gel showing the IRE1‐mediated cleavage pattern of the short XBP1‐BSL transcript of human origin.
-
B–DTBE–urea–PAGE gels showing the IRE1‐mediated cleavage pattern of human XBP1‐BSL cleavage‐site mutants. Red crosses over the schematic of the RNA structures on the right side of each gel indicate G‐to‐A substitutions in the IRE1 cognate sequence within the loops (see E for details).
-
ESecondary structure of cleavage‐site mutants of human XBP1‐BSL. Substituted bases are indicated in red. Arrowheads: scissile bonds.
-
FTBE–urea–PAGE gel showing that IRE1 does not cleave the spliced XBP1‐BSL RNA. The RNAs in (A–D, F) were incubated with 0.5 μM of IRE1α‐KR43 for the indicated times.
-
GSuperimposition of the predicted tertiary structures of unspliced and spliced XBP1‐BSL RNAs used in (A–D, F). Arrowheads: scissile bonds (unspliced structure); exon–exon junction (spliced structure). The intron is colored in grey, and the exons are colored in blue (unspliced structure) or gold (spliced structure). The guanosines 3′ of the scissile bond are colored in red.
-
HPictogram key.
Because of the repeated consensus motif at both splice junctions, IRE1 cleavage and exon–exon ligation restores this sequence element. Secondary structure prediction of the spliced XBP1‐BSL indicated that the newly generated CCG¦CAGC motif would be constrained in a 6‐mer loop rather than the conserved 7‐mer loop of the original splice junctions (Fig 1B). Importantly, IRE1α‐KR43 did not cleave an RNA probe containing the spliced XBP1 sequence (XBP1‐BSLspliced) (Fig 2F), demonstrating that, in order to be cleaved by IRE1α, the consensus sequence must be in the correct structural context. Ribonuclease T1 mapping further confirmed the presence of ES1 in the secondary structure of XBP1‐BSLspliced (Fig EV3B). In additional support of this notion, phylogenetically distant BSLs likewise predicted the restored consensus placed in 5‐ or 6‐membered loops after splicing (Fig EV1B). Thus, formation of ES1, which restrains this loop, serves to render the spliced product resistant to re‐cleavage by IRE1.
Formation of ES1 requires intron displacement and conformational rearrangements that may facilitate the completion of the splicing reaction after IRE1 cleavage. These changes are driven by the new base pairing between exons, which resembles a zipper‐like mechanism. Indeed, the predicted tertiary structure of the spliced RNA suggested that the conformational rearrangements introduced after formation of ES1 would allow the spliced XBP1 stem loop to sway away after its engagement with IRE1 to allow handover for ligation (Fig 2G). We reasoned that such zippering‐up of the exons during formation of ES1 might provide an RNA‐intrinsic switch important for intron removal.
To test this concept, we resolved the IRE1α‐KR43 cleavage products of XBP1‐BSL by native polyacrylamide gel electrophoresis (Fig 3A). Indeed, we observed that, upon IRE1α‐KR43‐catalyzed cleavage, the intron was ejected even in the absence of ligation, while the 5′ and 3′ exons remained base‐paired to each other (Fig 3B). Thus, the extended base pairing of the exons via formation of ES1 may serve as the driving force for melting the exon–intron base pairing in S2 and S3. Hence, exon–exon base pairing may have an additional function than promoting adherence of the ends to be joined together, as previously proposed 24.
Figure 3. Formation of ES1 is required for XBP1 mRNA intron ejection.
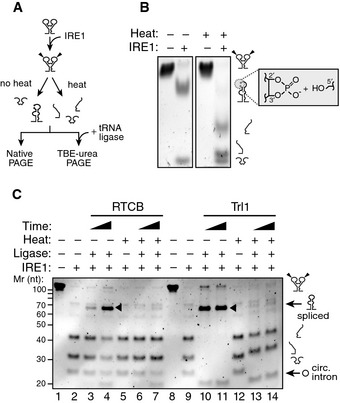
- Schematic of experimental steps for PAGE gels shown in (B, C).
- Native PAGE gel showing intron ejection and exon base pairing after IRE1 cleavage. The grey box indicates the unligated ends generated by IRE1 cleavage: 2′‐3′ cyclic phosphate and 5′ hydroxyl. The RNAs were incubated with 0.5 μM of IRE1α‐KR43 for 10 min prior to thermal denaturation.
- TBE–urea–PAGE gel showing the ligation of IRE1‐cleaved XBP1‐BSL after incubation with the RTCB complex or Trl1 ligase after thermal denaturation. Arrowheads: spliced products. Circ. intron: circularized intron. The RNAs were incubated with 0.5 μM of IRE1α‐KR43 for 5 and 30 min. All reactions with RTCB were supplemented with recombinant archease.
Formation of ES1 is required for intron ejection and efficient splicing of XBP1 mRNA
We next completed the RNA splicing reaction in vitro by inclusion of tRNA ligase (Fig 3A). First, we cleaved XBP1‐BSL with IRE1α‐KR43 and then added mammalian RTCB tRNA ligase complex (RTCB) or yeast tRNA ligase (Trl1) and confirmed the identity of the splice products by sequencing (Fig EV3C). We observed the production of circularized introns irrespective of thermal denaturation prior to ligase addition, indicating that the reaction conditions remained compatible with the ligation reaction (Fig 3C, lanes 4 and 7, and lanes 10–11, 13–14). By contrast, efficient exon–exon ligation only occurred in the absence of thermal denaturation, indicating that the exons must stay together for splicing to occur (Fig 3C, compare lanes 3–4 with 6–7 and 10–11 with 13–14).
While the reaction catalyzed by Trl1 requires a tripartite mechanism consisting of modifying the cleaved ends (i.e. opening the 2′–3′ cyclic phosphate produced by Ire1 and 5′‐end phosphorylation) followed by ligation 25, RTCB employs a direct mechanism where the ends generated by IRE1 are adjoined without prior modification 26. Regardless of the ligase we used, splicing was always abrogated after thermal denaturation, indicating that the base pairing of the exons is a prerequisite irrespective of the biochemistry of the ligation reaction (Fig 3C).
To explore the role of ES1 formation in these reactions, we engineered an XBP1‐BSL mutant, termed XBP1‐BSLNZ (“non‐zippering”), in which base substitutions prohibit the zippering of the exons and formation of ES1 after cleavage by IRE1, but maintain the predicted secondary and tertiary structures of XBP1‐BSL (Figs 4A and EV4A). Cleavage of XBP1‐BSLNZ by IRE1α‐KR43 was indistinguishable from that of the wild‐type XBP1‐BSL, validating the design (Fig 4B). Ribonuclease T1 mapping confirmed that the wild‐type XBP1‐BLS and XBP1‐BSLNZ assume similar structures, as predicted by our computational modeling (Fig EV4B). However, by contrast to the wild‐type XBP1‐BSL, the splicing efficiency of XBP1‐BSLNZ was severely compromised (Fig 4C and D, compare lanes 3–7 to lanes 10–14). This impairment occurred independent of which ligase we used.
Figure 4. Formation of ES1 is required for XBP1 mRNA splicing.
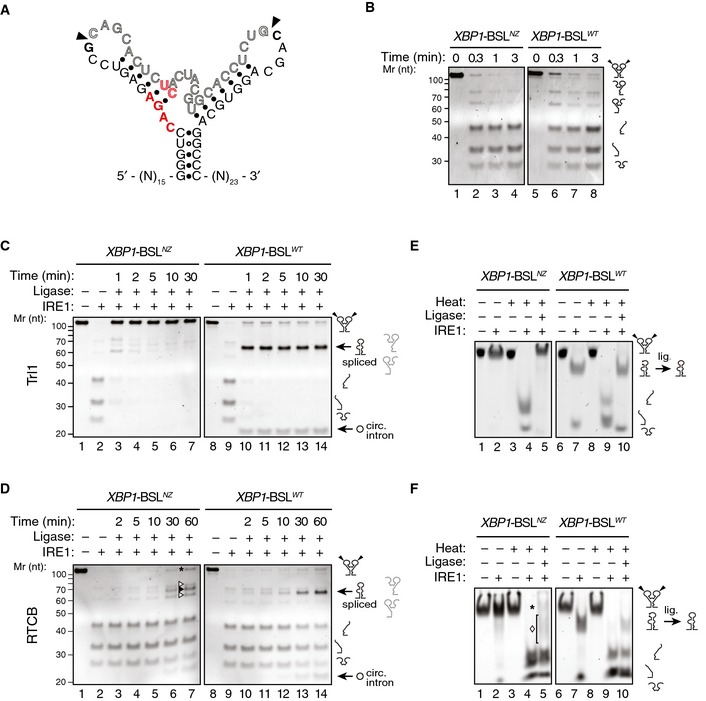
-
ASecondary structure of the mutant XBP1‐ BSLNZ RNA transcript. The base substitutions indicated in red disrupt exon–exon base pairing. Arrowheads: scissile bonds.
-
BTBE–urea–PAGE gels showing cleavage of the XBP1‐ BSLNZ or XBP1‐ BSLWT RNAs by IRE1. The RNAs were incubated with 0.5 μM of IRE1α‐KR43 for the indicated times.
-
C, DTBE–urea–PAGE gels showing IRE1‐mediated cleavage of the XBP1‐ BSLWT or XBP1‐ BSLNZ RNAs and their splicing after incubation of the cleaved RNAs with Trl1 (C), or the RTCB complex (D). All reactions with RTCB were supplemented with recombinant archease. Circ. intron: circularized intron. Asterisk: input RNA (both exons religated to intron). Open arrowheads: incomplete splice products (5′ or 3′ exons ligated to the intron). Closed arrowhead: spliced product. The RNAs in (C, D) were incubated with 0.5 μM of IRE1α‐KR43 for cleavage and with the depicted tRNA ligase for the indicated times. The light grey pictograms on the right of the gels indicate the partially cleaved or incompletely spliced RNAs (containing a single exon and the intron).
-
E, FNative PAGE gels showing IRE1‐mediated cleavage of the XBP1‐ BSLWT or XBP1‐ BSLNZ RNAs and their splicing after subsequent incubation of the cleaved RNAs with Trl1 (E) or the RTCB complex (F). The RNAs in (E, F) were incubated with 0.5 μM of IRE1α‐KR43 prior to thermal denaturation and ligation. The ligation reactions in (F) were supplemented with recombinant archease. Asterisk in (F): input RNA (both exons re‐ligated to intron). Diamond in (F): incompletely spliced products (a single exon ligated to the intron). Note that the spliced product (exon–exon ligation) co‐migrates with the incompletely spliced products, which makes it indistinguishable in native PAGE conditions.
Figure EV4. The non‐zipper mutant of XBP1‐BSL does not disrupt the secondary structure.
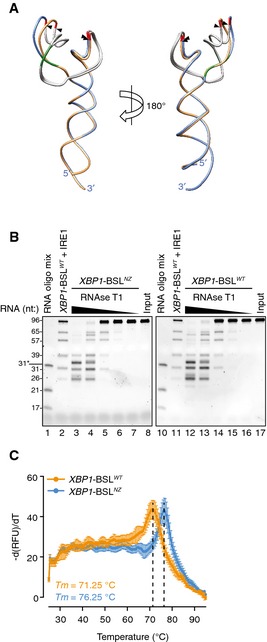
- Superimposition of the predicted tertiary structures of the non‐zipper mutant XBP1‐ BSLNZ (exons colored in blue, ΔG = −43.60 kcal/mol), and the corresponding wild‐type XBP1‐BSL (exons colored in gold, ΔG = −43.10 kcal/mol) RNAs used in this study. Arrowheads: scissile bonds. The intron is colored in grey. The guanosines 3′ of the scissile bond are colored in red. Mutant bases that disrupt the exon–exon base pairing after cleavage are colored in green.
- TBE–urea gels showing the RNase T1 mapping of the XBP1‐ BSLNZ or XBP1‐ BSLWT RNAs. RNA ladders: RNA oligo mix (lanes 1 and 10), and IRE1‐cleaved XBP1‐BSLWT (lanes 2 and 11). 31* indicates the migration of a 2′‐ACE‐(acid‐labile orthoester)‐protected RNA oligonucleotide. The XBP1‐BSL‐derived RNA ladder was cleaved with 0.5 μM of IRE1α‐KR43.
- Melting curves of the XBP1‐ BSLWT or XBP1‐ BSLNZ RNA transcripts. Mean ± SD.
Interestingly, we noted that, whereas ligation of the wild‐type XBP1‐BSL by Trl1 proceeded to the anticipated splicing product, XBP1‐BSLNZ was efficiently re‐ligated at the exon–intron boundaries to restore the input RNA (Fig 4C, lanes 3–7). By contrast, RTCB produced heterogeneous products, resulting from re‐ligation of either single exon to the intron (Fig 4D, lane 7, labeled by open arrowheads), both exons to the intron restoring the substrate (Fig 4D, lane 7, labeled by an asterisk), or exon‐to‐exon resulting in some fully spliced product (Fig 4D, lane 7, labeled by a closed arrowhead). We surmise that the difference between the ligases used may be due to the presence of the additional subunits in RTCB, which include DDX1, a helicase that might assist, albeit inefficiently, in intron removal even when ES1 cannot form.
To show directly that formation of ES1 is important for intron ejection, we next cleaved XBP1‐BSLNZ with IRE1α‐KR43 and analyzed the reaction products on native polyacrylamide gels (Fig 4E and F). Under these conditions, the cleavage products of XBP1‐BSLNZ co‐migrated at the same position as the input RNA unless the sample was heat‐denatured (Fig 4E and F, compare lanes 2 and 4), indicating that XBP1‐BSLNZ indeed failed to eject the intron. Adding Trl1 to XBP1‐BSLNZ in a subsequent reaction after the cleavage reaction was complete led to efficient re‐ligation to restore the input substrate (Fig 4E, lane 5). By contrast, when we used wild‐type XBP1‐BSL as the substrate, addition of Trl1 led to completion of the splicing reaction, consistent with the substrate's capacity to form ES1 and eject the intron (Fig 4E, lane 10). Adding the RTCB complex to XBP1‐BSLNZ after cleavage led to the production of incomplete splice products (Fig 4F, lane 5, labeled by a diamond) and restored some of the input (Fig 4F, lane 5, labeled by an asterisk), whereas the same experiment using wild‐type XBP1‐BSL as a substrate exclusively produced a spliced product, despite the reaction not reaching completion (Fig 4F, lane 10). These results are in agreement with those presented in Fig 4C and D.
Independent biochemical methods suggested that the XBP1‐BSL adopts a predominant structure (Figs EV2 and EV4B). However, computational predictions showed that even before IRE1 cleavage, XBP1‐BSL can adopt more than one secondary structure, which raises the possibility that additional conformers can establish an equilibrium between alternative structures (Fig EV5). By contrast, XBP1‐BSLNZ was predicted to assume only one conformation (Figs 4A and EV4A), locking it into a more rigid conformation that mimics the most stable XBP1‐BSL conformer (Fig EV4B). XBP1‐BSLNZ exhibited a melting temperature that was 5°C higher than that of wild‐type XBP1‐BSL (Fig EV4C). These results suggest that the added structural plasticity of XBP1‐BSL may be important to initiate the zippering of exons leading to ES1 formation and intron ejection. Similar analyses on the spliced XBP1‐BSL yielded an even higher melting temperature (~84°C), indicating the formation of a thermodynamically stable fold after splicing (Fig EV3D).
Figure EV5. Alternative secondary structures for XBP1‐BSL .
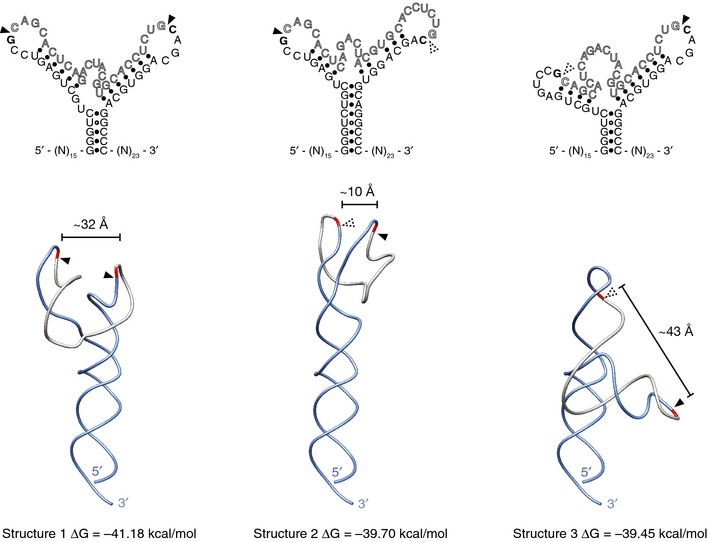
Predicted secondary (top) and tertiary (bottom) structures of the wild‐type human XBP1‐BSL RNA probe used throughout this study. Arrowheads: scissile bonds. Calculated distances between scissile bonds are indicated. Dashed arrowheads: scissile bonds placed in a context distinct from the 7‐mer loop recognized by IRE1.
Formation of ES1 is required for efficient XBP1 splicing in cells
We next asked if the RNA zippering mechanism leading to ES1 formation is important for XBP1 mRNA splicing in vivo. To this end, we introduced a XBP1::GFPvenus reporter bearing the non‐zippering mutations in the BSL into HEK293T cells (Fig 5A). Indeed, as measured by semi‐quantitative multiplex reverse transcription PCR, upon induction of ER stress by thapsigargin (which inhibits calcium re‐uptake into the ER) splicing was severely compromised in cells harboring the XBP1‐BSLNZ version of the reporter when compared to cells harboring the wild‐type version (Fig 5B, compare lanes 1–3 to 4–6). Note that PCR products derived from endogenous XBP1 mRNA provided a convenient internal control (Fig 5B, lanes 7–9). To ascertain the impact of the compromised splicing on the synthesis of the encoded protein product, we conducted immunoblot analyses of lysates of the cells transfected with the aforementioned constructs. Because the constructs encode N‐terminal FLAG epitope tags, we could analyze the accumulation of protein products encoded by the spliced and unspliced RNAs (Fig 5C). Cells expressing the mutant XBP1‐BSLNZ version of the reporter were compromised in generating the product encoded in the spliced mRNA and instead accumulated the product encoded in the unspliced one (Fig 5D, compare lanes 1–2 to 4–5). By late time points, the cells expressing the XBP1‐BSLNZ reporter showed a mild increase in the spliced product (Fig 5D, compare lanes 4–5 to lane 6); however, this accumulation was much reduced when compared to the product encoded by the wild‐type reporter (Fig 5D, compare lanes 3 and 6). This result is consistent with an inefficient conversion rate of the unspliced transcript into its spliced form. Thus, both in vitro and in vivo experiments converge in support of the model that an RNA conformational rearrangement via ES1 formation and intron ejection ensures efficient XBP1 mRNA splicing.
Figure 5. Formation of ES1 is required for efficient XBP1 mRNA splicing in vivo .

- Schematic of XBP1 reporters. Arrows: annealing positions of the oligonucleotides used for the semiquantitative multiplex RT–PCR.
- Agarose gel electrophoresis of multiplex RT–PCR products. Note the progressive loss of the non‐zipper XBP1 reporter mRNA upon ER stress (lanes 4–6), attributable to degradation of incomplete splice products.
- Schematic of the N‐terminus FLAG epitope‐tagged protein products encoded by the reporters in (A).
- Immunoblot analysis of the products encoded by the reporters in (A). Note the accumulation of unspliced product in the cells harboring the non‐zipper XBP1 reporter (lanes 4 and 5) compared to the cells harboring the wild‐type (lanes 1–3). GAPDH: loading control. Asterisk: non‐specific band.
Discussion
The non‐conventional splicing of XBP1 mRNA requires coordinated cleavage and ligation events. Since both exon–intron splice junctions are comprised of stem‐loop structures, base pairs need to be melted to eject the intron prior to ligation of the exons. Here, we show that an RNA‐intrinsic structural rearrangement allows the severed exons to engage in the pairing of bases (leading to the formation of ES1) that before cleavage were engaged in pairing to the intron (in stems S2 and S3). In this way, the exons zipper up into an extended stem (S1‐ES1), juxtaposing the RNA ends to be ligated but constraining the resulting loop so that no functional IRE1 cleavage‐site results. This mechanism ensures the correct RNA ends are presented to the ligase, suggesting that the ligase itself does not discriminate between exon and intron ends as corroborated by the observed exon–intron re‐ligation in the absence of intron ejection. Intron ejection occurs in the absence of ligase and was prevented in a mutant in which ES1 cannot form. Failure to form ES1 impaired mRNA splicing in vivo, indicating that the conformational rearrangements leading to intron ejection are important for XBP1 mRNA splicing in a living cell. These features are highly conserved throughout metazoan evolution, supporting their importance in orchestrating the splicing reaction with efficiency and fidelity.
The energetics of the RNA rearrangements are intriguing because, after IRE1‐cleavage, the XBP1‐BSL RNA settles into a conformation with fewer base pairs (5 base pairs in the newly formed ES1 versus the original 13 base pairs engaging the intron in the uncleaved XBP1‐BSL) that a priori would seem less stable. However, melting temperature analysis of the spliced BSL indicates that this is not the case and that the BSL adopts a thermodynamically stable fold after splicing. Analysis of the melting temperature and the prediction of a dynamic equilibrium between multiple alternative conformational states of the uncleaved XBP1‐BSL RNA suggest that intron ejection and ES1 formation are energetically favored because a more rigid, less plastic structure is formed. This could be attributable, at least in part, to the existence of conformers with partial zippering of the exons (see Fig EV5, middle structure, which shows incomplete formation of ES1). Moreover, both (i) the local concentration of the intron is reduced upon its ejection and, (ii) as suggested by our in vitro data, the intron is circularized by the tRNA ligase following its excision. Both mechanisms would render the back‐reaction substantially less favorable.
Structural models depicting the interaction of yeast Ire1 with substrate RNA postulate recognition of a single stem loop by an RNase‐active Ire1 dimer oriented in a back‐to‐back conformation 21. Both human IRE1 and yeast Ire1 possess high structural similarity 27, 28, suggesting a similar recognition mode, wherein each stem loop of the XBP1‐BSL could be recognized and cleaved by an active enzyme dimer. Our tertiary structure predictions place the scissile bonds in apposition and about 32 Å apart from each other (Fig 1C), suggesting that this particular RNA architecture allows coordination of both cleavage events by independent IRE1 dimers or higher‐order structures. This notion is in line with work from our laboratory, showing that higher‐order IRE1 assemblies are required for full activation of IRE1's ribonuclease activity in yeast and mammals 8, 29, 30. Moreover, as previously shown for yeast Ire1, only one stem loop can be accommodated in the dimeric RNase‐active site 21 and human IRE1 has a highly similar geometry (PDB ID 4Z7G) 31, further substantiating the need for independent RNase‐active sites. We speculate that the coordinated cleavage could be carried out by two adjacent dimers within an IRE1 oligomer in which the active sites are spaced by ~40 Å (yeast IRE1: PDB ID 3FBV) 8.
IRE1 exerts both corrective and pre‐emptive tasks through XBP1 mRNA splicing and RIDD, respectively. Both functions rely on the processing of ER‐bound mRNAs by IRE1: one involving a single or multiple non‐coordinated endonucleolytic cleavage events with low sequence or structural requirements leading to mRNA decay as occurs during RIDD, and the other requiring two precisely coordinated cleavage events that must present the correct substrate for the ligase to complete the XBP1 mRNA splicing reaction. Taken together, our data support the concept of an evolutionarily conserved structural RNA rearrangement that is hard‐wired in XBP1 mRNA as a fundamentally important element to shunt XBP1 mRNA into the splicing pathway. This defines XBP1 mRNA not as a static, passive substrate but as an active and dynamic element instrumental to the metazoan UPR.
Materials and Methods
Computational analyses
Homologous sequences containing the XBP1 BSL structure (intron sequences plus upstream and downstream flanking ~20‐mers) of 34 metazoans were retrieved from the National Center for Biotechnology Information (NCBI) using the Basic Local Alignment Search Tool (BLAST) 32 and assembled into a phylogenetically diverse list that includes vertebrates (mammals, birds, reptiles, amphibians and fishes), insects, and nematodes for multiple sequence alignment using the web‐based program ClustalW (European Molecular Biology Laboratory, European Bioinformatics Institute) 33. The intron–exon boundaries with the XBP1 BSL sequences were visually annotated by direct comparison to those in the XBP1 BSL of human origin. Secondary structure predictions of conserved BSL sequences were performed with the mFold web server (The RNA Institute, University at Albany, State University of New York) 34. We chose the secondary structures with the highest free energies computed as the sum of free energies assigned to all the loops and base pair stacks as determined by mFold for subsequent analyses. Secondary structure‐guided three‐dimensional structure predictions were performed using the RNAComposer modeling server (Bioserver, Institute of Computing Science, Poznan University of Technology and the Institute of Bioorganic Chemistry, Polish Academy of Sciences) 35. Parallel analysis of RNA structure (PARS) scores were computed for the transcript encoding unspliced XBP1 of human origin (Accession NM_005080) as previously described 22, 23 using the dataset in the Gene Expression Omnibus accession GSE50676. icSHAPE reactivities for mouse XBP1 mRNA were retrieved from the Gene Expression Omnibus accession GSE64169. The custom tracks (BigWig files) were uploaded into the University of California at Santa Cruz (UCSC) Genome browser (http://genome.ucsc.edu, and 36) and displayed on the 2011 mouse genome assembly (UCSC mm10; Genome Reference Consortium GRCm38).
Synthesis of short XBP1‐BSL transcripts
Long sense and antisense oligonucleotides containing a minimal T7 RNA polymerase promoter (5′‐TAATACGACTCACTATAGG‐3′) fused upstream of the sequence containing the XBP1 BSL of human origin (nucleotides 513–592 of NCBI Reference Sequence NM_005080.3) and an RNA polymerase III terminator sequence (5′‐TGGCTTTTT‐3′) of RNY4 of human origin (NCBI Reference Sequence NR_004393.1) fused downstream of the XBP1 BSL sequence and harboring 5′‐EcoRI and 3′‐BamHI overhangs were annealed and ligated into the cognate restriction sites of pUC19 (Invitrogen, Life Technologies). The resulting clone, pUC19‐T7‐hXBP1‐BSL‐Y4, was digested with BamHI, purified and used as a template for in vitro transcription reactions with T7 RNA polymerase using the HiScribe T7 high‐yield RNA synthesis kit (New England Biolabs) following the manufacturer's recommendations. The transcribed RNA was purified by urea‐polyacrylamide gel electrophoresis (urea–PAGE), and the RNA, recovered from gel fragments by the crush‐and‐soak method, was precipitated with 300 mM NaOAc and 1 volume of isopropanol. No co‐precipitants were employed. The precipitated RNA pellet was desalted by two washes with 70% ice‐cold ethanol, air‐dried and re‐suspended in an appropriate volume of either nuclease‐free water or RNA resuspension buffer (20 mM HEPES, 100 mM NaCl, 1 mM Mg(OAc)2). Mutant XBP1 BSL probes were constructed by standard site‐directed mutagenesis of the pUC19‐T7‐hXBP1‐BSL‐Y4 wild‐type clone using mutagenic oligonucleotides and Phusion high‐fidelity DNA polymerase (New England Biolabs). Mutant RNAs were transcribed in vitro and purified as described above.
RNA melting curves
Approximately 250–300 ng (for XBP1‐BSLWT or XBP1‐BSLNZ) or 500 ng (for XBP1‐BSLspliced) of in vitro transcribed RNAs was employed for melting curve analyses with 1× SYBR Gold nucleic acid stain (Invitrogen, Life Technologies) in 20 μl reactions using RNA resuspension buffer (20 mM HEPES, 100 mM NaCl, 1 mM Mg(OAc)2). The RNAs were heated to 90°C for 3 min and then cooled down to 25°C at a rate of 0.1°C/s. The RNAs were then melted in a CFX96 real‐time PCR thermocyler (BioRad), and melting curve data points were taken every 0.5°C. To find the temperature of dissociation, the negative of the first derivative was plotted as a function of temperature.
Protein expression and purification
The cytosolic kinase/ribonuclease domain construct of IRE1‐α (KR43) was expressed and purified as described previously 37.
The Chaetomium thermophilum tRNA ligase Trl1 (NCBI Entrez Gene ID: 18257519, CTHT_0034810 tRNA ligase‐like protein) was cloned from C. thermophilum cDNA into pET15b (EMD Millipore) using the restriction enzymes NdeI and EcoRV. Recombinant, His6‐tagged CtTrl1 was expressed in E. coli BL21‐CodonPlus (DE3)‐RIL (Agilent Technologies) and purified by Ni2+ affinity chromatography using a HisTrap FF column (GE Healthcare Life Sciences), followed by size‐exclusion chromatography using a HiLoad 16/60 Superdex200 pg column (GE Healthcare Life Sciences) in 20 mM Tris/HCl pH 7.1, 300 mM NaCl, and 1 mM MgCl2.
Human archease constructs were cloned as described elsewhere 38. His6‐tagged archease was expressed in E. coli BL21‐CodonPlus (DE3)‐RIPL (Agilent Technologies) and purified by Ni2+ affinity chromatography using a HisTrap FF column (GE Healthcare Life Sciences), followed by tag removal with thrombin (Sigma‐Aldrich) and a final size‐exclusion chromatography step using a HiLoad 16/60 Superdex200 pg column (GE Healthcare Life Sciences) in 25 mM Tris/HCl pH 7.5, 100 mM NaCl, 5% glycerol, and 1 mM TCEP. The human RTCB complex was immunoprecipitated from HEK293 cells expressing FLAG‐RTCB as described elsewhere 38.
All proteins were flash‐frozen in liquid nitrogen directly after purification and kept at −80°C.
In vitro cleavage and splicing assays
In vitro transcribed, PAGE‐purified, refolded RNAs (50 nM) were incubated with 0.5 μM IRE1α‐KR43 for the indicated times in RNA cleavage buffer (20 mM HEPES pH 7.5, 70 mM NaCl, 2 mM Mg(OAc)2, 1 mM TCEP, and 5% glycerol). Stop solution (10 M urea, 0.1% SDS, 1 mM EDTA, 0.05% xylene cyanol, 0.05% bromophenol blue) was added at five‐fold excess to stop the reactions followed by heating at 80°C for 3 min. The denatured samples were then loaded on 15% TBE–urea gels (Invitrogen, Life Technologies) and the gels stained with SYBR Gold nucleic acid stain (Invitrogen, Life Technologies).
For splicing assays, the cleavage reactions were performed as described above and subsequently stopped by addition of the IRE1 inhibitor 4μ8C (CAS No. 14003‐96‐4; Matrix Scientific, Cat. No. 037985) to a final concentration of 5 μM. Ligation of the cleaved RNA was initiated with 500 nM CtTrl1 (1 mM ATP, 1 mM GTP) or mammalian RTCB complex (1:5 dilution of FLAG‐RTCB eluate, 500 nM recombinant archease, 1 mM GTP, 250 μM MnCl2). Reactions were analyzed by denaturing urea–PAGE (after addition of stop solution, as described above) or native PAGE (see next paragraph).
Native polyacrylamide gel electrophoresis
Cleavage and splicing reactions were carried out as described above, and the reaction products were loaded on 6% Tris‐borate DNA retardation gels (Invitrogen, Life Technologies) using native RNA loading buffer (15% Ficoll w/v, 84% Tris‐borate‐EDTA (TBE), 0.5% bromophenol blue, 0.5% xylene cyanol). The gels were run in pre‐chilled 0.5× TBE at 100 V (constant voltage) and 4°C for 100 min. The gels were subsequently stained with SYBR Gold nucleic acid stain (Invitrogen, Life Technologies) to visualize the RNA‐containing complexes by ultraviolet trans‐illumination.
Sequencing of splicing products
Splicing reactions using the XBP1‐BSLWT RNA as a substrate were carried out and the samples separated in urea–PAGE gels as described in “In vitro cleavage and splicing assays”. A control reaction for sequencing the unspliced RNA was set up aside with no IRE1 and no ligase. The RNAs of interest (unspliced and complete splice product) were then cut out from the gels and purified by the crush‐and‐soak method followed by precipitation. GlycoBlue (Ambion, Life Technologies) was added as a co‐precipitant. The purified RNAs were resuspended in 20 μl of nuclease‐free water, and 9 μl of the RNA solution was used for reverse transcription using SuperScript III (Invitrogen, Life Technologies) in 20 μl reactions following the manufacturer's recommendations. The oligonucleotide for reverse transcription was T7‐hXBP1‐Y4_hyb_M13R: 5′‐ GTCGTGACTGGGAAAACGATCCAAAAAGCCAGT‐3′. About 50% of the cDNA was the used as template for PCR amplification using Phusion high‐fidelity DNA polymerase (New England Biolabs) and oligonucleotides T7‐hXBP1‐F: 5′‐ TAATACGACTCACTATAGGGAGGCCAGTGGC‐3′ and T7‐hXBP1‐Y4_hyb_M13R. The resulting PCR products were purified using DNA Clean & Concentrator‐5 columns (Zymo Research) and eluted in 12 μl. The purified PCR products (2 μl) were cloned into pCRII‐Blunt‐TOPO (Invitrogen, Life Technologies) following the manufacturer's recommendations and transformed into competent DH5α E. coli. About 10 colonies from each transformation were sequenced using the Sanger method.
RNA structure determination using ribonuclease T1
Approximately 25–30 ng of in vitro transcribed RNAs were resuspended in RNA re‐suspension buffer (20 mM HEPES, 100 mM NaCl, 1 mM Mg(OAc)2) and refolded. The RNAs were incubated with increasing concentrations of biochemistry grade RNase T1 (Ambion, Life Technologies) for 15 min at 20°C, and the reactions were immediately stopped with stop solution (10 M urea, 0.1% SDS, 1 mM EDTA, 0.05% xylene cyanol, 0.05% bromophenol blue) at 2‐fold excess followed by heating at 80°C for 3 min. The denatured samples were loaded on 15% TBE–urea gels (Invitrogen, Life Technologies) and the gels stained with SYBR Gold nucleic acid stain (Invitrogen, Life Technologies). The final RNase T1 concentrations were (in U/μl) as follows: 0.1, 0.03, 0.01, 0.003, and 0.001. RNA ladders included a mixture of three PAGE‐purified synthetic RNA oligonucleotides (a 31‐mer, GE Healthcare Dharmacon Inc.; a 21‐mer, Integrated DNA Technologies; and a 17‐mer, Integrated DNA Technologies) or IRE1‐cleaved XBP1‐BSLWT.
Mammalian expression constructs and cell transfection
The coding sequence of an ER stress reporter construct consisting of an N‐terminal FLAG epitope‐tagged partial coding sequence of XBP1 of human origin containing the IRE1‐cognate intron and the BSL structure, and fused to the venus variant of GFP (kind gift of Takao Iwawaki, RIKEN) 39, was used as a template to generate retroviral expression constructs containing both the wild‐type XBP1‐BSL and the non‐zipper XBP1‐BSLNZ mutant. The wild‐type sequence was amplified by PCR using Phusion high‐fidelity DNA polymerase (New England Biolabs) using oligonucleotides with 5′‐XhoI and 3′‐EcoRI engineered restriction sites. The resulting PCR product was cloned into the cognate sites of the mammalian expression vector pLPCX (Clontech). To generate the mutant XBP1‐BSLNZ construct, a fusion PCR strategy was employed. Left and right arm PCR products were generated with mutagenic oligonucleotides. The arms were then fused in a second PCR employing the outermost oligonucleotides with 5′‐XhoI and 3′‐EcoRI engineered restriction sites. The resulting PCR product harboring the mutant XBP1‐BSLNZ‐GFPvenus coding sequence was cloned into the cognate sites of the mammalian expression vector pLPCX (Clontech). HEK293T cells were transfected with 1 μg of either plasmid using Lipofectamine 2000 (Life Technologies) following the manufacturer's recommendations. The cells were diluted 1:3 24 h after transfection and re‐seeded onto 6‐well plates. After an additional 24 h, the cells were treated with the 200 nM of ER stress inducing agent thapsigargin (Sigma‐Aldrich) for 2 or 4 h. The sequences of the oligonucleotides used are the following (restriction enzyme sites and mutant bases are underlined): XhoI‐FLAG‐Hs‐XBP1‐Fwd: 5′‐ATTAATCTCGAGCCACCATGGACTACAAGGACGACGAT‐3′; GFPvenus‐EcoRI‐Rev: 5′‐GCCGGCGAATTCTTACTTGTACAGCTCGT‐3′; Hs‐XBP1‐BSLNZ‐Sense: 5′‐AGTGGCCGGGTCCAGAGAGTCCGCAGCACTCTCACTACGTGCACCT‐3′; and Hs‐XBP1‐BSLNZ‐Antisense: 5′‐AGGTGCACGTAGTGAGAGTGCTGCGGACTCTCTGGACCCGGCCACT‐3′.
cDNA generation and multiplex semi‐quantitative PCR
Transfected cells exposed to DMSO or thapsigargin were collected in 1 ml of TRIzol reagent (Life Technologies), and total RNA was extracted following the manufacturer's recommendations. To generate cDNAs, 500 ng of total RNA were reverse transcribed using the SuperScript VILO system (Life Technologies) following the manufacturer's recommendations. The resulting 20 μl reverse transcription reactions were diluted to 200 μl with 10 mM Tris–HCl pH 8.2, and 1% of this dilution was used for multiplex semi‐quantitative PCR. The multiplex PCR was set up using 1 μM of the common forward oligonucleotide and 0.5 μM of each of the gene‐specific reverse oligonucleotide, 0.4 units of Taq DNA polymerase (Thermo Scientific), 0.2 mM of each dNTP, and 1.5 mM MgCl2, in a 20 μl reaction using the following buffer system: 75 mM Tris–HCl pH 8.8, 20 mM (NH4)SO4, and 0.01% Tween‐20. The oligonucleotide sequences are the following: Hs_XBP1_Fwd: 5′‐GGAGTTAAGACAGCGCTTGG‐3′; Hs_XBP1_Rev: 5′‐ACTGGGTCCAAGTTGTCCAG‐3′; eGFP_Rev: 5′‐AAGTCGTGCTGCTTCATGTG‐3′. PCR products were amplified for 28 cycles and resolved on 2.5% agarose gels (1:1 mixture of regular and low‐melting point agarose) stained with ethidium bromide.
Immunoblotting
Total cell lysates were collected in SDS sample buffer (62.5 mM Tris pH 6.8, 10% glycerol, 2% SDS, 0.004% bromophenol blue). Lysates were sonicated for ~15 s to shear the genomic DNA. 2‐mercaptoethanol was added to a final concentration of 5% to the lysates just prior to boiling and loading on SDS–PAGE gels. Proteins were separated by electrophoresis and transferred onto 2.0 μm pore nitrocellulose membranes. The blocked membranes were probes with a mouse monoclonal anti‐FLAG antibody (clone M2, Sigma‐Aldrich F1804, 1:1,000) or an anti‐GAPDH rabbit polyclonal antibody (Abcam ab9485, 1:2,000). Immunoreactive bands were detected using HRP‐conjugated secondary antibodies (Amersham, GE Healthcare Life Sciences NA931, NA934, 1:5,000) and luminol‐based enhanced chemiluminescence substrates (SuperSignal West Dura Extended Duration Substrate, Pierce, Life Technologies) and exposed to radiographic film or imaged directly in a digital gel imager (Chemidoc XRS+, BioRad). Digital images were automatically adjusted for contrast using the photo editor Adobe Photoshop (Adobe Systems).
Author contributions
JP and DAA performed experiments and analyzed data. ASM purified human IRE1‐KR43 and helped with in vitro assays. JP, DAA, and PW designed the research and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We are grateful to Javier Martinez for providing us with a FLAG‐RTCB‐expressing HEK293 cell line; Howard Y. Chang and Ryan A. Flynn for help with PARS/icSHAPE data analysis; Voytek Okreglak for Chaetomium thermophilum cDNA and helpful advice; and Elif Karagöz, Silvia Ramundo, Weihan Li, Han Tran, and members of the Walter Laboratory for insightful discussions. This work was supported by a Long‐Term Postdoctoral Fellowship by the Human Frontiers Science Program (JP), and an Irvington Postdoctoral Fellowship of the Cancer Research Institute (DAA). PW is an Investigator of the Howard Hughes Medical Institute.
EMBO Reports (2015) 16: 1688–1698
References
- 1. Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086 [DOI] [PubMed] [Google Scholar]
- 2. Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519–529 [DOI] [PubMed] [Google Scholar]
- 3. Haze K, Yoshida H, Yanagi H, Yura T, Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10: 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature 397: 271–274 [DOI] [PubMed] [Google Scholar]
- 5. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP‐1 mRNA. Nature 415: 92–96 [DOI] [PubMed] [Google Scholar]
- 6. Shen X, Ellis RE, Lee K, Liu C‐Y, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K et al (2001) Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107: 893–903 [DOI] [PubMed] [Google Scholar]
- 7. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891 [DOI] [PubMed] [Google Scholar]
- 8. Korennykh AV, Egea PF, Korostelev AA, Finer‐Moore J, Zhang C, Shokat KM, Stroud RM, Walter P (2009) The unfolded protein response signals through high‐order assembly of Ire1. Nature 457: 687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Korennykh A, Walter P (2012) Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol 28: 251–277 [DOI] [PubMed] [Google Scholar]
- 10. Kawahara T, Yanagi H, Yura T, Mori K (1998) Unconventional splicing of HAC1/ERN4 mRNA required for the unfolded protein response sequence‐specific and non‐sequential cleavage of the splice sites. J Biol Chem 273: 1802–1807 [DOI] [PubMed] [Google Scholar]
- 11. Cox JS, Walter P (1996) A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87: 391–404 [DOI] [PubMed] [Google Scholar]
- 12. Sidrauski C, Cox JS, Walter P (1996) tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87: 405–413 [DOI] [PubMed] [Google Scholar]
- 13. Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, Heindl K, Hoffmann T, Busslinger M, Martinez J (2014) The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J 33: 2922–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kosmaczewski SG, Edwards TJ, Han SM, Eckwahl MJ, Meyer BI, Peach S, Hesselberth JR, Wolin SL, Hammarlund M (2014) The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep 15: 1278–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. u Y, Liang F‐X, Wang X (2014) A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell 55: 758–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Acosta‐Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD (2007) XBP1 controls diverse cell type‐ and condition‐specific transcriptional regulatory networks. Mol Cell 27: 53–66 [DOI] [PubMed] [Google Scholar]
- 17. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER‐associated degradation. Cell 101: 249–258 [DOI] [PubMed] [Google Scholar]
- 18. Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum‐localized mRNAs during the unfolded protein response. Science 313: 104–107 [DOI] [PubMed] [Google Scholar]
- 19. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS (2009) Regulated Ire1‐dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186: 323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hooks KB, Griffiths‐Jones S (2011) Conserved RNA structures in the non‐canonical Hac1/Xbp1 intron. RNA Biol 8: 552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Korennykh AV, Korostelev AA, Egea PF, Finer‐Moore J, Stroud RM, Zhang C, Shokat KM, Walter P (2011) Structural and functional basis for RNA cleavage by Ire1. BMC Biol 9: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E (2010) Genome‐wide measurement of RNA secondary structure in yeast. Nature 467: 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E et al (2014) Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505: 706–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gonzalez TN, Sidrauski C, Dörfler S, Walter P (1999) Mechanism of non‐spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J 18: 3119–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greer CL, Peebles CL, Gegenheimer P, Abelson J (1983) Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell 32: 537–546 [DOI] [PubMed] [Google Scholar]
- 26. Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Lührmann R, Söll D et al (2011) HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 331: 760–764 [DOI] [PubMed] [Google Scholar]
- 27. Ali MMU, Bagratuni T, Davenport EL, Nowak PR, Silva‐Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W et al (2011) Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J 30: 894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee KPK, Dey M, Neculai D, Cao C, Dever TE, Sicheri F (2008) Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132: 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aragón T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P (2009) Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 457: 736–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H, Korennykh AV, Behrman SL, Walter P (2010) Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci USA 107: 16113–16118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Joshi A, Newbatt Y, McAndrew PC, Stubbs M, Burke R, Richards MW, Bhatia C, Caldwell JJ, McHardy T, Collins I et al (2015) Molecular mechanisms of human IRE1 activation through dimerization and ligand binding. Oncotarget 6: 13019–13035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- 33. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- 34. Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Popenda M, Szachniuk M, Antczak M, Purzycka KJ, Lukasiak P, Bartol N, Blazewicz J, Adamiak RW (2012) Automated 3D structure composition for large RNAs. Nucleic Acids Res 40: e112–e112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D (2002) The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mendez AS, Alfaro J, Morales‐Soto MA, Dar AC, McCullagh E, Gotthardt K, Li H, Acosta‐Alvear D, Sidrauski C, Korennykh AV et al (2015) Endoplasmic reticulum stress‐independent activation of unfolded protein response kinases by a small molecule ATP‐mimic. eLife 4: e05434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Popow J, Jurkin J, Schleiffer A, Martinez J (2014) Analysis of orthologous groups reveals archease and DDX1 as tRNA splicing factors. Nature 511: 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iwawaki T, Akai R, Kohno K, Miura M (2004) A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med 10: 98–102 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File