

Abstract
c‐Myc is one of the major human proto‐oncogenes and is often associated with tumor aggression and poor clinical outcome. Paradoxically, Myc was also reported as a suppressor of cell motility, invasiveness, and metastasis. Among the direct targets of Myc are many components of the protein synthesis machinery whose induction results in an overall increase in protein synthesis that empowers tumor cell growth. At present, it is largely unknown whether beyond the global enhancement of protein synthesis, Myc activation results in translation modulation of specific genes. Here, we measured Myc‐induced global changes in gene expression at the transcription, translation, and protein levels and uncovered extensive transcript‐specific regulation of protein translation. Particularly, we detected a broad coordination between regulation of transcription and translation upon modulation of Myc activity and showed the connection of these responses to mTOR signaling to enhance oncogenic transformation and to the TGFβ pathway to modulate cell migration and invasiveness. Our results elucidate novel facets of Myc‐induced cellular responses and provide a more comprehensive view of the consequences of its activation in cancer cells.
Keywords: c‐Myc, transformation, metastasis, transcriptional responses, translational regulation
Subject Categories: Cancer, Protein Biosynthesis & Quality Control, Transcription
Introduction
c‐Myc (here termed Myc) is a major oncogene—it is overexpressed and/or activated in more than half of human cancers and is often associated with tumor aggression and poor clinical outcome 1, 2, 3, 4, 5, 6, 7. Myc activation enhances key processes that contribute to tumorigenesis, including cell proliferation and growth, persistent DNA replication, protein biogenesis, and angiogenesis 8, 9, 10, 11, 12. Suppression of Myc expression back to its physiological levels results in tumor regression in a wide variety of cancers, including hematopoietic, epithelial, and mesenchymal tumors 7, 13, 14, 15, 16. Paradoxically, notwithstanding its strong oncogenic role, Myc was recently indicated as a suppressor of cancer metastasis 17. This effect was attributed to Myc‐mediated repression of αV and β3 integrin subunits that results in reduced cell adhesion to ECM ligands, thus attenuating cell motility and invasiveness.
Myc is a basic helix–loop–helix (bHLH) transcription factor that forms a heterodimer with Max and binds to E‐box sequences (canonical consensus 5′‐CACGTG‐3′) near the promoter elements of actively transcribed genes 18. Many gene expression profiling studies have identified hundreds of Myc target genes in a variety of tumor cells 19, 20, 21, 22, 23. Recently, it was suggested that oncogenic Myc is an amplifier, rather than a specifier, of gene expression in cancer cells. This conclusion was based on the observation that in tumor cells expressing high levels of Myc, it amplifies the output of the existing gene expression program rather than inducing the expression of a new set of genes 24, 25, 26. In addition to regulation of transcription initiation, Myc was also shown to stimulate transcription elongation at certain genes 26, 27, 28, 29, 30, 31. Recently, Myc was reported to also directly repress transcription initiation of specific target genes, and it was suggested that this negative effect is mediated by complex formation of Myc and MIZ1 in promoters of the repressed genes 32.
Given its pivotal role in tumorigenesis, Myc is one of the most vastly studied human TFs, and the effect of its activation on cellular transcriptomes was thoroughly examined. In this study, we set out to elucidate novel facets of Myc‐induced responses and explore the effects of its activation on regulation of protein translation. Global enhancement of protein biogenesis upon Myc activation is well documented. Among the direct targets of Myc are many components of the protein synthesis machinery, including translation initiation and elongation factors, tRNA synthetases, and ribosomal proteins of the small and large ribosome subunits 33, 34. The induction of these target genes results in an overall increase in cellular protein synthesis that empowers cell growth 35. However, at present, it is largely unknown whether beyond this global effect on protein synthesis, Myc activation also results in translation modulation of specific transcripts, and how such responses might relate to its oncogenic and metastatic‐repressive effects. There are several preliminary and intriguing indications for transcription‐independent Myc effects on protein translation 36, 37, yet to date such possible functions of Myc have not been systematically explored.
Here, we applied, in parallel, RNA‐seq and Ribo‐seq (also known as ribosome profiling 38, 39) analyses and uncovered an extensive transcript‐specific regulation of protein translation induced by Myc, in addition to the induction of a wide transcriptional response network. Interestingly, we demonstrate that for dozens of Myc‐responsive transcripts (both induced and repressed), protein translation is modulated in a coordinated manner to augment the effect exerted on their transcription, indicating a mechanistic coupling between the regulation of the transcriptional and translational layers of gene expression upon Myc activation. This coordination between the transcriptional and protein translation responses reinforces Myc effects that on the one hand strongly enhance cancer transformation while on the other hand suppress cell motility and invasiveness.
Results
The effect of Myc activation on cancer‐cell transcriptomes was previously characterized by many studies. Here, we set out to systematically delineate transcript‐specific effects of Myc activation on protein translation. To this goal, we performed, in parallel, RNA‐seq and Ribo‐seq analyses on U2OS cells containing an inducible Myc expression vector (U2OS‐Mycind; 32) that were induced or mock‐treated in duplicates for 36 h. We selected this relatively late time point in order to obtain global snapshots of the Myc response network that is probably mediated by multiple secondary regulators. We confirmed that, at this time, Myc did not trigger phenotypic cellular changes such as cell cycle arrest or cell death. We carried out the experiment using independent duplicates. We readily detected the expression of 8,553 genes in the combined RNA‐seq and Ribo‐seq datasets (Tables EV1 and EV2) and observed high correlation between the Myc‐induced responses in the two repeats (see Materials and Methods and Fig EV1A–C). We also observed that the Myc response measured by Ribo‐seq was highly correlated with the response measured by RNA‐seq (Fig EV1D), indicating, as expected, that a large portion of the alterations in protein translation upon Myc induction reflected corresponding changes in mRNA levels of the encoding genes. Overall, we identified 724 and 616 genes that consistently responded to Myc induction in either the RNA‐seq or Ribo‐seq datasets, respectively (Fig EV1E). A total of 368 genes overlapped these two sets; thus, the combined dataset detected 972 unique genes that responded to Myc activation at either the RNA‐ or the protein translation level (or both).
Figure EV1. Analysis of Myc‐induced responses at the transcriptome, translatome and proteome levels.
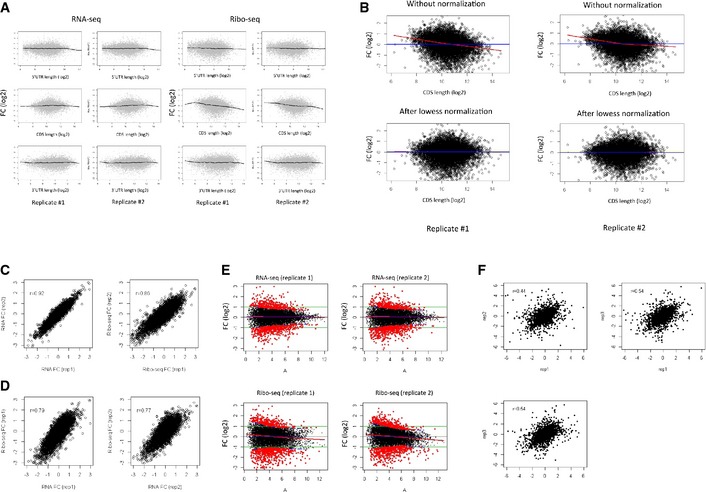
- Relationship between Myc‐induced gene responses measured by either RNA‐seq (left) or Ribo‐seq (right) and transcript 5′UTR (top), CDS (middle), and 3′UTR (bottom) lengths. A global relationship was detected between Ribo‐seq fold change (FC, in log2 scale) and CDS length.
- The technical relationship between Ribo‐seq fold change and transcripts CDS length was normalized using lowess normalization.
- Highly correlated Myc‐induced responses were observed for biologically independent duplicate RNA‐seq (left) and Ribo‐seq (right) experiments.
- The Myc‐induced response measured by Ribo‐seq was, as expected, highly correlated with the one measured by RNA‐seq.
- An adjusted fold‐change threshold was set to define Myc‐responsive genes: The standard deviation of fold change (at a log2 scale) was obtained as a function of expression level, and responsive genes (red dots) were defined as those whose fold change was at least 1.5‐fold the SD above mean (dashed blue lines). A total number of 724 and 616 genes consistently responded to Myc activation in the RNA‐seq (top) and Ribo‐seq (bottom) duplicate experiments, respectively (with an overlap of 368 genes).
- Correlations between Myc‐induced protein‐level fold‐change estimates obtained by three independent proteomic assays.
Next, we subjected the set of Myc‐responsive genes to cluster analysis. This analysis delineated two main regulatory patterns in both the Myc‐induced and repressed expression programs (Fig 1A; Table EV2). The first pattern (cluster #1 up and down) includes genes whose change in ribosome occupancy mirrored to a large extent the change observed in their mRNA level. Thus, in response to Myc activation, these genes were mainly regulated at the RNA level (i.e., regulation of gene transcription or transcript stability). The second pattern (cluster #2 up and down) includes genes whose alteration in ribosome occupancy was in the same direction but augmented compared to the change in their mRNA level. Interestingly, this response pattern suggests that Myc induction activates feedforward loops that couple the regulation of mRNA levels and translation rates (Fig 1B). To further characterize these two Myc‐induced regulatory modes, we calculated changes in transcripts' translation efficiency (TE) and compared them with alterations in mRNA level (Fig 1C). Genes assigned to cluster #1 (up or down) showed a significant change in mRNA level and only a negligible change in their TE, while genes assigned to cluster #2 showed coordinated changes in their mRNA level and TE. This observation suggests that despite the fact that genes assigned to cluster #2 showed reduced alteration in mRNA levels compared to genes assigned to cluster #1, at protein level they should show more similar response, as the difference in mRNA alteration should be compensated by the changes in TE of genes assigned to cluster #2. To examine this expectation, we carried out a proteomic analysis of the same biological conditions that were probed by RNA‐and Ribo‐seq. We performed the proteomic analysis using independent triplicates. (Proteomic measurements were noisier than RNA‐seq and Ribo‐seq ones and showed lower correlations between repeats (Fig EV1F).) The combined datasets containing measurements of the three techniques included 4,665 genes/proteins (Table EV3). The proteomic results confirmed our expectation and demonstrated that the difference in Myc‐induced protein‐level alterations between genes assigned to clusters #1 and #2 was narrowed compared to their difference in mRNA‐level responses (Fig 1D and E).
Figure 1. Regulatory patterns in the Myc response network.
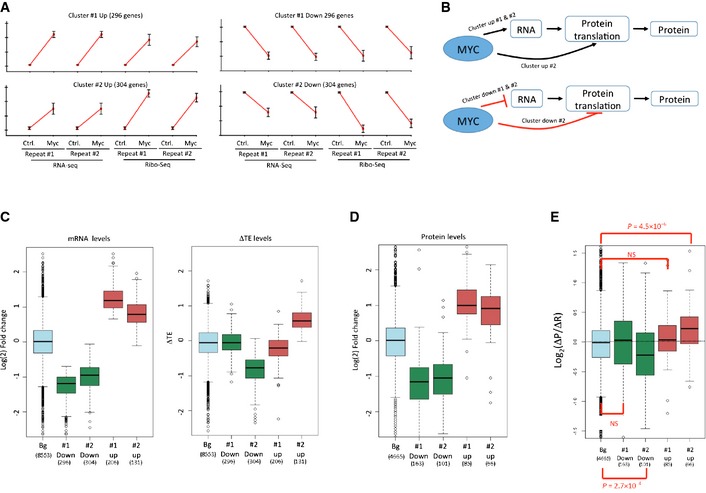
- Two main regulatory patterns detected by cluster analysis for the Myc‐induced (left) and Myc‐repressed (right) genes. For each gene, changes in RNA‐seq and Ribo‐seq levels were standardized to mean = 0 and SD = 1 prior to clustering, such that genes assigned to the same cluster share similar response patterns but might differ in their response magnitude. (y‐axis represents standardized levels.) Each cluster plot shows the mean standardized change in mRNA level and ribosome occupancy calculated over all the genes assigned to it (error bars equal SD calculated over the genes assigned to each cluster). Cluster #1 (for both induced and repressed genes) contains genes that showed similar change in mRNA level and ribosome occupancy; cluster #2 contains genes whose change in ribosome occupancy was in the same direction but amplified compared to the mRNA response.
- Cluster #2 (for both induced and repressed genes) indicates feedforward loops by which the response in mRNA level is further augmented by a coupled modulation of protein translation. Myc‐mediated effects could be direct or indirect.
- Distribution of alterations (log2 scale) in mRNA levels (left) and translation efficiencies (TE) (right) of genes assigned to the clusters shown in (A) (distribution of all other genes in the dataset (“Bg”) is shown as well in light blue). Consistent with the pattern shown in (A), genes assigned to cluster #1 responded at mRNA level with no or only marginal change in TE, while genes assigned to cluster #2 showed coordinated change in mRNA level and TE.
- Distribution of changes (log2 scale) in protein levels as measured by proteomic analysis for proteins encoded by the genes assigned to the clusters shown in (A). These results confirm that differences in protein response between clusters #1 and #2 are narrowed compared to their differences in mRNA response (C, left) as these differences are compensated by changes in TE (C, right).
- We compared changes in protein levels (ΔP; measured by proteomics) with changes in RNA levels (ΔR; measured by RNA‐seq) in response to Myc induction for the genes assigned to the four clusters shown in Fig 1A. As expected by the Ribo‐seq results, while no significant difference was observed for genes assigned to cluster #1 (for which the change in protein level is accounted for by a corresponding change in RNA level), change in protein levels significantly differed from change in RNA levels for genes assigned to clusters #2, as these genes are also subjected to modulation of translation efficiency upon Myc activation. P‐values were calculated using Wilcoxon's test (comparing each distribution to the background one).
To functionally characterize the biological programs that are exerted by the Myc‐induced network, we examined the enrichment of the above gene clusters for Gene Ontology (GO) functional categories (Table 1). We found that among the genes induced upon Myc activation, cluster #1 was significantly enriched for genes that function in ribosome biogenesis and for transcription factors. Cluster #2 was enriched for subunits of RNA polymerase II (RNAPII) and of the ribosome. Interestingly, this result indicates that upon Myc activation, subunits of both the transcription and translation core machineries (the RNA‐PolII and ribosome complexes) are coordinately induced at the layers of mRNA expression and translational efficiency. Among the genes that were repressed upon Myc activation, cluster #1 was enriched for cytoskeleton genes while cluster #2 was strikingly enriched for adhesion and extra cellular matrix (ECM) proteins. The repression of the network of ECM‐receptor interactions included numerous collagens, laminins, and integrin subunits (Fig EV2). This extensive suppressive response coordinated too the regulation of both mRNA and translation levels. Taken together, the Myc‐induced response enhances tumorigenesis while the Myc‐repressed response reduces cell adhesion to the ECM thus attenuating cellular motility and invasiveness.
Table 1.
Functional enrichments in gene clusters
| Term | Count | P‐value | FDR | Fold Enrichment | |
|---|---|---|---|---|---|
| up1 | GO:0042254 ‐ ribosome biogenesis | 18 | 8.7E‐07 | 1.4E‐03 | 4.2 |
| GO:0006364 ‐ rRNA processing | 15 | 3.1E‐06 | 2.4E‐03 | 4.6 | |
| GO:0043565 ‐ sequence‐specific DNA binding | 25 | 1.9E‐05 | 8.8E‐03 | 2.6 | |
| up2 | GO:0033279 ‐ ribosomal subunit | 6 | 1.2E‐03 | 1.9E‐02 | 7.5 |
| GO:0016591 ‐ DNA‐directed RNA polymerase II | 5 | 1.6E‐03 | 2.1E‐02 | 9.9 | |
| down1 | GO:0005856 ‐ cytoskeleton | 45 | 5.1E‐06 | 1.5E‐03 | 2.0 |
| Steroid biosynthesis | 7 | 2.5E‐05 | 4.1E‐03 | 11.8 | |
| lipid synthesis | 8 | 6.3E‐04 | 3.4E‐02 | 5.5 | |
| down2 | GO:0007155 ‐ cell adhesion | 49 | 8.5E‐17 | 1.9E‐13 | 4.0 |
| GO:0031012 ‐ extracellular matrix | 28 | 7.8E‐09 | 5.7E‐07 | 3.7 | |
| GO:0034329 ‐ cell junction assembly | 9 | 4.2E‐07 | 2.3E‐04 | 12.6 |
Figure EV2. Repression of an extensive network of ECM‐receptor interactions in response to Myc activation.
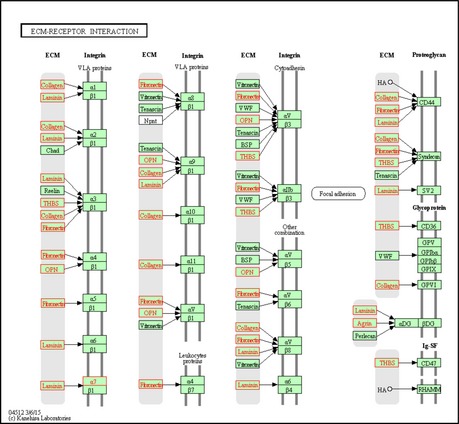
Shown is the KEGG 62 pathway map for ECM‐receptor interactions. Proteins whose encoding genes were assigned to the repressed cluster #2 are colored in red.
Our analysis discovered an intriguing coordination between the regulation of mRNA level and TE for dozens of Myc‐induced and repressed genes (see examples in Fig 2A). We experimentally examined this coupling for selected candidate genes. Changes in RNA level were measured using RT–PCR and alterations in TE were tested using polysome fractionation assay followed by RT–PCR to quantify, for each transcript, relative portion in each fraction. From the set of Myc‐induced genes, we selected, for examination, two subunits of RNAPII and its associated factor TAF1. These three genes showed a clear transcript shift from lower to higher polysome‐associated fractions upon Myc activation, confirming the increase in their TE (Fig 2B, left). From the set of Myc‐repressed genes, we selected three ECM proteins, all of them showed a transcript shift from higher to lower polysome‐associated fractions upon Myc activation, consistent with reduced TE (Fig 2B, right). These experimental validations confirm that Myc activation results in transcript‐specific modulation of protein translation which is coupled to and augments the Myc‐induced program that regulates mRNA levels.
Figure 2. Validation of Myc‐induced transcript‐specific modulation of translation efficiency.
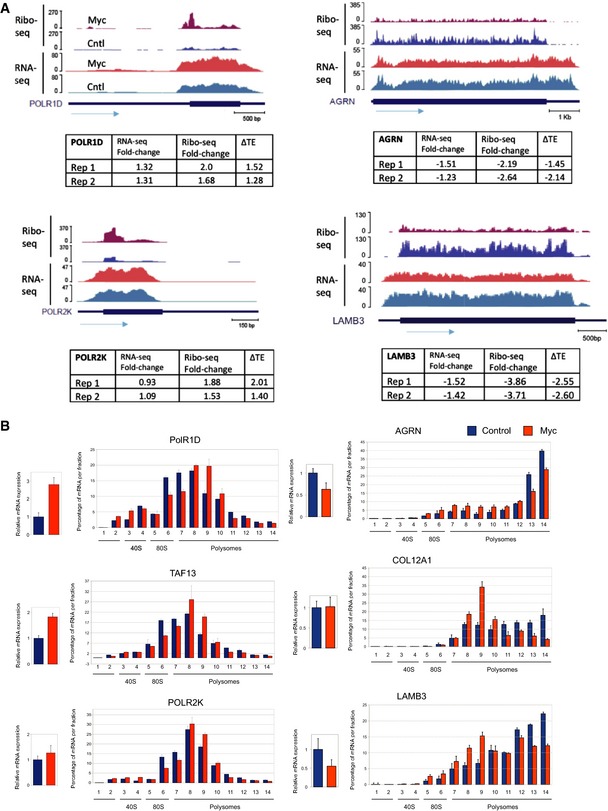
- Examples of genes that showed in the combined RNA‐ and Ribo‐seq analysis modulation of TE upon Myc activation. (RNA‐seq and Ribo‐seq experiments were carried out using independent duplicate samples).
- We selected for validation three candidate genes that showed increased (left) and decreased (right) TE, respectively. Changes in RNA level were measured using RT–PCR, and alterations in TE were tested using polysome fractionation assay followed by RT–PCR to quantify, for each transcript, relative portion in each fraction. Experiments were done in independent triplicates; error bars represent SD.
Factors that could potentially couple regulation of mRNA level and protein translation are microRNAs (miRs) and RNA‐binding proteins (RBPs). Binding of miRs and RBPs to 3′UTRs can modulate both transcript stability and translation rate 40. Alternatively, a mechanism that couples the regulation of transcription rate and translation efficiency is more puzzling, yet preliminary evidences suggest that Myc could play such a role 37. We therefore asked whether, for genes that showed a coordinated change in mRNA level and TE, the change in mRNA level was correlated with alterations in transcription rate. Alternatively, changes in mRNA levels of these genes could possibly be mainly due to modulation of their transcript stability. RNA‐seq measures steady‐state expression levels and consequently does not distinguish between changes in expression levels that stem from regulation of gene transcription or transcript stability. We therefore used global run‐on coupled with sequencing (GRO‐seq) analysis, which measures the expression of nascent transcripts and thus provides more direct estimates of gene transcription rates 41. We applied GRO‐seq analysis, in biologically independent duplicates, to Myc‐induced and control cells (Table EV4; Fig EV3A). These data clearly demonstrated that changes in mRNA levels (RNA‐seq data) were correlated with changes in transcription rates (GRO‐seq data) also for genes whose TE was modulated (that is, genes assigned to cluster #2) (Figs 3 and EV3B), indicating that genes that showed a coordinated change in mRNA level and TE were both transcriptionally and translationally regulated in response to Myc activation.
Figure EV3. Myc‐induced transcriptional responses.
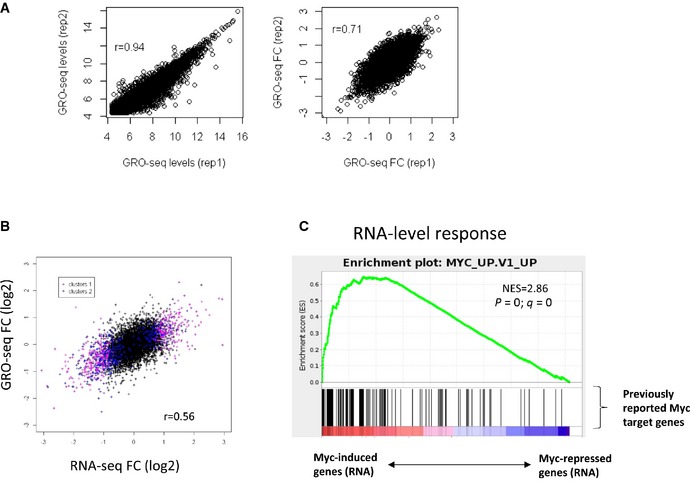
- Correlations between gene expression level (left) and gene response upon Myc activation (right) between two independent GRO‐seq measurements.
- Correlation between changes in mRNA levels (RNA‐seq) and transcription rates (GRO‐seq) induced by Myc. Genes assigned to clusters #1 and #2 are colored in pink and blue, respectively.
- GSEA analysis applied to genes sorted by their change in mRNA level upon Myc activation showed that the induced genes in our dataset were significantly enriched for Myc target genes reported by previous studies.
Figure 3. Myc‐induced changes in transcription rate.
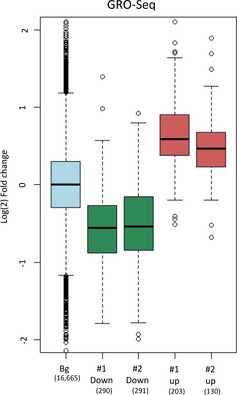
Distribution of changes in transcription rates upon Myc activation measured by GRO‐seq for the set of genes assigned to the clusters shown in Fig 1A. GRO‐seq experiments were done using independent duplicate samples. For the boxplots, the box indicates the 1st and 3rd quartiles; the horizontal band inside the box indicates the median. The whiskers extend to the most extreme data point which is no more than 1.5‐times the interquartile range from the box.
We next examined which of the four regulatory patterns delineated by clustering analysis in our dataset were likely to be controlled directly by Myc binding to target gene promoters. We analyzed a Myc ChIP‐seq dataset that was recorded in the same U2OS cellular system that was used in our experiments 32 and tested intersections between Myc direct target genes defined by ChIP‐seq and our gene clusters. We sorted the putative Myc target genes detected by ChIP‐seq according to the strength of Myc binding (binding affinity) to their promoters and found, as expected, that the cluster containing genes that were transcriptionally induced upon Myc activation (up cluster #1) was significantly enriched for high‐affinity Myc target genes (Table 2). The cluster of genes that were coordinately induced at the layers of transcription and translation was enriched too, albeit to a lesser extent, for high‐affinity Myc target genes. On the other hand, the clusters containing genes repressed upon Myc activation (cluster down #1 and #2) were significantly depleted of high‐affinity Myc targets (Table 2), indicating either secondary effects to Myc activation or effects specifically mediated by weak Myc binding. In line with previous reports and using MIZ1 ChIP‐seq data 32, we observed a statistical overrepresentation of putative MIZ1 targets among the repressed genes compared to the induced ones (33% vs. 26%, P = 0.04; chi‐squared test). In addition, computational motif analysis using iRegulon 42 detected that the promoters of genes assigned to cluster down #1 were significantly enriched for the binding motif of SP1 (P < 0.005; based on normal approximation for the enrichment scores). Interestingly, a previous report indicated that Myc activation suppresses the expression of CDKN1A (p21) by sequestering SP1 from the promoter of this gene (without direct binding of Myc to this promoter) 43. Seeking regulators of the Myc‐induced translation modulation, we searched for enriched sequence motifs in the 5′UTR and 3′UTR of genes assigned to cluster up/down #2, but we did not detect any, which may indicate a multifactorial mode of translation regulation.
Table 2.
Enrichment and depletion of Myc direct target genes (determined by ChIP‐seq) in the main response clusters
| Cluster | Top 1ka , b | Top 2k | Top 3k | Top 4k |
|---|---|---|---|---|
| Down_1 | −7.46E‐06 | −1.19E‐06 | −1.06E‐07 | −3.30E‐08 |
| Down_2 | −3.28E‐05 | −4.88E‐09 | −4.52E‐12 | −2.22E‐15 |
| Up_1 | 1.84E‐11 | 6.02E‐11 | 9.33E‐11 | 3.68E‐08 |
| Up_2 | 8.24E‐05 | 0.00015 | 0.00011 | 0.0025 |
P‐values calculated using the tail of hypergeometric distribution. Negative values indicate depletion of Myc direct target genes.
Myc target genes were ranked according to the strength of Myc binding to their promoter region. Intersections with genes assigned to the main response clusters in our dataset were examined for the top 1k, 2k, 3k and 4k Myc direct targets.
The clustering analysis described above was applied to the set of 972 genes that responded to Myc activation beyond certain cutoff values. To further functionally characterize the Myc response, we applied gene‐set enrichment analysis (GSEA) 44, which does not rely on any pre‐set cutoff levels, but instead is based on ranks of all genes detected in the data. For this analysis, we ranked the genes twice: first, by their change in mRNA levels upon Myc induction, and second, by the change in their TE in response to this treatment. Reassuringly, GSEA results showed that the set of genes whose mRNA level was up‐regulated in response to Myc was enriched for genes that were identified as Myc‐induced targets by previous transcriptomic studies (in other cellular systems) (Fig EV3C). With respect to genes regulated at the layer of protein translation, GSEA detected that the genes whose TE was elevated upon Myc induction were strongly enriched for ribosomal protein (RP) genes (Fig 4A). Individually, most RP genes showed only a modest increase in TE. Yet, as a set, this group of genes, encoding for ribosome constituents of both the large and small subunits, showed highly significant and coordinated elevation in TE. This result reflects one of the well‐characterized oncogenic effects of Myc induction, namely global enhancement of protein synthesis 45. Several Ribo‐seq studies previously showed that global increase in TE of RP genes is a molecular hallmark of mTOR activation 39, 46. This observation therefore suggests that the mTOR pathway mediates some of the translation modulation effects induced by Myc.
Figure 4. Effect of mTOR inhibition on the Myc‐induced response.
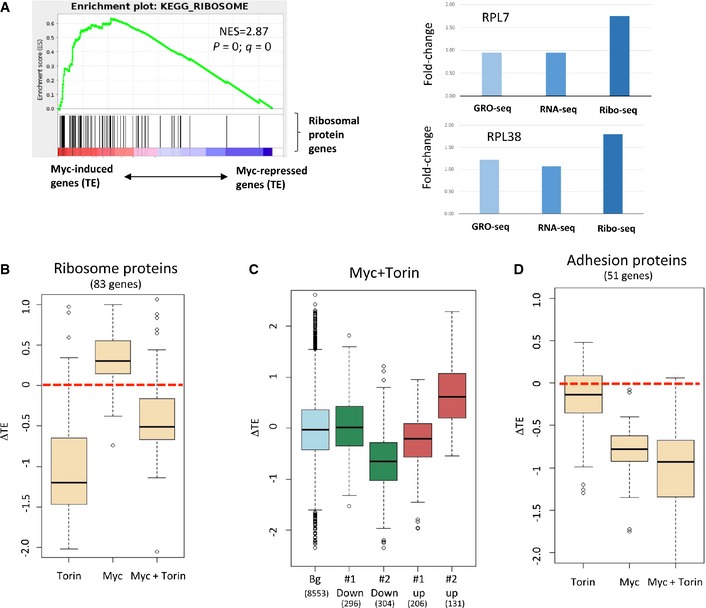
- Right: GSEA analysis demonstrated that the genes whose TE was elevated upon Myc activation were significantly enriched for ribosomal proteins (RPs). Genes were sorted from left to right according to the change in their TE in response to Myc induction. Vertical black bars indicate the location of RPs in this sorted list. (NES, normalized enrichment score; p and q, nominal and multiple‐testing corrected P‐values, respectively.) Left: example of two RP genes whose TE is induced by Myc activation.
- Distribution of changes in TE measured for the set of RPs (of both the large and small ribosome subunits) in response to treatment with Torin‐1, Myc, or Myc+Torin1. The results show that Torin‐1 treatment significantly attenuated the translation induction of the RP genes upon Myc activation.
- Distribution of changes in TE in response to Myc activation in the presence of Torin‐1 measured for the set of genes assigned to the clusters shown in Fig 1A. Unlike the drastic effect Torin‐1 had on the translation induction of RP genes, much of the Myc‐induced modulation of TE of genes assigned to these four clusters persisted in the presence of Torin‐1.
- Torin‐1 did not compromise the strong translation repression of adhesion proteins upon Myc activation (shown are results for 51 genes functionally annotated as encoding for adhesion proteins that were assigned to cluster down 2).
To explore this aspect, we repeated the Ribo‐seq and RNA‐seq experiments in the presence of Torin‐1, a potent inhibitor of mTOR 47. We found that the translational induction of RP genes upon Myc activation was significantly abolished by Torin‐1, indicating that this response was largely mediated through a Myc‐mTOR signaling axis (Figs 4B and EV4A). In contrast, other components of the Myc‐induced transcriptional and translational responses were not significantly affected by Torin‐1 (Figs 4C and EV4B and C), indicating that their regulation is not linked to the mTOR pathway. Importantly, the extensive Myc‐mediated translation repression of the network of ECM and adhesion proteins was not compromised by Torin‐1 treatment, showing that mTOR pathway is not involved in the regulation of this effect (Fig 4D). We further controlled whether Torin‐1 treatment affects Myc translation, as it was previously observed in another cellular system that mTOR inhibition using rapamycin resulted in a reduction in the amount of Myc mRNA associated with polysomes while total cellular Myc mRNA level and MYC protein stability remained unchanged 48. Using a polysome fractionation assay, we find that in our system Torin‐1 did not affect Myc translation (Fig EV4D). Overall, our results indicate that mTOR inhibition by Torin‐1 specifically represses the myc‐mediated translational induction of ribosomal proteins while having no marked effect on other parts of the Myc response network.
Figure EV4. Myc‐induced regulation of protein translation.
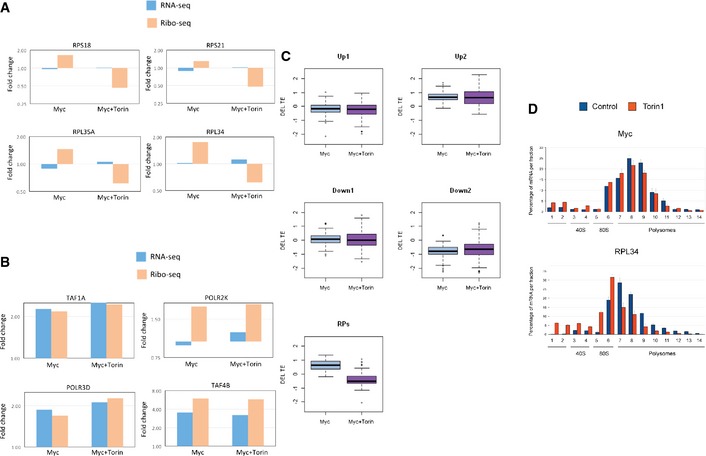
- Example of ribosomal protein genes whose Myc‐mediated translational induction was repressed and reversed by Torin‐1 treatment.
- Example of genes whose Myc‐mediated induction was not affected by Torin‐1 treatment.
- A global statistical analysis showing that in contrast to the significant effect that Torin‐1 had on the modulation of translation efficiency (TE) of ribosomal protein genes (RPs) upon Myc induction (bottom panel), Torin‐1 had no significant effect on the TE of other components of the Myc‐induced response network (divided by the clusters shown in Fig 1A). For the boxplots, the box indicates the 1st and 3rd quartiles; the horizontal band inside the box indicates the median. The whiskers extend to the most extreme data point which is no more than 1.5‐times the interquartile range from the box.
- Using polysome fractionation assay followed by RT–PCR to quantify relative portion of Myc mRNA in each fraction, we confirmed that Torin‐1 did not affect Myc TE. As a control, we used RPL34 whose TE is strongly repressed by Torin‐1 (see Fig EV4A). The repression of RPL34 is reflected by the strong shift of the signal toward the subpolysomal fractions. Experiments were done in independent triplicates; error bars represent SD.
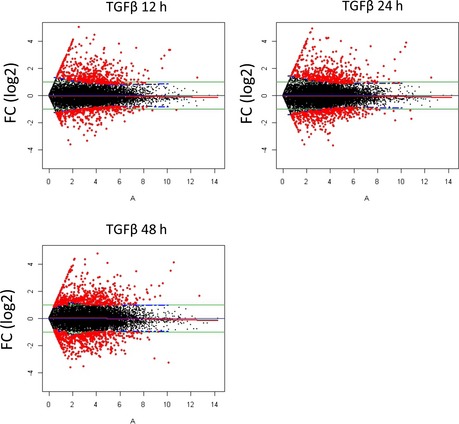
GSEA analysis also showed that the set of genes whose mRNA level was down‐regulated in response to Myc activation was enriched for a gene signature that is up‐regulated in invasive breast cancer and that the set of genes whose TE was attenuated upon Myc induction was strongly enriched for ECM and adhesion genes (Fig 5A). This observation suggests that the link between the Myc signaling pathway and cellular motility and invasiveness is wider than appreciated so far, and that it is mediated by Myc modulation of both gene transcription and protein translation. To further characterize possible functional links between Myc activity and cancer metastasis, we examined transcriptional and translational responses to TGFβ, a potent inducer of cell motility and invasiveness. Importantly, it was previously reported that TGFβ inhibits Myc expression 49. We therefore examined the effect of TGFβ treatment on the transcriptome of the human mammary epithelial MCF10A cells, a cellular system whose motility and invasiveness are strongly enhanced by this treatment 50. We probed the transcriptional responses to TGFβ at three time points (12 h, 24 h, and 48 h) and in accord with previous reports, confirmed that it resulted in a strong suppression of Myc expression (Fig 5B). Overall, we detected more than 1,600 genes that were induced or repressed upon TGFβ treatment (Fig EV5). Cluster analysis delineated two major response patterns containing the genes that were induced or repressed upon TGFβ treatment (Fig 5C). Concomitantly to Myc repression, the cluster of down‐regulated genes was significantly enriched for cell cycle genes reflecting the anti‐proliferative effect of TGFβ. The cluster of TGFβ‐induced genes was significantly enriched for adhesion and ECM‐protein encoding genes (Fig 5D), mirroring a reversed effect to the one observed in response to Myc activation. As cell invasion and migration depend on the interaction of the cell with ligands in the ECM, these results suggest that Myc repression contributes to the TGFβ‐mediated enhancement of cell motility and invasiveness of MCF10A cells.
Figure 5. TGFβ suppresses Myc‐induced responses.
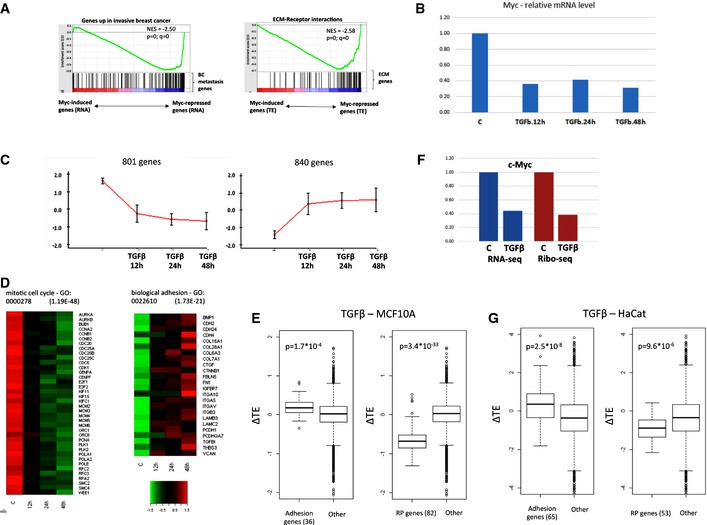
- GSEA analysis demonstrated that the genes repressed upon Myc activation were enriched for a gene signature that is up‐regulated in invasive breast cancer (defined by 61), and that the genes whose TE was reduced upon Myc activation were enriched for genes encoding for adhesion and ECM proteins. (NES, normalized enrichment score; p and q, nominal and multiple‐testing corrected P‐values, respectively.)
- Myc mRNA level in TGFβ‐treated MCF10A cells relative to its level in control sample as measured by RNA‐seq.
- Main expression patterns detected in MCF10A in response to TGFβ. Expression level of each gene was standardized to mean = 0 and SD = 1 prior to clustering. y‐axis shows standardized levels. Each cluster is represented by the average pattern calculate over all the genes it contains. Error bars represent ± SD, calculated over the genes assigned to each cluster.
- TGFβ response of representative cell cycle and adhesion genes selected, respectively, from the repressed and induced gene clusters.
- TGFβ treatment of MCF10A cells significantly increased TE of adhesion genes (left) and repressed TE of RP genes (right) (P‐values were calculated using Wilcoxon's test).
- Myc mRNA and translation levels in TGFβ‐treated HaCat cells relative to their levels in control HaCat sample as measured by RNA‐seq and Ribo‐seq.
- TGFβ treatment of HaCat cells also significantly increased the TE of adhesion genes (left) and repressed the TE of RP genes (right). P‐values were calculated using Wilcoxon's test.
Figure EV5. TGF‐beta‐induced transcriptional responses.
Similar to Fig EV1E, an adjusted fold‐change threshold was set to define TGFβ‐responsive genes in MCF10A cells. The union of the responsive genes identified by this analysis was subjected to cluster analysis shown in Fig 5C.
As we observed that both the oncogenic and metastasis‐repressive effects of Myc activation were carried out by coordinated transcriptional and translational responses, we examined the effect of TGFβ on the efficiency of protein translation using Ribo‐seq (48 h post‐TGFβ treatment) (Table EV6). We found that reduction in Myc activity due to TGFβ treatment was accompanied by significant increase in TE of adhesion genes and decrease in TE of ribosomal protein genes, here too, mirroring a reversed effect to the one we observed upon Myc activation (Fig 5E). To test the generality of our observations, we examined transcriptional and translational responses to TGFβ treatment in a second cellular system, the human‐immortalized keratinocyte HaCat cells, whose motility and invasiveness are enhanced by this treatment 51. Applying a combined RNA‐ and Ribo‐seq analysis, we found that, in this cellular system too, TGFβ treatment resulted in a strong repression of Myc expression (Fig 5F) that was accompanied by significant repression of cell cycle genes and induction of adhesion and ECM genes (Fig EV4B). Furthermore, in this cellular system too, TGFβ treatment resulted in increased TE of adhesion genes and attenuated TE of RP genes (Fig 5G). Taken together, our results pinpoint key roles of Myc in controlling oncogenesis and metastasis through modulation of gene transcription and translation and indicate its importance within the TGFβ response network.
Discussion
In this study, we systematically explored the effect of Myc activation on gene expression at the layers of transcription and protein translation. We unraveled that in addition to its known extensive effect on the cellular transcriptome, Myc induction also results in a broad transcript‐specific modulation of protein synthesis. Remarkably, we observed a widespread coordination between changes in RNA levels and protein translation efficiencies. Profiling of nascent‐RNA using GRO‐seq indicated that this effect results from a coupling between transcription and translation efficiencies that is exerted by Myc to enforce various cellular responses.
We found that the transcriptional response of dozens of Myc‐responsive genes is further amplified by a coupled translation response. This effect was observed for both induced and repressed genes. Such wide‐scale coordination between the regulation of gene transcription and protein translation is mechanistically puzzling, and detailed experimental follow‐up is required for its elucidation. Yet, an intriguing report has already indicated transcription‐independent function of c‐Myc in regulation of protein translation 36. Unexpectedly, it was shown that protein levels of several cyclin‐dependent kinases (CDKs) increased in response to Myc induction without any change in their mRNA levels. Mechanistically, it was demonstrated that Myc increases the translation of these specific mRNAs by promoting the methylation of their 5′ end (“cap methylation”) which is required for efficient translation. This model could explain the coupling we observed between the transcriptional and translational responses: Myc binding to promoters enhances gene transcription and then further, for a subset of transcripts, Myc enhances mRNA capping of the nascent transcripts, thereby increasing their translation efficiency. For transcripts that show an induction only in protein translation without a change in mRNA levels, it was suggested that Myc enhances mRNA capping without binding to the promoters thus functioning in these cases as transcription‐independent factor 37. Similar coupling between regulation of transcription and mRNA capping carried out by other factors could potentially explain the coordinated effect we observed for genes that were repressed upon Myc activation. Further studies are required to elucidate the mechanisms by which Myc activation modulates transcript‐specific protein translation, and whether these effects are directly regulated by Myc (in a transcription‐independent mode) or are secondary to its activation and mediated by Myc‐responsive regulators (e.g., RBPs, miRs).
From a functional perspective, our results further substantiate that Myc activation concomitantly enhances cell growth and oncogenic transformation while repressing cell migration and invasiveness. Interestingly, we demonstrate that both effects are enforced by coordinated transcriptional and translational responses (Fig 6). With respect to the oncogenic response arm, Myc activation results in a broad induction of both the transcriptional (RNAPII) and protein translation machineries. The induction of the translation of the translation machinery itself is a molecular hallmark of mTOR activation, and accordingly we show that mTOR inhibition by the ATP‐competitive Torin‐1 inhibitor significantly attenuates this effect of Myc. Importantly, mTOR‐mediated global enhancement of protein biogenesis is critical for Myc oncogenic function, and therefore, its inhibition was suggested as a strategy for treatment of Myc‐dependent human cancers. Significantly, a recent study uncovered a functional link between Myc activation and mTOR‐dependent phosphorylation of 4EBP1 protein 52, which is a key regulatory node of protein translation. Furthermore, that study showed that the use of another ATP‐competitive mTOR inhibitor, MLN0128, which is currently under clinical trials, attenuated 4EBP1 phosphorylation and had a remarkable therapeutic efficacy in Myc‐driven hematological cancers. As differences in biological effect exerted by competitive and allosteric mTOR inhibitors were documented in several systems (e.g., 53), it will be important to examine whether the effect we observed for Torin‐1 is elicited also by allosteric inhibition of mTOR using rapamycin.
Figure 6. A schematic depiction of Myc‐induced responses.
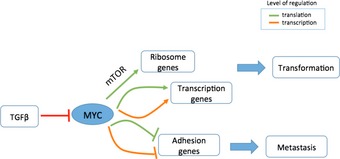
Myc coordinates extensive gene transcription and protein translation responses to enhance cell transformation and suppress cell invasiveness and metastasis.
With respect to the metastasis‐repressive response arm, Myc activation results in a global repression of an extensive network of ECM and adhesion proteins that govern cell migration and invasiveness. A recent study attributed Myc's metastasis‐repressive effect to its transcriptional suppression of the αV and β3 integrin subunits 17. We show that the repressive effect of Myc activation on cell interaction with the ECM is in fact much broader and encompasses a coordinated suppression of numerous ECM and adhesion proteins at both the transcriptional and translational layers. As TGFβ is a very potent inducer of cell motility and invasiveness that was reported to suppress Myc expression, we examined the effect of TGFβ treatment on gene expression and protein translation. Using two different cellular systems, we confirmed that TGFβ treatment strongly decreased Myc levels and demonstrated that this suppression was accompanied by transcriptional and translational responses reversed to the ones observed upon Myc activation, including TE modulation of ribosomal protein genes and of ECM and adhesion protein genes. These results suggest that Myc suppression contributes to the prometastatic function of the TGFβ signaling pathway. However, it is possible that this effect depends on cell‐specific factors, as there are cancer‐cell models in which Myc was reported to enhance TGFβ‐mediated epithelial‐to‐mesenchymal transition (EMT) 53. Further increasing the complexity of the cross talk between Myc and TGFβ signaling is tumor‐prompted stroma‐derived signals. For example, induction of Myc in a transgenic mouse lymphoma model stimulated surrounding macrophages to secrete TGFβ, which resulted in induction of cellular senescence 54.
Our current study reveals novel facets of the Myc‐induced network. Given its key role in tumorigenesis, therapies that target Myc are under development. Our results highlight the challenge and importance of developing novel therapeutic strategies specifically aimed at blocking the oncogenic arm of the Myc signaling pathway without affecting its metastasis‐suppressive effects. Targeting the mTOR pathway emerges as one such avenue that holds great promise for treatment of cancers in which Myc activity is amplified as we demonstrate that inhibition of mTOR strongly attenuates cell growth effectors induced by Myc but has no effect on the extensive Myc‐mediated repression of the network of ECM and adhesion proteins. Yet, as some components of the Myc‐induced network are cell‐specific, the generalizability of our observations should be examined by expanding our experiments to additional cancer‐cell models in which Myc activity is amplified.
Materials and Methods
Cell culture
Myc‐inducible U2OS cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat‐inactivated fetal calf serum in 5% CO2 at 37°C 32. HaCat cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. MCF10A cells were cultured in DMEM/F12 1:1 medium supplemented with 5% horse serum, EGF (10 ng/ml), insulin (10 μg/ml), cholera toxin (100 ng/ml), and hydrocortisone (500 ng/ml) in 5% CO2 at 37°C. For TGFβ1 treatment, MCF10a cells were treated with human recombinant TGFβ1 (10 ng/ml) for 48 h (R&D Systems); HaCat cell were treated with TGFβ1 (10 ng/ml) for 6 h.
RNA‐seq
RNA‐seq libraries were prepared using the TruSeq RNA Sample Preparation Kit (Illumina) following the manufacturer's instructions. Samples were sequenced using Hi‐Seq 2000 platform (single‐end reads; length of 50 nt).
Ribo‐seq
Cells were treated with cycloheximide (100 μg/ml) for 5 min, washed with ice‐cold phosphate‐buffered saline (cycloheximide, 100 μg/ml), pelleted, and lysed in buffer A (20 mM Tris–HCl, pH 7.8, 100 mM KCl, 10 mM MgCl2, 1% Triton X‐100, 2 mM DTT, 100 μg/ml cycloheximide, and 1× complete protease inhibitor). Lysates were centrifuged at 5,000 rpm, and the supernatant was treated with 2 U/μl of RNase I (Ambion) for 40 min at room temperature. Lysates were fractionated on a linear sucrose gradient (7 to 47%) using the SW‐41Ti rotor at 36,000 rpm for 2 h. Fractions enriched in monosomes were pooled and treated with proteinase K (Roche) in a 1% SDS solution. Released RNA fragments were purified using TRIsure reagent and precipitated in the presence of glycogen. For libraries preparation, RNA was gel‐purified on a denaturing 10% polyacrylamide urea (7 M) gel. A section corresponding to 30 to 33 nucleotides, the region where most of the ribosome‐protected fragments are comprised, was excised, eluted and ethanol precipitated. The resulting fragments were 3′‐dephosphorylated using T4 polynucleotide kinase (New England BioLabs, Inc.) for 6 h at 37°C in 2‐(N‐morpholino) ethanesulfonic acid (MES) buffer (100 mM MES‐NaOH, pH 5.5, 10 mM MgCl2, 10 mM β‐mercaptoethanol, and 300 mM NaCl). 3′ adaptor was added with T4 RNA ligase 1 (New England BioLabs, Inc.) for 2.5 h at 37°C. Ligation products were 5′‐phosphorylated with T4 polynucleotide kinase for 30 min at 37°C. 5′ adaptor was added with T4 RNA ligase 1 for 18 h at 22°C. Libraries from HaCat cells were sequenced in GAII and the MCF10a libraries in HiSeq 2000 (single‐end reads; length of 50 nt).
GRO‐seq
GRO‐Seq protocol was performed as previously described with minor modifications 41. Briefly, cells were treated with doxycyline for 5 h and 5 × 106 nuclei per condition were isolated and incubated 5 min at 30°C with equal volume of reaction buffer for the nuclear run‐on (10 mM Tris–Cl pH 8.0, 5 mM MgCl2, 1 mM DTT, 300 mM KCl, 20 units of SUPERase In, 1% sarkosyl, 500 μM ATP, GTP, and Br‐UTP, 0.2 μM CTP+32P CTP). The reaction was stopped and RNA extracted with TRIzol LS (Invitrogen) according to the manufacturer's instructions. Base hydrolysis was performed using RNA fragmentation reagents (Ambion), and the reaction was purified through p‐30 RNase‐free spin column (Bio‐Rad). BrUTP‐labeled nascent transcripts were immunoprecipitated with anti‐BrUTP agarose beads (Santa Cruz Biotech), washed once in binding buffer, once in low‐salt buffer (0.2× SSPE, 1 mM EDTA, 0.05% Tween‐20), once in high‐salt buffer (0.25× SSPE, 1 mM EDTA, 0.05% Tween‐20, 137.5 mM NaCl), and twice in TET buffer (TE with 0.05% Tween‐20). Then, BrUTP‐incorporated RNA was eluted with elution buffer (20 mM DTT, 300 mM NaCl, 5 mM Tris–Cl, pH 7.5, 1 mM EDTA, and 0.1% SDS) and RNA isolated with TRIzol LS. End‐repair of the enriched BrUTP‐incorporated RNAs was achieved by the treatment with tobacco acid pyrophosphatase (TAP; Epicenter) to remove 5′‐methyl guanosine caps and followed by T4 polynucleotide kinase treatment (PNK; NEB) to remove 3′‐phosphate group at low pH. Then, the BrU‐incorporated RNAs were treated with T4 PNK at high pH in the presence of ATP to add 5′‐phosphate group. The reaction was stopped, and RNA was extracted with TRIzol LS.
Library construction
Sequencing libraries were generated from two biological replicates using TruSeq Small RNA kit from Illumina. Final libraries were two times cleaned up and size‐selected by Agencourt AMPure XP (Beckman Coulter) and sequenced according to Illumina's protocol in a HiSeq 2000 platform.
Analysis of RNA‐seq and Ribo‐seq datasets
Sequenced reads were aligned to a reference set of human curated protein‐coding transcripts (plus the five human rRNA transcripts) using bowtie 55. This reference set of transcripts was based on Ensembl gene annotations (release 65). Alignment statistics are provided in Table EV1. For genes with multiple isoforms, the one with longest coding DNA sequence (CDS) region and, in case not unique, the one with longest UTRs among the ones with the longest CDS, was selected to represent the gene. Only uniquely mapped reads were used in subsequent analyses. RNA expression levels and ribosome occupancy were estimated by calculating reads per kilobase of mRNA per million reads (RPKM) per transcript, taking into account either all reads that map to the transcript (for estimation of RNA levels using RNA‐seq data) or only those mapping to its CDS (for estimation of ribosome occupancy). In estimation of ribosome occupancy in CDS, 5′ ends of reads were offset 12 nucleotides to the 3′ direction to match the P‐site location of ribosome 38. RNA‐seq and Ribo‐seq datasets were combined according to gene ID. Only genes with at least 40 reads in at least one RNA‐seq and one Ribo‐seq samples were included in subsequent analyses. The combined Myc‐U2OS dataset includes 8,553 (Table EV2). Fold change (FC) in mRNA level and ribosome occupancy upon treatment were calculated per gene (in log2). To avoid inflation of FC estimates due to low levels, RPKM levels below 1.0 were set to a “floor” value of 1.0. Examination of relationship between gene response to Myc induction and transcript features showed a global association between Ribo‐seq FC estimates and transcript CDS length (Fig EV1A). We normalized this technical effect using lowess normalization (Fig EV1B).
Seeking genes that responded to Myc activation in either the RNA‐seq or Ribo‐seq datasets, we set an adjusted fold‐change threshold that takes into account the higher measurement variability among lowly expressed genes (Fig EV1E). We detected 724 genes that consistently responded to Myc in the two RNA‐seq experiments, and 616 genes that consistently responded in the duplicate Ribo‐seq experiments. A total of 368 genes overlapped these two sets; thus, 972 unique genes were called responders in the combined dataset. This set of genes was subjected to cluster analysis using the CLICK algorithm implemented in the EXPANDER package 56. Translation efficiency (TE) was calculated for each gene per condition as the (log2) ratio of its ribosome occupancy and RNA level. Functional enrichment analysis was carried out using DAVID 57. All other statistical analyses were done in R.
Analysis of GRO‐seq dataset
Sequenced reads were aligned to the human genome (hg19) using bowtie 2, and a number of reads mapping to annotated genes (Ensembl v69) were counted by HTseq 58. Quantile normalization was then applied to allow comparisons between different samples. Only genes covered by at least 20 reads in at least one sample were included in the analysis (Table EV4).
Analysis of Myc ChIP‐seq dataset
We analyzed Myc ChIP‐seq data from 32 that were measured on the same U2OS system that we used in our RNA‐, Ribo‐, and GRO‐seq experiments. Fastq files were downloaded from GEO (accession number GSE44672) and were aligned to the human genome (hg19) using bowtie 2. We used Cisgenome 59 to detect genomic binding sites bound by Myc. The comparison between Myc and input samples detected 16,912 Myc binding sites at FDR cutoff of 1%. For intersection with our dataset, Myc binding site was associated with gene's promoter if it was located within 5 kbp from gene's transcription start site (TSS).
Isolation of polysome‐associated mRNA
Cells were lysed in buffer A containing 1 U of RNaseOUT (Invitrogen, Grand Island, NY, USA). Lysate was homogenized using a 26‐G needle, and the cytosolic extract was obtained by centrifugation at 1,300 g for 10 min. The extract was overlaid on a 7 to 47% linear sucrose gradient and centrifuged in a SW41Ti rotor (Beckman Coulter, California, USA) at 36,000 rpm for 2 h at 4°C. Fourteen fractions were collected from the gradients, and RNA was isolated from each using TRIsure reagent (Bioline). Reverse transcription was performed using SuperScript III cDNA synthesis kit (Life Technologies) following the manufacturer's instructions.
Real‐time PCR
One microgram of total RNA was reverse transcribed using the SuperScript III first‐strand synthesis system (Life Technologies). Real‐time PCR was performed using the SensiFAST SYBR real‐time PCR kit (Bioline) in the LightCycler 480 System (Roche). Primers used in this study are listed in Table EV5.
Proteomics analysis
Sample preparation and offline HpH‐RP fractionation
Myc‐induced and mock‐treated U2OS cell lysates were sonicated in lysis buffer (8 M urea in 50 mM ammonium bicarbonate (pH 8.5)) in the presence of protease inhibitors (Complete Mini tablets, Roche) and subsequently cleared by centrifugation. Protein concentration was determined with a 2D Quant Kit (GE Healthcare), according to the manufacturer's instructions. Aliquots corresponding to 70 μg protein were first reduced with DTT and alkylated with iodoacetamide, before proteolytic digestion with Lys‐C (Wako) for 4 h at 37°C, enzyme/substrate ratio 1:75. The mixture was then diluted fourfold to 2 M urea and digested overnight at 37°C with sequencing grade trypsin (Promega) in enzyme/substrate ratio 1:100. Digestion was quenched by the addition of formic acid (final concentration 10%), after which the peptides were simultaneously desalted and stable isotope dimethyl labeled on a Sep‐Pak C18 column (Waters, Massachusetts, USA). For 2 out of 3 biological replicates, mock samples were labeled with “light” and Myc samples with “heavy” label, whereas labels were swapped for the third replicate. For each replicate, the light and heavy sample was mixed in a 1:1 ratio, dried down in a speed vacuum centrifuge, and stored at −80°C until offline peptide fractionation. Basic reversed‐phase (HpH‐RP) high‐performance liquid chromatography separation of labeled peptides was employed for offline peptide fractionation. Dried peptides were reconstituted in 10 mM ammonium hydroxide (NH4OH, solvent A) and loaded onto a Phenomenex Gemini C18 analytical column (100 mm × 1 mm, particle size 3 μm, 110 Å pores) coupled to an Agilent 1260 HPLC system consisting of a nanopump, autosampler, multiple wavelength detector, and 96‐well plate fraction collector. Per replicate, 120 μg peptides were eluted at a constant flow of 100 μl/min in a gradient containing a 30‐min linear increase from 0 to 10% solvent B (90% acetonitrile (ACN)/10% NH4OH), a further increase to 23% solvent B at t = 50 min, 30% solvent B at t = 61 min, and finally a 5‐min wash with 85% solvent B. A total number of 67 HpH‐RP fractions were collected, which were concatenated to 12 fractions and subsequently dried in a vacuum centrifuge. Prior to mass spectrometry analysis, the peptides were reconstituted in 10% formic acid.
Mass spectrometry
Peptides were separated using the Proxeon nLC 1000 system (Thermo Scientific, Bremen) fitted with a trapping (ReproSil‐Pur 120 C18‐AQ 3 μm (Dr. Maisch GmbH, Ammerbuch, Germany); 100 μm × 30 mm) and an analytical column (ReproSil‐Pur 120 C18‐AQ 2.4 μm (Dr. Maisch GmbH); 75 μm × 500 mm), both packed in‐house. The outlet of the analytical column was coupled directly to a Thermo Orbitrap Fusion hybrid mass spectrometer (Q‐OT‐qIT, Thermo Scientific) using the Proxeon nanoflex source. Nanospray was achieved using a distally coated fused silica tip emitter (generated in‐house, o.d. 375 μm, i.d. 20 μm) operated at 2.1 kV. Solvent A was 0.1% formic acid/water and solvent B was 0.1% formic acid/ACN. Aliquots of concatenated HpH‐RP fractions were eluted from the analytical column at a constant flow of 250 nl/min in a 140‐min gradient, containing a 121‐min linear increase from 7 to 25% solvent B, followed by a 18‐min wash at 80% solvent B. Survey scans of peptide precursors from m/z 375–1,500 were performed at 120‐K resolution with a 4 × 105 ion count target. Tandem MS was performed by quadrupole isolation at 1.6 Th, followed by HCD fragmentation with normalized collision energy of 33 and ion trap MS2 fragment detection. The MS2 ion count target was set to 104, and the max injection time was set to 50 ms. Only precursors with charge state 2–6 were sampled for MS2. Monoisotopic precursor selection was turned on; the dynamic exclusion duration was set to 30 s with a 10 ppm tolerance around the selected precursor and its isotopes. The instrument was run in top speed mode with 3‐s cycles.
Data analysis
Raw data files were processed using Proteome Discoverer (PD, version 1.4.1.14, Thermo Fisher Scientific). MS2 spectra were searched against the Swissprot database (release 2014_08, 546,238 entries) using Mascot (version 2.5, Matrix Science, UK) and Homo sapiens as taxonomy filter (20,194 entries). Carbamidomethylation of cysteines was set as fixed modification and oxidation of methionine; dimethyl “light” label (K and N‐term) and dimethyl “heavy” label (K and N‐term) were set as a dynamic modifications. Trypsin was specified as enzyme and up to two miscleavages were allowed. Data filtering was performed using percolator, resulting in 1% peptide false discovery rate (FDR), and Mascot peptide ion score > 20 was set as additional filter. Protein [heavy/light] ratios were log2‐transformed and normalized on protein median.
Data availability
Raw sequence data of our study are deposited at GEO; accession number GSE: GSE66929. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium 60 via the PRIDE partner repository with the dataset identifier PXD002073.
Author contributions
RE, FLP, and RA conceived the study and wrote the manuscript. RE did the bioinformatics data analysis; FLP carried out the RNA‐seq and Ribo‐seq experiments and their validations; GK, RL, and PCvB did the GRO‐seq experiment. OBB and AFMA performed the proteomic analysis. EW, FL, and ME generated the Myc construct and contributed reagents.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Review Process File
Acknowledgements
This work was supported by funds from the Fundação para a Ciência e a Tecnologia, Portugal to R.L. SFRH/BD/74476/2010, the project Proteins At Work, financed by the Netherlands Organization for Scientific Research (NWO) as part of the National Roadmap Large‐scale Research Facilities of the Netherlands (project number 184.032.201) for the proteomics, as well as the Dutch Cancer Society (KWF), the VICI program of the NWO, and the European Research Council (ERC) Advanced Grant EnhReg to R.A. A.F.M.A. acknowledges additional support by the NWO through a VIDI grant (723.012.102). We also thank all members of the Agami group for helpful discussions and Liesbeth Hoekman for technical assistance.
EMBO Reports (2015) 16: 1723–1736
References
- 1. Escot C, Theillet C, Lidereau R, Spyratos F, Champeme MH, Gest J, Callahan R (1986) Genetic alteration of the c‐myc protooncogene (MYC) in human primary breast carcinomas. Proc Natl Acad Sci USA 83: 4834–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ladanyi M, Park CK, Lewis R, Jhanwar SC, Healey JH, Huvos AG (1993) Sporadic amplification of the MYC gene in human osteosarcomas. Diagn Mol Pathol 2: 163–167 [PubMed] [Google Scholar]
- 3. Gamberi G, Benassi MS, Bohling T, Ragazzini P, Molendini L, Sollazzo MR, Pompetti F, Merli M, Magagnoli G, Balladelli A et al (1998) C‐myc and c‐fos in human osteosarcoma: prognostic value of mRNA and protein expression. Oncology 55: 556–563 [DOI] [PubMed] [Google Scholar]
- 4. Kawate S, Fukusato T, Ohwada S, Watanuki A, Morishita Y (1999) Amplification of c‐myc in hepatocellular carcinoma: correlation with clinicopathologic features, proliferative activity and p53 overexpression. Oncology 57: 157–163 [DOI] [PubMed] [Google Scholar]
- 5. Stock C, Kager L, Fink FM, Gadner H, Ambros PF (2000) Chromosomal regions involved in the pathogenesis of osteosarcomas. Genes Chromosom Cancer 28: 329–336 [DOI] [PubMed] [Google Scholar]
- 6. Boxer LM, Dang CV (2001) Translocations involving c‐myc and c‐myc function. Oncogene 20: 5595–5610 [DOI] [PubMed] [Google Scholar]
- 7. Gabay M, Li Y, Felsher DW (2014) MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 4: pii: a014241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Felsher DW (2003) Cancer revoked: oncogenes as therapeutic targets. Nat Rev Cancer 3: 375–380 [DOI] [PubMed] [Google Scholar]
- 9. Shachaf CM, Felsher DW (2005) Rehabilitation of cancer through oncogene inactivation. Trends Mol Med 11: 316–321 [DOI] [PubMed] [Google Scholar]
- 10. van Riggelen J, Felsher DW (2010) Myc and a Cdk2 senescence switch. Nat Cell Biol 12: 7–9 [DOI] [PubMed] [Google Scholar]
- 11. Bachireddy P, Rakhra K, Felsher DW (2012) Immunology in the clinic review series; focus on cancer: multiple roles for the immune system in oncogene addiction. Clin Exp Immunol 167: 188–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dang CV (2012) MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Felsher DW, Bishop JM (1999) Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 4: 199–207 [DOI] [PubMed] [Google Scholar]
- 14. Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, Felsher DW (2002) Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 297: 102–104 [DOI] [PubMed] [Google Scholar]
- 15. Pelengaris S, Khan M, Evan GI (2002) Suppression of Myc‐induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 109: 321–334 [DOI] [PubMed] [Google Scholar]
- 16. Marinkovic D, Marinkovic T, Mahr B, Hess J, Wirth T (2004) Reversible lymphomagenesis in conditionally c‐MYC expressing mice. Int J Cancer 110: 336–342 [DOI] [PubMed] [Google Scholar]
- 17. Liu H, Radisky DC, Yang D, Xu R, Radisky ES, Bissell MJ, Bishop JM (2012) MYC suppresses cancer metastasis by direct transcriptional silencing of alphav and beta3 integrin subunits. Nat Cell Biol 14: 567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blackwood EM, Eisenman RN (1991) Max: a helix‐loop‐helix zipper protein that forms a sequence‐specific DNA‐binding complex with Myc. Science 251: 1211–1217 [DOI] [PubMed] [Google Scholar]
- 19. Schuhmacher M, Kohlhuber F, Holzel M, Kaiser C, Burtscher H, Jarsch M, Bornkamm GW, Laux G, Polack A, Weidle UH et al (2001) The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res 29: 397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV (2003) An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol 4: R69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schlosser I, Holzel M, Hoffmann R, Burtscher H, Kohlhuber F, Schuhmacher M, Chapman R, Weidle UH, Eick D (2005) Dissection of transcriptional programmes in response to serum and c‐Myc in a human B‐cell line. Oncogene 24: 520–524 [DOI] [PubMed] [Google Scholar]
- 22. Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F (2006) The c‐Myc target gene network. Semin Cancer Biol 16: 253–264 [DOI] [PubMed] [Google Scholar]
- 23. Kim YH, Girard L, Giacomini CP, Wang P, Hernandez‐Boussard T, Tibshirani R, Minna JD, Pollack JR (2006) Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 25: 130–138 [DOI] [PubMed] [Google Scholar]
- 24. Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA (2012) Transcriptional amplification in tumor cells with elevated c‐Myc. Cell 151: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R et al (2012) c‐Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151: 68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA (2010) c‐Myc regulates transcriptional pause release. Cell 141: 432–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eberhardy SR, Farnham PJ (2001) c‐Myc mediates activation of the cad promoter via a post‐RNA polymerase II recruitment mechanism. J Biol Chem 276: 48562–48571 [DOI] [PubMed] [Google Scholar]
- 28. Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM (2003) c‐Myc recruits P‐TEFb for transcription, cellular proliferation and apoptosis. Oncogene 22: 5707–5711 [DOI] [PubMed] [Google Scholar]
- 29. Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M (2004) Myc‐induced proliferation and transformation require Akt‐mediated phosphorylation of FoxO proteins. EMBO J 23: 2830–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gargano B, Amente S, Majello B, Lania L (2007) P‐TEFb is a crucial co‐factor for Myc transactivation. Cell Cycle 6: 2031–2037 [DOI] [PubMed] [Google Scholar]
- 31. Bres V, Yoshida T, Pickle L, Jones KA (2009) SKIP interacts with c‐Myc and Menin to promote HIV‐1 Tat transactivation. Mol Cell 36: 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay‐Odelot H, Karim S, Bartkuhn M et al (2014) Activation and repression by oncogenic MYC shape tumour‐specific gene expression profiles. Nature 511: 483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003) Genomic targets of the human c‐Myc protein. Genes Dev 17: 1115–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC et al (2006) Global mapping of c‐Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci USA 103: 17834–17839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, Rao PH, Ruggero D (2008) Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456: 971–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cowling VH, Cole MD (2007) The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy‐terminal domain independently of direct DNA binding. Mol Cell Biol 27: 2059–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cole MD, Cowling VH (2008) Transcription‐independent functions of MYC: regulation of translation and DNA replication. Nat Rev Mol Cell Biol 9: 810–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS (2009) Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loayza‐Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R (2013) p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol 14: R32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eulalio A, Huntzinger E, Izaurralde E (2008) Getting to the root of miRNA‐mediated gene silencing. Cell 132: 9–14 [DOI] [PubMed] [Google Scholar]
- 41. Core LJ, Waterfall JJ, Lis JT (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janky R, Verfaillie A, Imrichova H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D et al (2014) iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol 10: e1003731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, Tyner AL (2001) Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci USA 98: 4510–4515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iritani BM, Eisenman RN (1999) c‐Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci USA 96: 13180–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ et al (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP‐competitive mammalian target of rapamycin inhibitor reveals rapamycin‐resistant functions of mTORC1. J Biol Chem 284: 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wall M, Poortinga G, Hannan KM, Pearson RB, Hannan RD, McArthur GA (2008) Translational control of c‐MYC by rapamycin promotes terminal myeloid differentiation. Blood 112: 2305–2317 [DOI] [PubMed] [Google Scholar]
- 49. Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF (2004) Transforming growth factor beta‐mediated transcriptional repression of c‐myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol 24: 2546–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wendt MK, Smith JA, Schiemann WP (2010) Transforming growth factor‐beta‐induced epithelial‐mesenchymal transition facilitates epidermal growth factor‐dependent breast cancer progression. Oncogene 29: 6485–6498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Naz S, Ranganathan P, Bodapati P, Shastry AH, Mishra LN, Kondaiah P (2012) Regulation of S100A2 expression by TGF‐beta‐induced MEK/ERK signalling and its role in cell migration/invasion. Biochem J 447: 81–91 [DOI] [PubMed] [Google Scholar]
- 52. Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D (2013) Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc‐driven cancers. Proc Natl Acad Sci USA 110: 11988–11993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hayman TJ, Kramp T, Kahn J, Jamal M, Camphausen K, Tofilon PJ (2013) Competitive but Not Allosteric mTOR Kinase Inhibition Enhances Tumor Cell Radiosensitivity. Transl Oncol 6: 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reimann M, Lee S, Loddenkemper C, Dorr JR, Tabor V, Aichele P, Stein H, Dorken B, Jenuwein T, Schmitt CA (2010) Tumor stroma‐derived TGF‐beta limits myc‐driven lymphomagenesis via Suv39h1‐dependent senescence. Cancer Cell 17: 262–272 [DOI] [PubMed] [Google Scholar]
- 55. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ulitsky I, Maron‐Katz A, Shavit S, Sagir D, Linhart C, Elkon R, Tanay A, Sharan R, Shiloh Y, Shamir R (2010) Expander: from expression microarrays to networks and functions. Nat Protoc 5: 303–322 [DOI] [PubMed] [Google Scholar]
- 57. da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- 58. Anders S, Pyl PT, Huber W (2015) HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH (2008) An integrated software system for analyzing ChIP‐chip and ChIP‐seq data. Nat Biotechnol 26: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N et al (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32: 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, Walter M, Fehm T, Solomayer E, Riess O, Wallwiener D et al (2006) Progression‐specific genes identified by expression profiling of matched ductal carcinomas in situ and invasive breast tumors, combining laser capture microdissection and oligonucleotide microarray analysis. Cancer Res 66: 5278–5286 [DOI] [PubMed] [Google Scholar]
- 62. Tanabe M, Kanehisa M (2012) Using the KEGG database resource. Curr Protoc Bioinformatics Chapter 1 Unit 1: 12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Review Process File
Data Availability Statement
Raw sequence data of our study are deposited at GEO; accession number GSE: GSE66929. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium 60 via the PRIDE partner repository with the dataset identifier PXD002073.