

Abstract
Neuropeptides are small neuronal signaling molecules that act as neuromodulators for a variety of neural functions including analgesia, reproduction, social behavior, learning, and memory. One of the endogenous neuropeptides—Met-Enkephalin (Met-Enk), has been shown to display an inhibitory effect on cell proliferation and differentiation. Here, a novel lipid-modification approach is shown to create a small library of neuropeptides that will allow increased bioavailability and plasma stability after systemic administration. It is demonstrated, on an experimental model of human pancreatic adenocarcinoma, that lipid conjugation of Met-Enk enhances its tumor suppression efficacy compared to its nonlipidated counterparts, both in vitro and in vivo. More strikingly, the in vivo studies show that a combination therapy with a reduced concentration of Gemcitabine has suppressed the tumor growth considerably even three weeks after the last treatment.
Keywords: lipids, met-enkephalin, neuropeptides, pancreatic cancer, squalene
1. Introduction
Central nervous system (CNS) neurons corelease a variety of neuropeptides during neurotransmission. They are small neuronal signaling molecules that act as neuromodulators for a variety of neural functions including analgesia, reproduction, metabolism, social behavior, learning, and memory.[1] Presently, about 100 known endogenous neuropeptides exist in the CNS such as oxytocin, vasopressin, endorphins, endomorphins, enkephalins, etc.[2] Although peptide-based therapy has gained great interest recently due to their therapeutic effects against various diseases, their wide use as active therapeutics is largely hindered by their short plasma life-time.[3]
We have previously developed a unique lipid-modification of small hydrophilic drug molecules with very short plasma half-life (such as gemcitabine, doxorubicin, and adenosine) to improve their efficacy in vivo.[4] This new concept was achieved by coupling a natural and biocompatible terpene- squalene (SQ)- to molecules of great therapeutic values to form SQ-conjugated prodrugs.[5] These bioconjugates, due to their inherent amphiphilic nature, spontaneously self-organized as nanoassemblies (NAs) in water.[6] Furthermore, SQ-modification allowed for a chemical encapsulation, leading to NAs with very high drug loading (around 60%) and absence of burst release, compared to the conventional physical encapsulation into nanocarriers such as liposomes or polymeric vesicles (around 5%).[7] However, it has been a great challenge to extend the SQ-based bioconjugation approach to peptide molecules with higher molecular weight (Mw), due to various hurdles in performing successful conjugation reactions. Thus, the context of the present study is to establish a generic lipid-modification method that could apply to various active therapeutic peptides in order to design a library of peptide-based nanomedicines. As a proof of concept, four different endogenous neuropeptides [Met-Enkephalin (Met-Enk), Leu-Enkephalin (Leu-Enk), Endomorphin-1 (EM-1), and Endomorphin-2 (EM-2)] were used in this study due to their great potential as biotherapeutic agents but whose application in pharmacy is dramatically hindered by rapid metabolism and low bioavailability after administration.[8]
Here, we developed simple bioconjugation methods using SQ, the precursor in cholesterol biosynthesis, to lipidate different neuropeptides. This approach yielded amphiphilic SQ-neuropeptide bioconjugates that could potentially act as prodrugs-based nanoparticles with pharmacological activity.
2. Results and Discussion
Scheme 1 depicts the bioconjugation approaches that were typically employed for the lipid modification of neuropeptides using appropriate SQ derivatives. Since solid-supported peptides pose the risk of possible SQ degradation during Trifluoroacetic acid (TFA) treatment that is commonly employed for the peptide–resin cleavage, a postsynthetic strategy was designed. The bioconjugation was carried out using peptide coupling reactions by either (i) activating the –COOH group of the neuropeptide (Leu-Enk and Met-Enk) with a potent coupling reagent such as HATU (due to the steric hindrance imparts from the coiled structure of SQ) to react with SQ–NH2 (Scheme 5A) or (ii) via the –NH2 of the neuropeptides (EM-1 and EM-2) to react with an amine reactive SQ–NHS under slight basic conditions (Scheme 5B). To avoid a self-condensation of Leu- or Met-Enk (Scheme 5A), the primary amino group of the peptides was protected with a Fmoc group. After 48 h of reaction at room temperature under inert atmosphere, the N-terminal deprotection of the bioconjugate was carried out by adding piperidine to the reaction mixture. The resulting SQ–neuropeptides had a final yield of around 50% and were purified using silica column and characterized using NMR and MS (see the Experimental Section and Supporting Information).
Figure 5. Scheme.
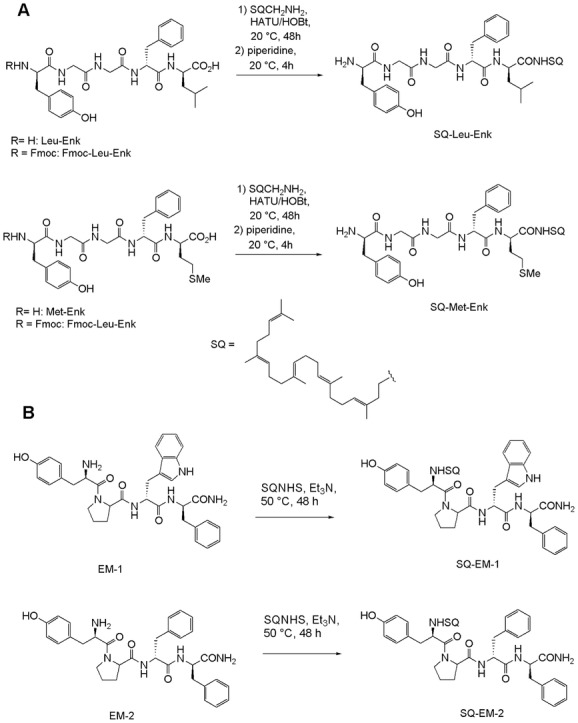
Scheme illustrating the chemical bioconjugation steps used in obtaining SQ–neuropeptides. Synthesis routes for Leu- and Met-Enkephalins (A) and for Endomorphin-1 and 2 (B).
2.1. Nanoassembly Formation and Physical Characterization
The SQ–neuropeptide bioconjugates spontaneously formed NAs in water, resulting in self-assembled neuropeptide (SAN) structures (Scheme 2). The size of NAs varied from 100 to 150 nm, depending on the peptide and the conjugation site (Table 1). Noteworthy, from the ratio between the Mw of the peptide (drug) and the Mw of SQ (drug transporter), the drug loading could be calculated and expressed in % (Table 1). SQ-modification of peptides allowed the formation of NAs with higher drug loading (from 59% to 62%) compared to conventional drug carriers, when peptides are only physically encapsulated (generally drug loading is lower than 10%). Figure 1 shows representative cryo-EM images of the SAN NAs formed at concentrations of 2 mg mL−1 in an aqueous solution of 5% dextrose. They displayed spherical and monodisperse structures (size discrepancy between the Cryo-EM and DLS methods could be attributed to the known hydrodynamic radius-related differences) with net surface charges ranging from +36 to −58 mV (Table 1). The difference in surface charges on the SANs could be related to the nature of amino acids that are present after the bioconjugation. For example, in the case of Leu- and Met-Enk, the SQ bioconjugation on the –COOH site leaves one primary –NH2 group available (Scheme 5A), which leads to a net positive surface charge. However, the surface charge was inversed when the conjugation with SQ was performed on the –NH2 group. On the contrary, SQ modification of EM-1 and 2 peptides leaves an amide terminal group available (Scheme 5B) that leads to a nearly neutral surface charge. SAN NAs were stable for up to a month at 4 °C when formed in 5% dextrose at 2 mg mL−1 concentration.
Figure 6. Scheme.
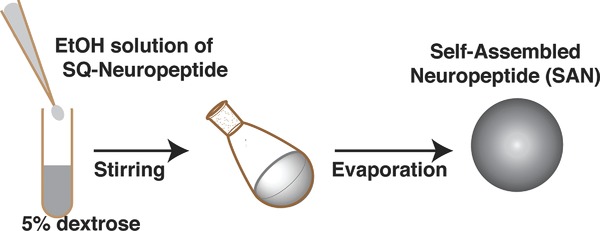
Sketch (not to scale) depicting the procedure of preparing SAN NAs from SQ–neuropeptide bioconjugates.
Table 1.
Table outlining the physical/chemical parameters of SAN NAs
| Neuropeptide | Conjugation sitea) | Sizeb) [nm] | PDIb,c) | Charged) | Drug loading |
|---|---|---|---|---|---|
| Leu-enkephalin | –COOH | 147 | 0.174 | +21 mV | 60% |
| Met-enkephalin | –COOH | 130 | 0.181 | +36 mV | 61% |
| Endomorphin-1 | –NH2 | 117 | 0.079 | −0.62 mV | 62% |
| Endomorphin-2 | –NH2 | 103 | 0.107 | −0.14 mV | 59% |
| Leu-enkephalin | –NH2 | 136 | 0.079 | −58 mV | 59% |
Detailed structure in the Supporting Information;
polydispersity index;
DLS method;
Zeta potential measurements.
Figure 1.
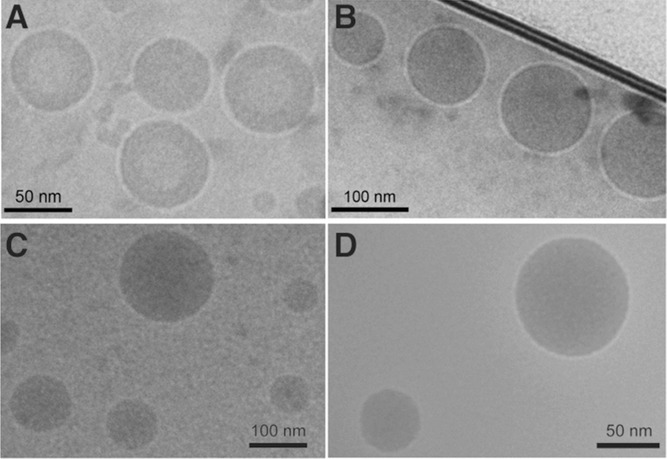
Representative Cryo-EM images showing the formation of SAN NAs from different SQ–neuropeptides. A) SQ–Leu-Enk, B) SQ–Met-Enk, C) SQ–EM-1, and D) SQ–EM-2. Scale bars = 50 nm (A,D) and 100 nm (B,C).
2.2. Neuropeptides and their Anticancer Activity
Though these neuropeptides are primarily known for their opioid activities due to their high affinity toward specific opioid receptors,[9] Met-Enk has been identified as an inhibitory growth factor in cancer and wound healing.[10] Due to its unique cell growth regulation properties, in addition to the classic agonist activities toward mu-opioid (MOP) and delta-opioid (DOP) receptor subtypes, this pentapeptide (Tyr–Gly–Gly–Phe–Met) is termed as opioid growth factor (OGF).[11] OGF functions in direct combination with its receptor, the opioid growth factor receptor (OGFr). OGFrs are found to be overexpressed in the nuclear membranes of several cancer cells and immune cells such as T-cells, macrophages, and dendritic cells.[10] It has been shown that there is an elevated level of Met-Enk in the plasma of cancer patients, suggesting their functional roles in such disease states.[11] This is particularly true in the case of human pancreatic cancer in which both OGF and OGFr have been detected at higher levels.[12]
Several groups have already suggested the use of Met-Enk in the treatment of various types of human cancers including pancreatic, ovarian, colon, and breast cancers.[13] Since the mechanism of action of Met-Enk is entirely different from standard chemotherapy agents and is nontoxic and nonapoptotic, the treatments using Met-Enk are called “biotherapy.”[14] On the other hand, the widely used standard chemotherapies are extremely toxic and have several side effects that affect the quality of life of the patients even after potential recovery from disease. Thus, any effort to find and/or improve novel biotherapy treatments for cancer and immunodeficient diseases are of paramount importance.[15] Even though Met-Enk had been shown to enhance some efficacy in the treatment (alone or in combination therapy)[14] of various types of cancers, its anticancer activity remains modest due to already mentioned short plasma half-life and susceptibility to enzymes. Therefore, it is of great importance to develop drug delivery strategies to improve the stability and bioavailability of OGFs. As a first proof of concept of the pharmacological activity of our SAN NAs, we tested self-assembled Met-Enk (SAME) NAs as biotherapy for pancreatic tumor, an extremely devastating disease.
2.3. In Vitro Cell Proliferation Studies
Prior to assess the anticancer activity of SAME NAs, we checked the expression level of the OGFrs in the Mia PACA-2 human pancreatic cancer cell line by using OGFr polyclonal primary antibodies in combination with Alexa-555 coupled secondary antibodies (red). Immunocytochemistry experiments performed on fixed cells revealed abundance of these receptors in the cytosol of Mia PACA-2 (Figure 2A,C). Image shown in panels B and D followed similar experimental steps, except that the cells were incubated with 50 × 10−6m SAME NAs for 24 h prior to fixation. The nuclei were labeled with Hoechst stain (blue) before mounting the coverslips. The nuclei in the latter case showed violet purplish color, which suggests that some of the OGFrs had been translocated into the nuclei upon binding to the Met-Enk. The mechanism that is operative here is in good agreement with what has been suggested in literature regarding the functioning of OGF–OGFr axis to control the DNA synthesis in the nucleus, which arrests the cell proliferation.[16]
Figure 2.
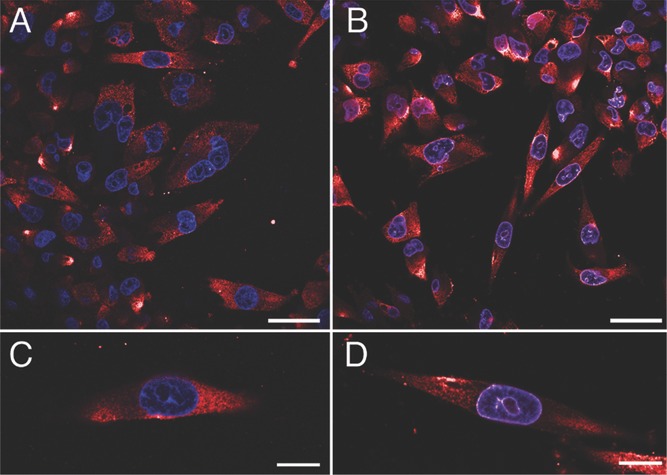
Representative laser scanning confocal images showing the abundance of OGFrs in MiaPACA-2 cells. The fluorescence is derived from anti-Rabbit Alexa-555 secondary antibodies (red) that are species-specifically bound to primary OGFr polyclonal antibodies. The nucleus was stained using Hoechst 33342 (blue). The cells before (A) and 24 h after (B) treatment with SAME NAs are shown. Panels C and D are zoomed-in views of panels (A) and (B), respectively. Scale bars = 10 µm (A,B) and 5 µm (C,D).
Standard MTT assay was used to determine the in vitro anticancer activity of SAME NAs. To be noted that unlike typical anticancer drugs usually employed in similar experiments, SAME NAs do not exert any cytotoxicity leading to cell death. The cell viability data presented thus indicate the percentage of cells that were multiplied over a three days period and not the percentage of cells that survived a toxic insult. Figure 3A shows the cell viability (%) of free Met-Enk (green) and SAME NAs (red) after 72 h of incubation at concentrations ranging from 100 × 10−12m to 100 × 10−6m. SAME NAs were significantly more efficient than free Met-Enk in reducing cell viability from a concentration of 1 × 10−9m (p < 0.01) until 100 × 10−6m (p < 0.001) where cell viability was only 25% for SAME and 90% for free Met-Enk. All control experiments including 5% dextrose solution, squalenic acid nanoassemblies (SQ NAs), etc., have shown no effect in the cell proliferation (see Figure SI.1, Supporting Information) in similar conditions. The antiproliferative activity of SAME NAs was further confirmed on human breast (MCF-7), ovarian (SK-OV-3), and colon (HT-29) cancer cell lines (Figure SI.2, Supporting Information).
Figure 3.

In vitro antiproliferative activity of SAME NAs. Cell viability of MiaPACA-2 cells was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye in standard MTT tests. A) Viability of Mia PACA-2 against a range of concentrations of SAME NAs (red) indicates that lipid bioconjugation improves the effectiveness of OGF–OGFr axis as compared to their nonlipidated counterparts (green). B) Combined treatment of SAME NAs (at 50 and 100 × 10−6m) with a range of concentrations of Gem (from 1 to 100 × 10−9 m). At 50 × 10−6m concentration, SAME NAs display a combined anticancer activity for Gem concentrations of 10 and 50 × 10−9 m, by comparison with the cell viability of individual treatments (A). Statistical significance with confidence levels of >95% (Student's t-test with Bonferroni correction for multiple comparisons) are indicated by * (p < 0.01), ** (p < 0.005), and *** (p < 0.001).
We further tested the combined efficacy of SAME NAs in combination with gemcitabine (Gem), an anticancer compound prescribed as a first line treatment in the pancreatic cancer. Practically, a range of concentrations of Gem were supplemented with SAME NAs (50 and 100 × 10−6m) and incubated with MiaPACA cells (Figure 3B). Remarkably, an important synergistic antiproliferative activity of SAME NAs was noted, especially at the lowest concentration of Gem (i.e., 1 and 10 × 10−9m). The order of addition of either component used in the combined therapy did not alter the in vitro anticancer efficacy.
2.4. In Vivo studies
Finally, the in vivo anticancer efficacy of SAME NAs was performed on the human pancreatic carcinoma xenograft model of Mia PACA-2. As depicted in Figure 4A, after tumors had grown up to 100 mm3, the first group of animals received 15 mg kg−1 (eq. weight of Met-Enk) of SAME NAs by i.v. injections daily from day 0 to 4, followed by single injections on days 7 and 10. For comparison, a second group received Met-Enk injections at same doses and intervals. A third group received same treatments of SAME NAs as the first group, but with additional injections of 7 mg kg−1 of Gem (just after the first injection of NAs) on days 1, 4, 7, and 10. A similar schedule was used for the treatment with free Met-Enk associated with Gem. Finally, treatment with Gem alone was also performed at 7 mg kg−1 (days 1, 4, 7, and 10). The controls were saline and SQ NAs. The dose schedules for all these treatments did not show any toxicity in healthy mice (Figure SI.3, Supporting Information).
Figure 4.
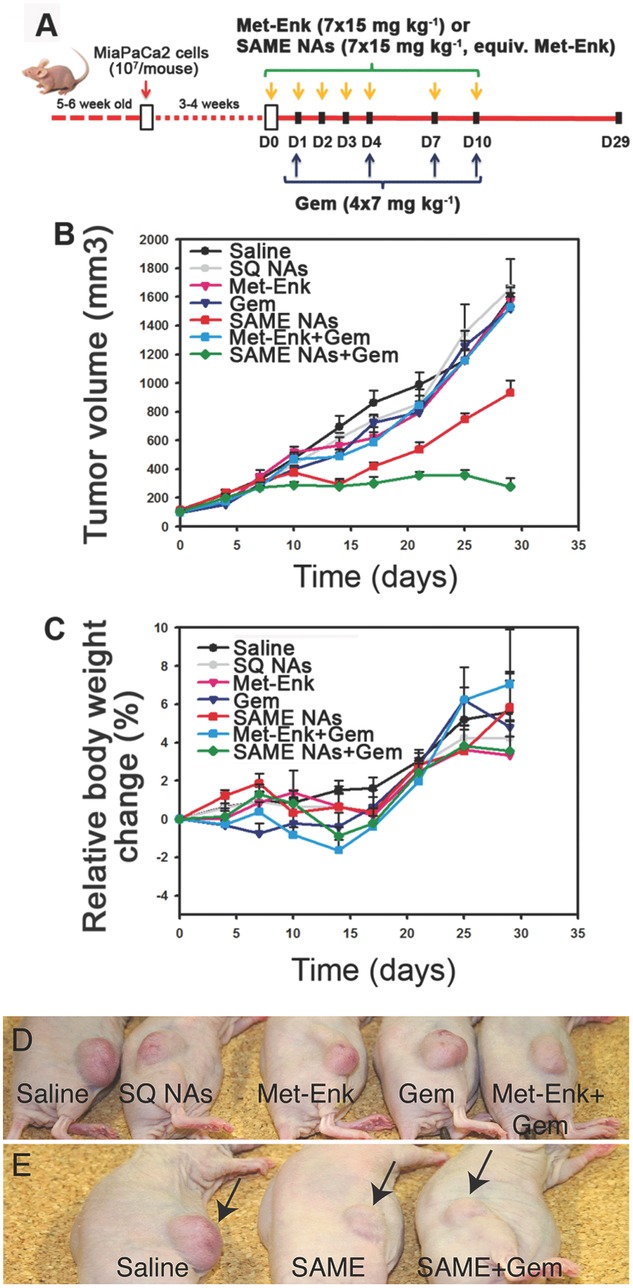
In vivo anticancer activity of SAME NAs alone or in combination with Gem (A) administration schedules and doses (B). Tumor inhibition after treatment with SAME NAs, SAME NAs + Gem, Met-Enk, Met-Enk + Gem, or Gem alone. Controls are saline and drug-free squalenic acid nanoassemblies (SQNAs). Only the groups treated either with SAME NAs (red) or SAME NAs+Gem (green) showed tumor volume inhibition. C) Relative weight change showing absence of toxicity for all treatments. D,E) Representative photographs showing the mice that underwent anticancer treatment. The control groups (D) showing visible tumor growth on day 29 post-injection. The mice that were administered with SAME NAs or SAME NAs + Gem are shown along with the reference group that received only saline (E). The suppression of tumor volume is clearly visible in the latter case.
Figure 4B shows the absolute tumor volume growth over a period of 29 days postinjection. In all the control groups, tumors (saline and SQ NAs) grew progressively to reach ≈1500 mm3 at the end of the experiment. The group of animals received SAME NAs alone (Figure 4B; red curve) showed a significant inhibition in tumor growth (nearly 50%) between days 10 and 15 before regrown to reach a final volume of about 50% of that of the control groups. Interestingly, the combination therapy of SAME NAs and Gem (Figure 4B; green curve) displayed a remarkable tumor inhibition even after the injections have ended (day 10), whereas Gem alone (Figure 4B, dark blue), Met-Enk alone (Figure 4B, pink), or the combined treatment of Gem and Met-Enk (Figure 4B, blue) did not display any anticancer activity. Moreover, the relative weight change as seen in Figure 4C indicates absence of toxicity. The images of the model animals that underwent the treatments are shown (Figure 4D,E) for a clear visual understanding of the difference in tumor growth in different experimental groups. Furthermore, since neuropeptides are active functional neuromodulators, we performed some basic behavioral studies on mice injected with SAN NAs as a function of dose and time. When injected (i.v. and i.p.) with concentrations ranging from 5 to 20 mg kg−1, the SAN NAs-treated mice did not show any significant change in their response to external stimuli such as light, heat, or sound. Further studies are however required to completely rule out any potential neurological damages and/or any positive neurological effects resulting from SAN NAs.
Progression of cancer diseases is generally characterized by various mechanisms, acting concomitantly. Therefore, the treatment of neoplastic diseases is often inefficient when using a single anticancer compound - either antimitotic or antiangiogenic. In addition, a single treatment may also allow for an easier emergence of resistances to the treatment.[17] This is the reason why it is now well documented that the combination of drugs with complementary pharmacological activity may significantly improve anticancer efficacy.[18] A typical example is given here with the association of (i) Met-Enk prodrug nanoparticles, a nanoformulation of an autocrine-produced peptide that interacts with the nuclear receptor (OGFr), allowing to inhibit cell proliferation and (ii) Gem, a nucleoside analogue, with cytotoxic activity resulting from DNA insult. From a toxicological point of view, this approach has some obvious advantages over the more classical combination of two cytotoxic drugs, since through its adjuvant activity Met-Enk may enhance the anticancer activity of Gem without adding any toxicity to the treatment.
3. Conclusion
In summary, this study demonstrates that the linkage of different neuropeptides to squalene in various chemical positions allows the production of a library of nanoassemblies. Since neuropeptides are very unstable molecules, highly sensitive to enzymatic degradation, it was expected that this approach will improve the delivery and hence the pharmacological activity of such fragile molecules. We provide here a first proof of concept with Met-Enkephalin, a ligand of the opioid growth factor receptor involved in tumor progression. It was found that when linked to squalene and administered as nanoparticles, Met-Enkephalin was capable to induce significant inhibition of human experimental pancreatic cancer. More strikingly, when used in combination with a classical chemotherapy, like Gem, an additive pharmacological activity could be obtained. This is an important observation, which could allow to reduce the doses of chemotherapeutics, thus also decreasing toxicity and side effects which represent a major limitation in cancer treatment. Since neuropeptides have great potential in treating various CNS disorders, including pain, further experiments are ongoing to establish the potential of these new SAN nanoassemblies for drug delivery across the blood brain barrier.
4. Experimental Section
General: Chemicals obtained from commercial suppliers were used without further purification. SQ, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), 1-hydroxybenzotriazole (HOBt) were obtained from Sigma-Aldrich. Sodium acetate (VWR France), hydroxylamine.HCl (VWR France), N,N-diisopropylethylamine (DIPEA) (Sigma-Aldrich), lithium aluminum hydride (Sigma-Aldrich), Fmoc–Met-Enkephain (Fmoc–Met-Enk) was purchased from GeneCust Europe (Luxembourg). The 1H and 13C NMR spectra were recorded on Bruker AC 200 P (200 and 50 MHz for 1H and 13C, respectively) or Bruker Avance-300 (300 and 75 MHz for 1H and 13C, respectively) or Bruker ARX 400 (400 and 100 MHz for 1H and 13C, respectively) spectrometers. Recognition of methyl, methylene, methine, and quaternary carbon nuclei in 13C NMR spectra rests on the J-modulated spin-echo sequence. Mass spectra were recorded on a Bruker Esquire-LC. Analytical thin-layer chromatography (TLC) was performed on Merck silica gel 60F254 precoated glass plates (0.25 mm layer). Column chromatography was performed on Merck silica gel 60 (230–400 mesh ASTM). Tetrahydrofuran (THF) was distilled from sodium/benzophenone ketyl and DMF was distilled from calcium hydride, under a nitrogen atmosphere. All reactions involving air- or water-sensitive compounds were routinely conducted in glassware, which was flame-dried under a positive pressure of nitrogen.
Bioconjugation of Neuropeptides
a) Synthesis and Characterization of SQ–NH2: 1′,2-trisnorsqualenamine (SQ–NH2) was synthesized from 1,1′,2-trisnorsqualene aldehyde (SQ–CHO)[19] that was readily available in a few reaction steps starting from SQ by oxidation/reduction sequence. (See the Supporting Information for the details.)
b) Biomodification of Met-Enk: In a typical peptide coupling reaction, SQ–NH2 (200 mg, 0.518 mmol) was reacted with Fmoc–Met-Enk (206.4 mg, 0.259 mmol) in the presence of HATU (118.2 mg, 0.310 mmol), HOBt (47.5 mg, 0.310 mmol), and DiPEA (33.5 mg, 0.259 mmol) in anhydrous DMF for 48 h under nitrogen atmosphere at RT. The progress of the reaction was monitored using TLC (eluent: DCM/MeOH 90:10). After 72 h, 20% v/v piperidine was added and the reaction mixture was stirred during 4 h and then concentrated under reduced pressure. The crude product was rinsed (three times each) with n-pentane, Et2O, and EtOAc in the respective order and then further purified using a silica column. The pure product SQ–Met-Enk was obtained with 55% of yield. 1H NMR (400 MHz, DMSO) δ: 9.20 (broad s, 1H, OH), 8.20–8.06 (broad, 3H, 3NH), 7.63 (broad s, 1H, NH), 7.23–7.25 (m, 4H, CHPhe), 7.17 (m, 1H, CHPhe) 6.99 (d, J = 8.4 Hz, 2H, CHTyr), 6.67 (d, J = 8.4 Hz, 2H, CHTyr), 5.10–5.02 (m, 5H, HC=C(CH3)), 4.58–4.48 (m, 1H, CHPhe), 4.32–4.24 (m, 1H, CHMet), 3.80–3.50 (m, 4H, 2 CH2Gly), 3.39 (m, 1H, CHTyr), 3.10–2.75 (m, 5H, CH2CH2CH2NHCO, CH2Phe, CH2Tyr), 2.41–2.35 (m, 2H, CH2CH2SCH3), 2.10–1.87 (m, 23H, =C(CH3)CH2CH2, CH2CH2CH2NH, CH2CH2SCH3, CH2CH2SCH3), 1.62 (s, 3H, =C(CH3)2), 1.54 (s, 15H, =C(CH3)CH2), 1.50–1.43 (m, 2H, CH2CH2CH2NH). 13C NMR (75 MHz, DMSO) δ: 174.9 (C, CONH), 170.9 (C, CONH), 170.5 (C, CONH), 169.3 (C, CONH), 168.8 (C, CONH), 155.8 (C, COHTyr), 137.8 (C, CPhe), 134.4 (C, HC=C(CH3)), 134.3 (C, HC=C(CH3)), 134.2 (C, HC=C(CH3)), 134.0 (C, HC=C(CH3)), 130.6 (C, =C(CH3)2), 130.1 (2CH, CHortho Tyr), 129.2 (2CH, CHortho Phe), 128.5 (C, CTyr), 128.0 (2 CH, CHmeta Phe), 126.2 (CH, CHpara Phe), 124.0 (3CH, HC=C(CH3)), 123.9 (CH, HC=C(CH3)), 115.0 (2 CH, CHmeta Tyr), 56.3 (CH, CHTyr), 54.1 (CH, CHPhe), 52.1 (CH, CHMet), 42.1 (CH2, CH2Gly), 42.0 (CH2, CH2Gly), 39.2 (CH2, =C(CH3)CH2CH2), 39.1 (CH2, =C(CH3)CH2CH2), 38.2 (CH2, CH2CH2CH2NHCO), 37.2 (CH2, CH2Phe), 36.3 (CH2, CH2CH2CH2NHCO), 31.8 (CH2, CH2CH2S), 29.6 (CH2, CH2CH2S), 27.7 (2CH2, =C(CH3)CH2CH2), 27.3 (CH2, CH2CH2CH2NHCO), 26.2 (2CH2, =C(CH3)CH2CH2), 26.1 (CH2, =C(CH3)CH2CH2), 26.0 (CH2, =C(CH3)CH2CH2), 25.4 (CH3, =C(CH3)2), 17.5 (CH3, =C(CH3)CH2), 15.8 (2CH3, =C(CH3)CH2), 15.7 (CH3, =C(CH3)CH2), 14.6 (CH3, SCH3). MS (+ESI) m/z (%): 1005.6 (11) [M + Na + CH3CN]+, 963.6 (15) [M + Na]+, 941.6 (100) [M + H]+; HRMS (+ESI): calc. for C51H81N6O6S: 941.5938; found 941.5983.
c) Biomodification of Leu-Enk: In a similar reaction, SQ–NH2 (100 mg, 0.259 mmol) was reacted with Fmoc–Leu-Enk (100.8 mg, 0.130 mmol) in the presence of HATU (59.32 mg, 0.156 mmol), HOBt (23.89 mg, 0.156 mmol), and DiPEA (16.80 mg, 0.130 mmol) in anhydrous DMF for 72 h under nitrogen atmosphere at RT. After 48 h, 20% v/v piperidine was added and the reaction mixture was stirred during 4 h and then concentrated under reduced pressure. The progress of the reaction was monitored using TLC (eluent: DCM/MeOH 90:10). The crude product was rinsed (three times each) with n-pentane, followed by Et2O and then further purified using a silica column. The pure product SQ–Leu-Enk was obtained with 50% of yield. 1H NMR (400 MHz, MeOD) δ: 7.31–7.15 (m, 5H, CHPhe), 7.11 (d, J = 8.5 Hz, 2H, CHortho Tyr), 6.78 (d, J = 8.5 Hz, 2H, CHmeta Tyr), 5.21–5.05 (m, 5H, HC=C(CH3)), 4.64 (dd, J = 5.5 Hz, J = 8.8 Hz, 1H, CHPhe), 4.33 (dd, J = 5.6 Hz, J = 9.0 Hz, 1H, CHLeu), 4.06 (dd, J = 6.9 Hz, J = 7.8 Hz, 1H, CHTyr), 3.96 (d, J = 16.5 Hz, 1H, CH2Gly), 3.89 (d, J = 16.7 Hz, 1H, CH2Gly), 3.79 (d, J = 16.5 Hz, 1H, CH2Gly), 3.76 (d, J = 16.7 Hz, 1H, CH2Gly), 3.22–2.92 (m, 6H, CH2Tyr, CH2Phe, CH2CH2CH2NHCO), 2.14–1.92 (m, 18H, =C(CH3)CH2CH2, CH2CH2CH2NHCO), 1.66 (s, 3H, =C(CH3)2), 1.64–1.53 (m, 20H, =C(CH3)CH2, CH2CH2CH2NHCO, CH2CH(CH3)2, CH2CH(CH3)2), 0.94 (d, J = 6.0 Hz, 3H, CH2CH(CH3)2), 0.90 (d, J = 6.0 Hz, 3H, CH2CH(CH3)2). 13C NMR (100 MHz, MeOD) δ: 174.2 (CONH), 173.3 (C, CONH), 171.5 (C, CONH), 171.4 (C, CONH), 171.0 (C, CONH), 158.3 (C, COHTyr), 138.2 (C, CPhe), 135.9 (2C, HC=C(CH3)), 135.8 (C, HC=C(CH3)), 135.2 (C, HC=C(CH3)), 132.0 (C, HC=C(CH3)2), 131.5 (2CH, CHortho Tyr), 130.3 (2CH, CHortho Phe), 129.5 (2CH CHmeta Phe), 127.8 (CH, CHpara Phe), 126.1 (C, CHTyr), 126.0 (CH, HC=C(CH3)), 125.5 (2CH, HC=C(CH3)), 125.4 (2CH, HC=C(CH3)), 116.9 (2CH, CHmeta Tyr), 56.2 (CH, CHNH2Tyr or CHNHPhe), 56.1 (CH, CHNHPhe or CHNH2Tyr), 53.4 (CH, CHNHLeu), 43.7 (CH2, CH2Gly), 43.4 (CH2, CH2Gly), 42.0 (CH2, CH2CH(CH3)2), 40.8 (3 CH2, =C(CH3)CH2CH2), 40.2 (CH2, CH2CH2CH2NHCO), 38.6 (CH2, CH2Phe), 37.9 (CH2, NHCH2CH2CH2 or CH2Tyr), 37.8 (CH2, NHCH2CH2CH2 or CH2Tyr), 29.2 (2 CH2, =C(CH3)CH2CH2), 28.6 (CH2, NHCH2CH2CH2), 27.8 (CH2, =C(CH3)CH2CH2), 27.7 (CH2, =C(CH3)CH2CH2), 27.5 (CH2, =C(CH3)CH2CH2), 25.9 and 25.8 (CH, CH3, CH2CH(CH3)2, =C(CH3)2), 23.4 (CH3, CH2CH(CH3)2), 22.1 (CH3, CH2CH(CH3)2), 17.7 (CH3, =C(CH3)CH2) 16.1 (3 CH3, =C(CH3)CH2), 16.0 (CH3, =C(CH3)CH2). MS (+ESI) m/z (%): 945.6 (35) [M + Na]+, 923.6 (100) [M + H]+; HRMS (+ESI): calc. for C55H83N6O6: 923.6374; found 923.6407. In an alternate approach, SQ–NHS (180 mg, 0.359 mmol) was used to couple Leu-Enk (100 mg, 0.178 mmol) through a typical amine-reactive crosslinking chemistry in the presence of Et3N (0.25 mL, 0.18 mmol) in anhydrous DMF for 72 h under nitrogen atmosphere at 50 °C. The progress of the reaction was monitored using TLC (eluent: DCM/MeOH 90:10). The crude product was rinsed (three times each) with n-pentane, followed by Et2O and then further purified using a silica column. The pure product SQ–Leu-Enk was obtained with 50% of yield. 1H NMR (400 MHz, MeOD) δ: 7.27–7.23 (m, 4H, 2HAr-ortho Phe, 2HAr-meta Phe), 7.19 (m, 1H, HAr-para Phe), 7.04 (d, 2H, HAr-ortho Tyr, J = 8.4 Hz), 6.70 (d, 2H, HAr-meta Tyr, J = 8.4 Hz), 5.13–5.08 (m, 5H, HC=C(CH3)), 4.67 (dd, 1H, CHPhe, J = 4.8 Hz, J = 9.2 Hz), 4.47 (m, 1H, CHTyr), 4,43 (m, 1H, CHLeu), 3.83 (m, 1H, CHaHbGly), 3.78 (m, 2H, CH2Gly), 3.73 (m, 1H, CHaHbGly), 3.20 (dd, 1H, CHaHbPhe, J = 4.8 Hz, J = 14.5 Hz), 3.02 (m, 1H, CHaHbTyr), 2.97 (m, 1H, CHaHbPhe), 2.85 (dd, 1H, CHaHbTyr, J = 8.0 Hz, J = 14.0 Hz), 2.30 (m, 2H, CH2–CH2–CO), 2.18 (m, 2H, CH2–CH2–CO), 2.13–1.93 (m, 16H, 8CH2–HC = C(CH3)), 1.66 (m, 4H, HC=C(CH3), CH2–CH(CH3)2Leu), 1.64 (m, 2H, CH2–CH(CH3)2Leu), 1.59 (s, 12H, 4HC=C(CH3)), 1.58 (s, 3H, HC=C(CH3)), 0.94 (d, 3H, CH2–CH(CH3)2Leu, J = 6.2 Hz), 0.90 (d, 3H, CH2–CH(CH3)2Leu, J = 6.2 Hz). 13C NMR (75 MHz, MeOD) δ: 176.2 (CONH), 175.9 (CONH), 174.8 (CONH), 173.3 (CONH), 172.1 (CONH), 171.2 (CONH), 157.4 (CAr-para Tyr), 138.5 (CAr Phe), 136.0 (3HC=C(CH3)), 134.8 (HC=C(CH3)), 132.1 (HC=C(CH3)2), 131.3 (2CHAr-ortho Tyr), 130.4 (2CHAr-ortho Phe), 128.9 (CAr Tyr), 129.4 (2CHAr-meta Phe), 127.7 (CHAr-para Phe), 126.2 (HC=C(CH3)), 125.6 (HC=C(CH3)), 125.5 (HC=C(CH3)), 125.4 (2HC=C(CH3)), 116.3 (2CHAr-meta Tyr), 57.0 (CHTyr), 55.9 (CHPhe), 52.4 (CHLeu), 43.9 (CH2Gly), 43.4 (CH2Gly), 41.7 (CH2–CH(CH3)2Leu), 38.7 (CH2Phe), 37.9 (CH2Tyr), 36.5 (CH2–CH2–CO), 35.8 (CH2–CH2–CO), 29.2 (3CH2–HC=C(CH3)), 27.8 (3CH2–HC=C(CH3)), 27.5 (CH2–HC=C(CH3)), 26.2 (CH2–HC=C(CH3)), 25.9 (CH2–CH(CH3)2Leu, HC=C(CH3)), 23.4 (CH2–CH(CH3)2Leu), 21.9 (CH2–CH(CH3)2Leu), 17.7 (HC=C(CH3)), 16.2 (3HC=C(CH3)), 16.0 (HC=C(CH3)). MS (+APCI) m/z (%): 938.7 (100) [M + H]+, 920.6 (35), 807.7 (30); HRMS (+APCI): calc. for C55H80N5O8: 938.6001; found 938.6038.
d) Biomodification of EM-1: In a typical amine-reactive crosslinking chemistry, SQ–NHS (163 mg, 0.327 mmol) was reacted with EM-1 (100 mg, 0.164 mmol) in the presence of Et3N (0.14 mL, 0.164 mmol) in anhydrous DMF for 72 h under nitrogen atmosphere at 50 °C. The progress of the reaction was monitored using TLC (eluent: DCM/MeOH 90:10). After 48 h, the product was concentrated under reduced pressure. The crude product was then purified using chromatography over silica gel eluting with an increasing ratio of MeOH (from 0.5% to 10%) to obtain the pure SQ–EM-1 with 46% of yield and characterized by 1H NMR (400 MHz, MeOD): two isomers; major (75%) + minor (25%). δ: 7.57 (m, 1H, HAr-4 Trp), 7.35–6.95 (m, 11H, 5HAr Phe, 2HAr-ortho Tyr, 4HAr Trp), 6.70 (d, 2H, HAr-meta Tyr, J = 8.3 Hz Majo), 6.68 (d, 2H, HAr-meta Tyr, J = 8.3 Hz mino), 5.27–5.03 (m, 5H, HC=C(CH3)), 4.68–4.49 (m, 3H, CHTrp, CHTyr, CHPhe), 4.43 (m, 1H, CHPro mino), 4,32 (m, 1H, CHPro Majo), 3.66 (m, 1H, CH2–CH2–CHaHb–NPro Majo), 3.52 (m, 1H, CH2–CH2–CHaHb–NPro mino), 3.27–2.57 (m, 7H, CH2–CH2–CHaHb–NPro, CH2Trp, CH2Tyr, CH2Phe), 2.3–1.90 (m, 21H, 10 CH2–HC=C(CH3), CHaHb–CH2–CH2–NPro), 1.78 (m, 2H, CH2–CH2–CH2–NPro), 1.68 (m, 1H, CHaHb–CH2–CH2–NPro), 1.68–1.54 (m, 15H, HC=C(CH3)). MS (+ESI) m/z (%): 1094.9 (24) [M + DMSO + Na]+, 1016.1 (100) [M + Na]+; HRMS (+ESI): calc. for C61H81N6O6: 993.6212; found 993.6188.
e) Biomodification of EM-2: In a similar reaction as for EM-1, SQ–NHS (174 mg, 0.35 mmol) was reacted with EM-2 (100 mg, 0.175 mmol) in the presence of Et3N (0.2 mL, 0.175 mmol) in anhydrous DMF for 72 h under nitrogen atmosphere at 50 °C. The progress of the reaction was monitored using TLC (eluent: DCM/MeOH 90:10). After 48 h, the product was concentrated under reduced pressure. The crude SQ–EM-2 was then rinsed (three times each) with n-pentane, followed by Et2O. The pure product was obtained with 40% of yield and characterized by 1H NMR (400 MHz, MeOD). Two isomers; major (57%) + minor (43%) δ: 7.3–7.15 (m, 10H, 5HAr Phe), 7.04 (d, 2H, 2HAr-ortho Tyr), 6.71 (d, 2H, 2HAr-meta Tyr, J = 8.4 Hz majo), 5.23 (m, 1H, HC=C(CH3) mino), 5.18–5.05 (m, 5H majo, 4H mino, HC=C(CH3)), 4.73–4.52 (m, 2H, CHPhe, CHTyr), 4.48 (m, 1H, CHPhe), 4.34 (dd, 1H majo, CHPro, J = 4.7 Hz, J = 8.3 Hz), 4.28 (dd, 1H mino, CHPro, J = 5.8 Hz, J = 10.2 Hz), 3.69 (m, 1H, CH2–CH2–CHaHb–NPro majo), 3.54 (m, 1H, CH2–CH2–CHaHb–NPro mino), 3.32 (m, 1H, CH2–CH2–CHaHb–NPro), 3.25–2.6 (m, 6H, 2CH2Phe, CH2Tyr), 2.45–1.92 (m, 21H, 10 CH2–HC=C(CH3), CHaHb–CH2–CH2–NPro), 1.82 (m, 2H, CH2–CH2–CH2–NPro), 1.73 (m, 1H, CHaHb–CH2–CH2–NPro), 1.55–1.63 (m, 18H, 5HC=C(CH3)). MS (+ESI) m/z (%): 1055.9 (5) [M + DMSO + Na]+, 977.0 (100) [M + Na]+; HRMS (+ESI): calc. for C59H80N5O6: 954.6103; found 954.6075.
2. Preparation of NAs: 500 μL EtOH solution of SQ–neuropeptide bioconjugates at a concentration of 2 mg mL−1 was added dropwise to 1 mL of 5% dextrose aqueous solution under continuous stirring. The solution became turbid upon addition of the SQ-bioconjugate. The solvent was then evaporated using a rotavapor at −5 °C and 50 mbar. The resulting self-assembled neuropeptides were characterized using DLS and Zeta potential (Nano ZS, Malvern; 173° scattering angle at 25 °C) for primary evaluation about the size and surface charges. The measurements were performed in triplicate following appropriate dilution of the NAs in water (DLS) or 1 × 10−3m NaCl (Zeta potential).
3. Cryo-EM: 5 μL solution of SQ–neuropeptides was added to Lacey Formvar/Carbon 300 mesh copper grids (Tedpella, CA, USA). The specimens were frozen hydrated by plunge-freezing into a liquid ethane slush in liquid nitrogen (LEICA EM CPC, Wien, Austria). The cryofixed specimens were mounted into a Gatan cryoholder (Gatan, Inc., PA, USA) for direct observation at −180 °C in a JEOL 2100HC cryo-EM operating at 200 kV with a LaB6 filament. Images were recorded in zero-loss mode with a Gif Tridiem energy-filtered-CCD camera equipped with a 2k × 2k pixel-sized chip (Gatan, Inc., PA, USA). Acquisition was accomplished with the Digital Micrograph software (versions 1.83.842, Gatan, Inc., PA, USA).
4. Cell Proliferation Studies: Mia Paca-2 cells were defrosted and grown in culture at 37 °C/5% CO2 humidified chambers in cell culture medium DMEM (Lonza), supplemented with 10% FBS, 2.5% horse serum, and 0.5% PenStrep. The cells were passaged every two days and cells at passage 3 or later were used for the experiments. For the MTT experiments, cells were plated at 10 000 cells/well in 96-well plates and were grown in culture for 24 h before adding the molecules for the cytotoxicity test. After adding the drugs of interest at the respective final concentrations, the cells were left in the incubator for 72 h until performing the MTT procedure. In a typical experiment, the MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma-Aldrich) was prepared at 5 mg mL−1. 20 μL of this solution was added to each of the wells and incubated for 90 min at 37 °C/5% CO2 atmosphere. The cells were then removed from the incubator and the entire medium containing the MTT reagent was aspirated. 200 μL of DMSO (prewarmed to 55 °C) was added into each well and was agitated gently in a shaker. The plates were read using a bottom-read plate reader (LT-5000MS ELISA Reader, Labtech, East Sussex, UK) at λ = 570 nm. In these experiments, the first and last rows in the 96-well plates were filled only with PBS and the well A1 was used as the blank for the reading. Manta data analysis software (associated with the instrument) was used for the read-outs.
5. Immunostaining and Confocal Microscopy: Cells were seeded at 50 000 cells/well in a 6-well plate containing 22 mm glass coverslips. After 72 h in culture, the cells were fixed with 4% paraformaldehyde in phosphate buffer (PB), pH 7.4, for 20 min, washed (three times) with PBS and treated with 0.2% Triton X-100 for 5 min. The cells were rinsed (three times) with PBS and incubated in blocking solution (BSA) for 1 h, followed by incubation in primary antibodies diluted in antibody dilution buffer (1% BSA in 100 mL PBS) overnight at 4 °C. Cells were washed three times in PBS, incubated in alexa-555 coupled secondary antibodies, washed 3× in PBS, and mounted on glass slides. Rabbit polyclonal anti-OGFr primary antibody (Proetintech, IL, USA) was used at a dilution of 1:25 and the secondary antibody (anti-Rabbit, highly cross-adsorbed IgG; Invitrogen, USA) was used at a dilution of 1:200. Before mounting, the coverslips were incubated for 5 min with nuclear stain (Hoescht 33342, Sigma Aldrich) at 1:1000 dilution and washed three times using PBS. The stained samples (on glass coverslips) were mounted using geltol on microscopic slides and sealed. In the case of Met-Enk treated cells, 50 × 10−6m (final concentration) of Met-Enk was added to the cells after 48 h in culture and then fixed 24 h later. Fluorescence images were recorded on a TCS SP8 STED 3× microscope (Leica Microsystems, Mannheim, Germany) in the confocal mode, using a Leica HC PL APO 100×/1.40 oil immersion objective. The samples were excited at λex 543 nm/λem > 565 nm for observing the Alexa 555 tagged secondary antibodies and at λex 405 nm/λem 425–490 nm for the nuclear stain. The images were taken in a sequential mode using Leica SP8 LAS AF software (version 3.6).
6. In Vivo Drug Treatments: The antitumor efficacy of SAME NAs and their combination with Gem was investigated on the human pancreatic carcinoma xenograft model Mia PACA-2. Briefly, 200 μL of the Mia PACA-2 cell suspension, equivalent to 1 × 107 cells, were injected subcutaneously into nude mice toward the upper portion of the right flank, to develop a solid tumor model. Tumors were allowed to grow to a volume of ≈100 mm3 before initiating the treatment. Tumor length and width were measured with calipers, and the tumor volume was calculated using the following equation: Vtumor = length × width2/2. Tumor-bearing nude mice were randomly divided into seven groups of seven each and all groups received intravenous injections of (i) 0.2 mL of sterile saline daily (i.e., on days 0–4, 7, and 10), (ii) 7 mg kg−1 Gem on days 1, 4, 7, and 10, (iii) 15 mg kg−1 Met-Enk on days 0–4, 7, and 10, (iv) SAME NAs (15 mg kg−1 eq. Met-Enk) on days 0–4, 7, and 10, (v) 15 mg kg−1 Met-Enk on days 0–4, 7, and 10 and 7 mg kg−1 Gem on days 1, 4, 7, and 10, (vi) SAME NAs (15 mg kg−1 eq. Met-Enk) on days 0–4, 7, and 10 and 7 mg kg−1 Gem on days 1, 4, 7, and 10, and (vii) 5 mg kg−1 SQ NAs daily (i.e., on days 0–4, 7, and 10). On the days when both drugs were given together, SAME NAs or Met-Enk was administrated first, immediately followed by Gem injection. The injected volume was 10 μL g−1 of the body weight. The mice were monitored regularly for changes in tumor size and weight.
Acknowledgments
G.G. is a recipient of “Research Based University Chairs of Excellence—Universities of Paris (RBUCE-UP)” independent research grant from European Research Council (ERC). This work was funded by the RBUCE-UP grant agreement (#00001002483/78) between the ERC and Université Paris Sud. Part of this research was also funded by the ERC under the Framework Program FP7/2007–2013 (Grant Agreement N°249835). UMR 8612 (Couvreur team) is a member of the laboratory of excellence NANOSACLAY. The authors thank the service of the animal experimentation from the IFR141 IPSIT (Châtenay Malabry). Ms. Julie Mougin (UMR 8612) and Mrs. Ghislaine Frébourg (Electron Microscopy Facility/FR 3631–CNRS-UPMC) are acknowledged for their contribution to Cryo-EM. Ms. Claire Troufflard and Ms. Camille Dejean (BioCIS: Châtenay Malabry) are acknowledged for help with the NMR interpretations.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1a.Hokfelt T, Millhorn D, Seroogy K, Tsuruo Y, Ceccatelli S, Lindh B, Meister B, Melander T, Schalling M, Bartfai T. Experimetia. 1987;43:768. doi: 10.1007/BF01945354. [DOI] [PubMed] [Google Scholar]
- 1b.Hokfelt T, Broberger C, Xu ZQD, Sergeyev V, Ubink R, Diez M. Neuropharmacology. 2000;39 doi: 10.1016/s0028-3908(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 1c.Mollereau C, Roumy M, Zajac JM. Curr. Med. Chem. 2005;5 doi: 10.2174/1568026053544515. [DOI] [PubMed] [Google Scholar]
- 2a.Meyer-Lindenberg A, Domes G, Kirsch P, Heinrichs M. Nat. Rev. Neurosci. 2011;12:524. doi: 10.1038/nrn3044. [DOI] [PubMed] [Google Scholar]
- 2b.Donaldson ZR, Young LJ. Science. 2008;322 doi: 10.1126/science.1158668. [DOI] [PubMed] [Google Scholar]
- 2c.Bodnar RJ. Peptides. 2013;50 doi: 10.1016/j.peptides.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Sato AK, Viswanathan M, Kent RB, Wood CR. Curr. Opin. Biotechnol. 2006;17:638. doi: 10.1016/j.copbio.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 4a.Maksimenko A, Alami M, Zouhiri F, Brion J-D, Pruvost A, Mougin J, Hamze A, Boissenot T, Provot O, Desmaële D, Couvreur P. ACS Nano. 2014;8:2018. doi: 10.1021/nn500517a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b.Maksimenko A, Dosio F, Mougin J, Ferrero A, Wack S, Reddy LH, Weyn A-A, Lepeltier E, Bourgaux C, Stella B, Cattel L, Couvreur P. Proc. Natl. Acad. Sci. U.S.A. 2014;111 doi: 10.1073/pnas.1313459110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c.Gaudin A, Yemisci M, Eroglu H, Lepetre-Mouelhi S, Turkoglu OF, Dönmez-Demir B, Caban S, Sargon MF, Garcia-Argote S, Pieters G, Loreau O, Rousseau B, Tagit O, Hildebrandt N, Le Dantec Y, Mougin J, Valetti S, Chacun H, Nicolas V, Desmaële D, Andrieux K, Capan Y, Dalkara T, Couvreur P. Nat. Nanotechnol. 2014;9 doi: 10.1038/nnano.2014.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Couvreur P, Stella B, Reddy LH, Hillaireau H, Dubernet C, Desmaële D, Lepêtre-Mouehli S, Rocco F, Dereuddre-Bosquet N, Clayette P, Rosilio V, Marsaud V, Renoir J-M, Cattel L. Nano Lett. 2006;6:2544. doi: 10.1021/nl061942q. [DOI] [PubMed] [Google Scholar]
- 6.Harrisson S, Nicolas J, Maksimenko A, Bui DT, Mougin J, Couvreur P. Angew. Chem. Int. Ed. 2013;52:1678. doi: 10.1002/anie.201207297. [DOI] [PubMed] [Google Scholar]
- 7a.Gardikis K, Tsimplouli C, Konstantinos D, Micha-Screttas M, Demetzos C. Int. J. Pharm. 2010;402:231. doi: 10.1016/j.ijpharm.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 7b.Yadav A, Mishra P, Jain S, Mishra P, Mishra AK, Agrawal GP. J. Drug Target. 2008;16 doi: 10.1080/10611860802095494 . [DOI] [PubMed] [Google Scholar]
- 8a.Bird AP, Faltinek JR, Shojaei AH. J. Controlled Release. 2001;73:31. doi: 10.1016/s0168-3659(01)00246-2. [DOI] [PubMed] [Google Scholar]
- 8b.Varamini P, Toth I. Front. Pharmacol. 2013;4 doi: 10.3389/fphar.2013.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Fichna J, Janecka A, Costentin J, Do Rego J-C. Pharmacol. Rev. 2007;59:88. doi: 10.1124/pr.59.1.3. [DOI] [PubMed] [Google Scholar]
- 9b.Okada Y, Tsuda Y, Salvadori S, Lazarus LH. Int. J. Med. Chem. 2012;2012 doi: 10.1155/2012/715123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zagon IS, McLaughlin PJ. World J. Gastroenterol. 2014;20:2218. doi: 10.3748/wjg.v20.i9.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a.Schäfer M, Mousa SA. Adv. Palliative Med. 2009;8:53. [Google Scholar]
- 11b.Zagon IS, Verderame MF, McLaughlin PJ. Brain Res. Rev. 2002;38 doi: 10.1016/s0165-0173(01)00160-6. [DOI] [PubMed] [Google Scholar]
- 12.Zagon IS, Hytrek SD, Smith JP, McLaughlin PJ. Cancer Lett. 1997;112:167. doi: 10.1016/s0304-3835(96)04566-1. [DOI] [PubMed] [Google Scholar]
- 13a.Plotnikoff NP, Faith RE, Murgo AJ, Herberman RB, Good RA. Clin. Immun. Immunother. 1997;82:93. doi: 10.1006/clin.1996.4287. [DOI] [PubMed] [Google Scholar]
- 13b.Li W, Chen W, Herberman RB, Plotnikoff NP, Youkilis G, Griffin N, Wang E, Lu C, Shan F. Cancer Lett. 2014;344 doi: 10.1016/j.canlet.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 13c.Donahue RN, McLaughlin PJ, Zagon IS. J. Cancer Ther. 2011;2 [Google Scholar]
- 13d.Ibegbu AO, Mullaney I, Fyfe L, MacBean D. Br. J. Pharmacol. Toxicol. 2011;2 [Google Scholar]
- 14.Zagon IS, Jaglowski JR, Verderame MF, Smith JP, Leure-Dupree AE, McLaughlin PJ. Cancer Chemother. Pharmacol. 2005;56:510. doi: 10.1007/s00280-005-1028-x. [DOI] [PubMed] [Google Scholar]
- 15a.Zagon IS, Donahue RN, Bonneu RH, McLaughlin PJ. Immunobiology. 2011;216:173. doi: 10.1016/j.imbio.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 15b.Zunich KM, Kirkpatrick CH. J. Clin. Immunol. 1988;8 doi: 10.1007/BF00917896. [DOI] [PubMed] [Google Scholar]
- 16.McLaughlin PJ, Zagon IS. Biochem. Pharmacol. 2012;84:746. doi: 10.1016/j.bcp.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 17.Rosenzweig SA. Biochem. Pharmacol. 2012;83:1041. doi: 10.1016/j.bcp.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerber HP, Ferrara N. Cancer Res. 2005;65:671. [PubMed] [Google Scholar]
- 19.van Tamelen EE, Curphey TJ. Tetrahedron Lett. 1962;3:121. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary